Abstract
We investigated whether long-term administration of exogenous corticosterone (CST) or vehicle as daily treatment induces changes in rat behavior and in gene expression of the rat brain insulin signaling pathway and the formation of tau protein. Two groups of male adult rats received daily subcutaneous injections of 26.8 mg/kg CST (CST stress group) or vehicle-sesame oil (injection stress group) for 60 days while the third group was taken as untreated controls (n = 8 each). Body weight and plasma CST were measured and psychometric investigations were conducted using a rat holeboard test system before and after the treatment. Gene expression analyzes were performed by RT-PCR in cerebral cortical tissue for insulin genes 1 and 2, insulin receptor (IR), insulin degrading enzyme (IDE), and tau protein. Daily injections of CST for 60 days induced a significant, 2-fold increase in rat plasma CST concentrations in comparison to untreated controls. Significantly reduced behavioral abilities in CST-treated rats were associated with reduced gene expression of insulin 1 ( − 20%), IDE ( − 23%), and IR ( − 26%), indicating an insulin-resistant brain state, followed by increased tau protein (+28%) gene expression. In summary, chronic CST administration affects gene expression in the brain IR signaling cascade and increases tau gene expression, which is associated with reductions in cognition capacity in rats.
Introduction
It is well known that steroid hormones influence different functions of the nervous system (Melcangi and Panzica Citation2003). Sustained high glucocorticoid (GC) levels may be associated with the progression of CNS diseases such as stroke, depression, and sporadic Alzheimer's disease (sAD; Stratakis and Chrousos Citation1995; O'Brien et al. Citation1996; Plotsky et al. Citation1998; Murialdo et al. Citation2000; Dai et al. Citation2004; Marklund et al. Citation2004). Additionally, high levels of circulating GCs can also adversely affect cognition and reduce memory capacity (Newcomer et al. Citation1994; Young et al. Citation1999; Vanitallie Citation2002; Wolf Citation2003). In line with that, increased basal activity of the hypothalamic–pituitary–adrenal (HPA) axis associated with a higher concentration of cortisol has been found in neurodegenerative diseases such as sAD, (Holsboer and Barden Citation1996). Additionally, a polymorphism in the gene coding for 11β-hydroxysteroid dehydrogenase 1, which reduces cortisone to the active hormone cortisol, is associated with a 6-fold increased risk for a predominating late-onset, sporadic type of AD (sAD) (De Quervain et al. Citation2004). Furthermore, a growing body of evidence shows that the basal tonus of the HPA axis also increases with aging, promoting hypercortisolemia (Lupien et al. Citation2005), which is of interest as regards aging being recognized as a risk factor for sAD (McDowell Citation2001).
Hypercortisolemia has been shown to compromise the function of the insulin/insulin receptor (IR) system (Landfield et al. Citation2007). Interestingly, recent data indicate that sAD is associated with brain insulin deficiency and an insulin-resistant brain state (Hoyer Citation2004; Salkovic-Petrisic et al. Citation2006, Grünblatt et al. Citation2007) and that low insulin levels found post mortem in the brains of patients with sAD could contribute to sAD neuropathology (Steen et al. Citation2005). This is unsurprising since brain insulin has been found to have an important role in learning and memory functions (Park Citation2001; Zhao et al. Citation2004). An interaction between insulin/IR and GCs has been suggested based on reports of cortisol acting as the inhibitor of IR tyrosine kinase activity, which consequently causes downstream IR signaling dysfunction (Giorgino et al. Citation1993). Namely, GCs influence the insulin signaling cascade at the level of IR, whereby phosphorylation of the tyrosine residue is dysregulated and resistance thus induced (Giorgino et al. Citation1993), and GCs also impair the translocation of insulin-sensitive glucose transporter 4 (GLUT4; Piroli et al. Citation2007). Another study shows that corticosterone (CST) is also an important mediator in diabetes-induced cognitive deficit and impaired hippocampal synaptic plasticity (Stranahan et al. Citation2008). Although the exact mechanisms underlying these effects are still unknown in detail, they may be related to brain IR signaling downstream of the phosphatidyl-inositol-3 kinase pathway. Here, glycogen synthase kinase-3 beta isoform may regulate the tau protein phosphorylation/dephosphorylation balance (Hoyer Citation2004; Salkovic-Petrisic et al. Citation2006), which, if disturbed, represents one of the neuropathological hallmarks in AD (Blennow et al. Citation2006).
Bearing in mind that GC and insulin interact at the molecular (IR signaling inhibition) and functional (physiological antagonism, e.g., catabolism and hyperglycemia vs. anabolism and hypoglycemia, respectively) levels, an imbalance in insulin–cortisol activity may play an important role in the pathogenesis of sAD (Landfield et al. Citation2007).
Thus, we hypothesize that chronic administration of exogenous GCs as one element of the stress circuit may decrease activity of the brain insulin/IR system. As a result and in line with the physiological role of GCs and insulin in learning and memory, this may impair cognitive functions and thus pave the way for sAD to develop.
Materials and methods
Animals
Adult (12-month-old) male Wistar rats (n = 24, breeder: Thomae, Biberach, Germany, mean body weight 576 ± 64 g) were housed in individual cages in a temperature-controlled animal room (22.0 ± 0.5°C) with a reversed 12 h:12 h light/dark cycle (lights on at 7 p.m.). Psychometric experiments were conducted in normal lighting during the dark part of the cycle after 2 weeks of adaptation. Free access to food and water was given throughout the experimental period from day 1 until day 57 and on day 60 after testing. All rats underwent psychometric testing (Lannert and Hoyer Citation1998; Plaschke et al. Citation1999) with handling three times within 5 days before the injection period started. During the habituation (5 days), training (3 days), and testing period (1 day), food was restricted (5 g/day) to enhance the rats' motivation to perform the holeboard tests. After the first handling and training period, rats were divided into three subgroups: controls (without any treatment and handling, stress-free group), vehicle-sesame oil placebo group (injection stress group with daily injection of placebo), and exogenous CST group (daily injection of CST). Psychometric testing in the second period was performed in a shortened form before the end of the experiments at days 58–60 after the first injection (see below).
Over the entire experimental period body weight was measured regularly in all rats.
At the beginning of the injections and before the rats were killed, blood samples were taken from the tail vein between 1 and 2 p.m. under 1.5 vol.% halothane anesthesia and nitrous oxide/oxygen (70:30). At the end of the experiments, the rats were decapitated under anesthesia with 2.5 vol.% halothane and nitrous oxide/oxygen (70:30). Brains were rapidly removed, frozen in isopentane for 10 min, and stored at − 80°C until analysis.
The right and left hemispheres of the cerebral cortex were divided along the midline in a cryostat (Brandel, Darmstadt, Germany) at − 20°C and stored at − 80°C until analysis.
Exogenous CST administration
To induce high plasma concentrations of CST, rats received subcutaneous (s.c.) injections of either CST (Sigma-Aldrich, Steinheim, Germany) at 26.8 mg/kg (Stein-Behrens et al. Citation1994) body weight (equivalent to 10 mg/day, in 1 ml sesame oil), or vehicle (1 ml sesame oil alone) daily at 9 a.m. over a period of 60 days. We chose the respective dose of CST in accordance with references cited herein and with our previous experiments (Plaschke et al. Citation2006). Based on the studies of Woolley et al. (Citation1990) and Coburn-Litvak et al. (2003), this dose results in peak plasma CST concentrations comparable to stress levels, which then fall to non-stress levels within 24 h (Haugher et al. Citation1987). For daily injections, the rats were anesthetized with 1.5 vol.% halothane (O2: N2O = 30:70) using a vaporizer for a short period (1–2 min). As previously published, a significant effect of daily short halothane anesthesia on CST and on the HPA axis can be essentially excluded (Karuri et al. Citation1998; De Haan et al. Citation2002). The control group did not receive daily injections or halothane anesthesia.
Psychometric test parameters
To measure memory and cognition capacities and rat behavior, the psychometric holeboard memory test was performed according to Lannert and Hoyer (Citation1998) and Plaschke et al. (Citation1999). Briefly, habituation (7 days), training (5 days), and retesting (2 days) of memory measurements were performed in a defined holeboard box before daily injections were started. A square closed-field area (70 × 70 × 40 cm) contained 16 holes on the flat in a 4 × 4 array. Each hole contained a metal cup (3.5 cm in diameter, 3 cm deep) which had a perforated bottom. Each hole supplied with food pellets was covered by a false bottom (a metal cup, 3 cm deep) to mask potential odor cues emanating from the ‘reward’ in the baited holes. Thus, rats were unable to distinguish between baited and unbaited holes by olfactory stimuli.
On one side of the wall, a starting box was attached and separated from the testing area by means of a guillotine door which could be operated from a distance. For habituation, the rats were placed into the starting box and allowed to enter the testing area to explore the holeboard in which all 16 holes were baited with 50 mg of food pellet (Altromin, standard no. 1320, Lage, Germany). A trial started when the door was opened and ended when the rat had dipped into all 16 holes. Even if the rat could not find all food pellets, the trial ended after 10 min. After a habituation process, the rats were trained to search in 4 out of 16 baited holes in a fixed order. The trial was terminated when the rat had found all food pellets or when 5 min had elapsed, whatever occurred first. Two training trials were performed each day. For (re)testing values, the rats were tested by the same procedure as during training, but different combinations of food holes were selected to avoid the possibility of habituation as experienced for the baited set of food holes during the training period. Working memory ratio [(number of food rewarded visits/number of visits and revisits to the baited set of holes) × 100] and reference memory [(number of visits and revisits to the baited set of holes/number of visits and revisits to all holes) × 100] were determined at the beginning and after the end of the experimental period and were calculated according to van der Staay et al. (Citation1990). In addition, the latency time (time from the start of the experiment until the first food-rewarded hole was visited) and the whole run time (time to fulfil the task) were determined as behavioral determinants of locomotor activity.
All behavioral tests in the second psychometric testing period (days 58–60) were carried out at a minimum of 3 h after giving the daily injections in the early afternoon, at about 1 p.m. Immediately after the last psychometric test, food and water were given before blood samples were taken about 1 h later (see below). In detail, on days 58 and 59 rats were trained and on day 60 the retesting was done. Longitudinal changes in behavioral parameters were evaluated by comparing data before injections (T1) with data after 60 days of chronic injections (T2). Additionally, differences between the experimental groups were analyzed at T2.
CST determination
Rat blood samples were drawn from the tail vein in heparinized vials under 1.5 vol.% halothane anesthesia and nitrous oxide/oxygen (70:30) after 2 p.m. To determine CST concentrations, the blood was rapidly centrifuged at 4000 rpm for 10 min at 4°C, the supernatant separated and stored at − 80°C until CST analysis.
Plasma CST concentration was measured with a specific radioimmunoassay (RIA) after extraction according to a method published previously (Vecsei Citation1979; Vollmayr et al. Citation2001). Briefly, 10 μl of plasma was supplemented with 100 μl of 5% aqueous ethanol, tritium-labeled CST (to determine individual loss), and the mixture was extracted with 1 ml of cyclohexane/dichloromethane (2:1, v/v). The organic extract was separated, evaporated to dryness, dissolved in 1 ml of 5% aqueous ethanol, and quantified with a specific RIA. Intra-assay variation was 12.4%, inter-assay variation 14.3%. Each result was corrected for individually determined procedural loss. The antisera used for the RIA were raised in this laboratory and have been extensively characterized, especially for cross-reactivity with potentially interfering endo- and exogenous steroids.
Quantitative real-time RT-PCR
Total RNA extraction
Total RNA was isolated separately for each rat cerebral cortex sample by using the RNeasy midi kit (Qiagen GmbH, Hilden, Germany). The original protocol (Qiagen GmbH) was changed by adding an additional step in order to obtain more pure DNA-free total RNA. The total RNA on the column was pretreated with DNase I (Qiagen GmbH) and the original protocol was continued in order to isolate total RNA from the tissue.
Rt-pcr
Before gene expression profiling with real time RT-PCR, the total mRNA from each sample was reversely transcribed with random hexamer and oligo–dT primers using the iScript™ cDNA Synthesis Kit (BioRad Labaratories, Munich, Germany). Transcribed cDNA was then employed to analyze the gene expression profiles of IR (Qiagen, Cat. No. QT00198968), insulin-1 (Qiagen, Cat. No. QT00373303), insulin-2 (Qiagen, Cat. No. QT00177380), insulin degrading enzyme (IDE; Qiagen, Cat. No. QT 00191464), and tau (Qiagen, Cat. No. QT00174797). The house-keeping genes used to normalize the gene expression profile in the cortex of rats treated with CST were: actin (ACTB Qiagen, Cat. No. QT00193473), glyceraldehyde-3-phosphate dehydrogenase (GAPDH Qiagen, Cat. No. QT00199633), beta-2-microglobulin (B2m Qiagen, Cat. No. QT00176295), and ubiquitin (UBC Qiagen, Cat. No. QT00372596). All the house-keeping genes were tested for stability with the geNorm program (Vandesompele et al. Citation2002), and a normalization factor from the four genes together was also calculated according to the geNorm program. This normalization factor was than applied to normalize gene expression for all genes of interest.
For the RT-PCR, the iCycler iQ system (BioRad Co., Hercules, CA, USA) was used as described previously (Svaren et al. Citation2000). Transcribed cDNA from each sample was mixed with gene-specific primer (200 nmol/l final concentration) and the QuantiTect SYBR Green PCR Kit (Qiagen GmbH). Real-time PCR was subjected to PCR amplification, starting with one cycle at 95°C for 15 min and continuing with 34–45 cycles at 94°C for 15 s, and for annealing and to detecting specific fluorescent color at 55°C for 30 s and extension at 76°C for 30 s. At the end of each amplification reaction the melting curve was determined to ensure that all samples had a similar melting temperature. The PCR reactions were run in triplicate. The comparative threshold cycle (CT) method was used to quantify the amplified transcripts and then analyzed with BioRad iCycler iQ system program. Standard cDNAs were isolated from agarose gel using the MiniEluet™ gel extraction kit (Qiagen Inc) or the Bio Rad Freeze N' Squeeze Kit. For each amplification product, standard curves were generated from the 10-fold diluted pooled cDNA amplicons to determine efficiency and for quantification purposes.
Statistics
Psychometric testing and plasma CST measurements
Results from the physiological and blood parameters and from holeboard tests are expressed as mean ± standard deviation (SD). Differences within or between the normally distributed data were analyzed by analysis of variance (ANOVA) for multiple measurements using SPSS 15.0 version followed by post hoc Tukey test. A value of p < 0.05 was considered statistically significant.
Gene expression determination
Differences in gene expression in the cerebral cortex were compared between the groups using the Kruskal–Wallis ANOVA median test, followed by Mann–Whitney U-test. A value of p < 0.05 was considered statistically significant for both tests (Stat View 5.0. software; SAS Institute Inc. Cary, NC, USA). Data are expressed as mean ± SEM.
Results
Plasma CST concentrations and rat body weight
Sixty days of daily CST injection resulted in an approximately 2-fold increase (p = 0.02) in plasma CST concentrations. In the placebo group receiving vehicle on a daily basis, plasma CST concentrations nonsignificantly increased (p = 0.059), perhaps indicating that the daily injection procedure per se had evoked a small hormonal stress response. However, the differences between vehicle-treated placebo and basal concentrations did not reach statistical significance ().
Table I. Rats' body weight and plasma CST levels.
The body weight of rats after 60 days of daily CST administration was significantly reduced. Despite this body weight reduction, the general condition of these rats was not markedly impaired. Significant changes in rat body weight in untreated controls and vehicle-treated placebo rats were not observed ().
Psychometric parameters
Daily CST injection caused different changes in behavior after 60 days. In relation to locomotor activity, CST-injected rats took more time to fulfill the psychometric task. In detail, the total running time for the food search was significantly increased about 2.5-fold in comparison to the untreated controls, from 99 ± 33 to 248 ± 47 s after CST administration (p = 0.009). In addition, the latency time increased (p = 0.025) from 3.9 ± 3.0 to 12.9 ± 7.3 s after 60 days of daily exogenous CST administration.
Consistent with a proposed moderate stress response to daily injection, vehicle-treated placebo rats also demonstrated a significant increase in total run time from 99 ± 33 to 262 ± 49 s (p = 0.015), while the latency time increase in vehicle-treated rats from 3.9 ± 3.0 to 10.4 ± 3.9 s (p = 0.08) was not statistically significant, in comparison to the untreated controls.
The changes observed in working and reference memories are illustrated in . As can be seen from this figure, working and reference memories were significantly (p < 0.05) reduced by nearly 50% in the rats receiving CST as compared with vehicle-treated placebo and control groups.
Figure 1 Working memory [(number of food rewarded visits/number of visits and revisits to the baited set of holes) × 100] and reference memory [(number of visits and revisits to the baited set of holes/number of visits and revisits to all holes) × 100] ratios were determined at the beginning and after the end of the CST-treatment and were calculated according to van der Staay et al. (Citation1990). T1, before daily injections (n = 24), T2, after 60 days of daily injections (n = 8 per group). CST, corticosterone treatment. Data are expressed as mean ± SD. Significant differences (p < 0.05): *between CST-/placebo-treated and control groups (p = 0.008); #between the CST-group at T2 as compared to T1 (p = 0.018).
![Figure 1 Working memory [(number of food rewarded visits/number of visits and revisits to the baited set of holes) × 100] and reference memory [(number of visits and revisits to the baited set of holes/number of visits and revisits to all holes) × 100] ratios were determined at the beginning and after the end of the CST-treatment and were calculated according to van der Staay et al. (Citation1990). T1, before daily injections (n = 24), T2, after 60 days of daily injections (n = 8 per group). CST, corticosterone treatment. Data are expressed as mean ± SD. Significant differences (p < 0.05): *between CST-/placebo-treated and control groups (p = 0.008); #between the CST-group at T2 as compared to T1 (p = 0.018).](/cms/asset/8aaf4c2a-1e51-4f20-8a93-420e91926be9/ists_a_408210_f0001_b.gif)
Rt-pcr
The effect of daily injections of CST for 60 days on the gene expression profile of IR, insulin 1, insulin 2, IDE, and tau protein mRNA in the rat cortex is shown in . IR mRNA expression was significantly down-regulated in CST-treated rats by 25.5% as compared to the untreated (stress-free) controls (p < 0.0014). However, no significant difference was found between the vehicle-treated placebo (control with injection procedure-induced stress) and rats receiving CST, while a significant difference was found between the untreated (stress-free) control and the vehicle-treated placebo group (25.5%, p = 0.034). Insulin 2 mRNA expression remained unchanged among all groups studied while CST administration caused a significant decrease of 57.5% (p < 0.01) in the expression of insulin 1 mRNA. A significant decrease in IDE mRNA expression was observed in rats receiving CST (20.6%, p = 0.02) in comparison to the vehicle-treated placebo group, and also a decreasing tendency in comparison to the untreated controls. However, due to a high intra-group variability in the untreated controls, this did not reach statistical significance. In both untreated (stress free) and vehicle-treated controls (with moderate stress) no changes in the expression of IDE mRNA were observed after 60 days in comparison to the values on day 1.
Figure 2 Normalized mRNA expression (means and SEM) for IR, insulin (ins) 1, insulin 2, and IDE in the cortex of CST-treated rats, vehicle-treated placebo rats (injection-stress group) and untreated control (stress-free group). Kruskal–Wallis ANOVA test, *p < 0.05, n = 8 per group. p(IR) = 0.025, p(Ins1) = 0.032, p(IDE) = 0.05.
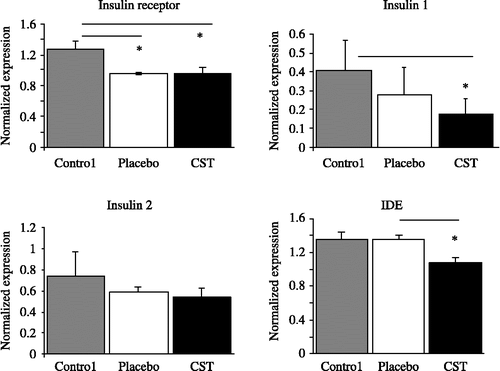
Figure 3 Normalized total mRNA expression (means and SEM) for tau protein in rat cortex after 60 days of CST treatment as compared to vehicle-treated placebo rats (injection-stress group) and untreated control. Kruskal–Wallis ANOVA test *p < 0.05, n = 8 per group, p = 0.034.
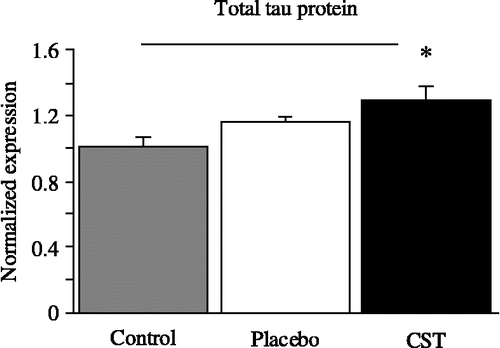
After CST administration, an increase of 28% (p < 0.019) in tau protein mRNA expression was found as compared to the untreated controls (), whereas the vehicle-treated placebo group did not show significant differences either from untreated controls or rats receiving CST.
Discussion
A chronically maintained increase in plasma CST concentration induced by the daily injection of CST was accompanied by a loss in body weight, which is an expected physiological response. In terms of behavior, both working and reference memory declined. At the molecular level, insulin 1, IR, and IDE mRNA levels were reduced in the rat cerebral cortex whereas tau protein mRNA was upregulated as compared to the untreated control group. To our knowledge this is the first time that such reduced expression has been demonstrated in this rat model.
Because CST was dissolved in sesame oil, a placebo-treated group was comprised of the rats that received daily injections of sesame oil. This procedure did not significantly increase plasma CST concentrations and did not cause behavioral disturbances. However, a significant decrease in IR gene expression was observed compared to controls, suggesting that stress due to the injection procedure per se and handling of the animals may have a likewise stressful effect (Horner et al. Citation1991). Thus, in order to consider the specific role of GCs, some nonspecific stress effects had to be taken into account. However, the changes seen in IR gene expression following placebo injections may indicate that stress, regardless of its duration, impairs the IR signaling cascade from the very beginning, while no abnormalities were found in the gene expression of insulin and IDE.
Normal secretion of GCs shows a daily rhythm, with peak concentrations in the late afternoon (De Leon et al. Citation1988). This daily rhythm can be disturbed as the result of increased basal levels of cortisol, e.g., as found in sAD patients (De Leon et al. Citation1988). Chronic elevation of cortisol may disturb negative feedback regulation in the HPA axis. We know from previous studies that a marked atrophy of the adrenal glands develops after chronic CST administration; the weight of the adrenal glands was significantly reduced by about 50% compared to the respective control and placebo groups. Published histopathological reports showed that atrophy mainly affected the zona fasciculata (Plaschke et al. Citation2006). In addition, chronic CST may lead to a functional disruption of the insulin signaling cascade at the level of IR tyrosine kinase activation (Giorgino et al. Citation1993; Hoyer and Froelich Citation2006).
In the present study, the chronically elevated plasma CST concentrations downregulated the gene expression of insulin 1, IR, and IDE mRNAs. Together with the inhibition of the IR tyrosine kinase activity, the existence of an exogenous CST-induced insulin-resistant brain state may be assumed (Salkovic-Petrisic and Hoyer Citation2007), causing different downstream abnormalities in oxidative/energy metabolism (Hoyer and Lannert Citation2008). Two out of several other disturbances may be of particular significance. The reduced capacity of the glycolytic chain (Plaschke and Hoyer Citation1993) forms the basis for the decrease in serum concentrations of the glycolytic compound fructose-6-phosphate, which represents the source for the generation of UDP-N-acetylglucosamine that is needed for Glc-NA cyclation of tau protein, thus inhibiting its hyperphosphorylation (Hart Citation1997). The reduction in fructose-6-phosphate levels may thus pave the way for tau protein hyperphosphorylation. Additionally, the ATP-dependent protein kinases PKerk36 and PKerk40 also control normal tau protein phosphorylation (Roder and Ingram Citation1991). It may be assumed that the reduction in available ATP after chronic CST administration disturbs this process (Hoyer and Lannert Citation2008) and favors hyperphosphorylation of tau protein. In the present study, expression of tau protein mRNA was elevated. It is tempting to assume that this abnormality is the first step of a process that lasts for a longer time and leads to the formation of hyperphosphorylated tau protein and beta-amyloid (Grünblatt et al. Citation2007).
Interestingly, triple transgenic mice that received dexamethasone injections for 7 days showed pathologically increased levels of beta-amyloid and tau protein (Green et al. Citation2006). In conjunction with increased beta-amyloid concentrations, the downregulation of IDE mRNA expression after CST administration may also be of significance. IDE degrades beta-amyloid at an elevated km of 2.0 (Pérez et al. Citation2000). Reduced IDE protein concentrations, which can be expected when IDE gene expression is downregulated, may cause beta-amyloid to accumulate; in nonhuman primates (Macacanemestrina) a reduction in both IDE mRNA and protein was observed after exposure to high-dose hydrocortisone acetate (Kulstad et al. Citation2005).
There is evidence that a significant linear correlation exists between the concentration of cerebral energy-rich phosphates and memory capacities in rats (Plaschke et al. Citation1999). In addition to the decline in available energy, some additional processes and parameters may play a role in the generation of CST-induced memory disturbances.
As for exogenous CST administration, most studies were performed on rat hippocampal tissue, reporting dendritic remodeling/atrophy (McEwen Citation1999), up-regulated expression of both NR2A and NR2B subunit mRNA of glutamate NMDA receptors (Weiland et al. Citation1997), and down-regulation of 5HT1A mRNA in the dentate gyrus (Meijer and de Kloet Citation1998). In addition to a cognitive decline, suppressed glucose reuptake in the temporal lobe was also observed following a bolus injection of cortisol (De Kloet et al. Citation1998). Furthermore, GCs have been found to inhibit glucose transport by approximately 15–30% in both primary and secondary hippocampal astrocyte cultures (Virgin et al. Citation1991). Cognitive decline found in humans treated with GCs and in individuals with Cushing syndrome suggests that stress levels of GCs do indeed affect cognition (De Kloet et al. Citation2006). Although we did not measure insulin in rat plasma and brain, it should be noted that an activated insulin/IR signaling pathway increases glucose reuptake in the brain (Hoyer Citation2004) and has a beneficial effect on learning and memory functions (De Leon et al. Citation1988; Hoyer and Froelich Citation2006). In addition, we could conclude that the missing link between the high GC levels and the cognitive decline that are both found in sAD might be caused by alterations in the brain insulin/IR system. A group of authors recently found that hippocampal insulin pathway genes were down-regulated in aged, cognitively impaired rats, suggesting that insulin/IR may be decreased as the result of increased GC action associated with the aging process (Landfield et al. Citation2007). We showed here for the first time that insulin 1 and IDE as well as IR gene expression were also significantly disturbed in the cerebral cortex of 12-month-old rats, a cerebral area with high GC receptor immunoreactivity (Morimoto et al. Citation1996).
In summary, the data presented here showed that a chronically elevated plasma concentration of CST has significant effects on the brain insulin system in rat cerebral cortex and leads to an increase in tau protein mRNA. Therefore, exogenous GC-mediated changes can be regarded as a major risk factor, inducing brain insulin signaling disturbances and thus generating abnormalities in brain metabolism and behavior indicative of sAD.
Ethics. The experimental protocol was approved by the appropriate review committee of the Medical Faculty of the University of Heidelberg, Germany, and complied with the guidelines of the responsible government agency. All animal experiments were carried out in accordance with the European Communities Council Directives of 24 November 1986 (86/609/EEC) or the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80 23, revised 1978).
Acknowledgements
This work was supported in part by the Croatian Ministry of Science, Sports and Education (108-1080003-0020), and DAAD.
We want to thank PD Dr Sabina Lewicka from the Department of Pharmacology, University of Heidelberg, Germany, for her support in corticosterone determination.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Blennow K, de Leon MJ, Zetterberg H. 2006. Alzheimer's disease: Seminar. Lancet. 368:387–403.
- Dai J, Buijs R, Swaab D. 2004. Glucocorticoid hormone (cortisol) affects axonal transport in human cortex neurons but shows resistance in Alzheimer's disease. Br J Pharmacol. 143:606–610.
- De Haan M, van Herck H, Tolboom JB, Beynen AC, Remie R. 2002. Endocrine stress response in jugular-vein cannulated rats upon multiple exposure to either diethyl-ether, halothane/O2/N2O or sham anaesthesia. Lab Anim. 36:105–114.
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. 1998. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 19:269–301.
- De Kloet CS, Vermetten E, Geuze E, Kavelaars A, Heijnen CJ, Westenberg HG. 2006. Assessment of HPA-axis function in posttraumatic stress disorder: Pharmacological and non-pharmacological challenge tests, a review. J Psychiatr Res. 40:550–567.
- De Leon MJ, McRae T, Tsai JR, George AE, Marcus DL, Freedman M, Wolf AP, McEwen B. 1988. Abnormal cortisol response in Alzheimer's disease linked to hippocampal atrophy. Lancet. 2:391–392.
- De Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A. 2004. Glucocorticoid-related genetic susceptibility for Alzheimer's disease. Hum Mol Genet. 13:47–52.
- Giorgino F, Almahfouz A, Goodyear LJ, Smith RJ. 1993. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo. J Clin Invest. 91:2020–2030.
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. 2006. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J Neurosci. 26:9047–9056.
- Grünblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S. 2007. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem. 101:757–770.
- Hart GW. 1997. Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu Rev Biochem. 66:315–335.
- Haugher RL, Millan MA, Catt KJ, Aguilera G. 1987. Differential regulation of brain and pituitary corticotropin-releasing factor receptors by corticosterone. Endocrinology. 120:1527–1533.
- Holsboer F, Barden N. 1996. Antidepressants and hypothalamic–pituitary–adrenocortical regulation. Endocr Rev. 17:187–205.
- Horner CH, O'Regan M, Arbuthnott E. 1991. Neural plasticity of the hippocampal (CA1) pyramidal cell—Quantitative changes in spine density following handling and injection for drug testing. J Anat. 174:229–238.
- Hoyer S. 2004. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 490:115–125.
- Hoyer S, Froelich L. 2006. Chapter 11. In: Sun MK, editors. Research progress in Alzheimer's disease and dementia. New York, NY: Nova Science.
- Hoyer S, Lannert H. 2008. Long-term effects of corticosterone on behavior, oxidative and energy metabolism of parietotemporal cerebral cortex and hippocampus. J Neural Transm. 115:1241–1249.
- Karuri AR, Engelking LR, Kumar MS. 1998. Effects of halothane and methoxyflurane on the hypothalamic–pituitary–adrenal axis in rat. Brain Res Bull. 47:205–209.
- Kulstad JJ, McMillan PJ, Leverenz JB, Cook DG, Green PS, Peskind ER, Wilkinson CW, Farris W, Mehta PD, Craft S. 2005. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurol. 64:139–146.
- Landfield PW, Blalock EM, Chen KC, Porter MM. 2007. A new glucocorticoid hypothesis of brain aging: Implications for Alzheimer's disease. Curr Alzheimer Res. 4:205–212.
- Lannert H, Hoyer S. 1998. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci. 112:1199–1208.
- Lupien SJ, Fiocco A, Wan N, Maheu F, Lord C, Schramek T, Tu MT. 2005. Stress hormones and human memory function across the lifespan. Psychneuroendocrinology. 30:225–242.
- Marklund N, Peltonen M, Nilsson TK, Olsson T. 2004. Low and high circulating cortisol levels predict mortality and cognitive dysfunction early after stroke. J Intern Med. 256:15–21.
- McDowell I. 2001. Alzheimer's disease: Insights from epidemiology. Aging. 13:143–162.
- McEwen BS. 1999. Stress and hippocampal plasticity. Annu Rev Neurosci. 22:105–122.
- Meijer OC, de Kloet ER. 1998. Corticosterone and serotonergic neurotransmission in the hippocampus: Functional implications of central corticosteroid receptor diversity. Crit Rev Neurobiol. 12:1–20.
- Melcangi RC, Panzica G. 2003. Steroids and the nervous system. Ann NY Acad Sci. 1007:1–5.
- Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. 1996. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: An immunohistochemical and in situ hybridization study. Neurosci Res. 26:235–269.
- Murialdo G, Nobili F, Rollero A, Gianelli MV, Copello F, Rodriguez G, Polleri A. 2000. Hippocampal perfusion and pituitary–adrenal axis in Alzheimer's disease. Neuropsychobiology. 42:51–57.
- Newcomer JW, Craft S, Hershey T, Askins K, Bardgett ME. 1994. Glucocorticoid-induced impairment in declarative memory performance in adult humans. J Neurosci. 14:2047–2053.
- O'Brien JT, Ames D, Schweitzer I, Colman P, Desmond P, Tress B. 1996. Clinical and magnetic resonance imaging correlates of hypothalamic–pituitary–adrenal axis function in depression and Alzheimer's disease. Br J Psychiatry. 168:679–687.
- Park CR. 2001. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev. 25:311–323.
- Pérez A, Morelli L, Cresto JC, Castaño EM. 2000. Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res. 25:247–255.
- Piroli GG, Grillo CA, Reznikov LR, Adams S, McEwen BS, Charron MJ, Reagan LP. 2007. Corticosterone impairs insulin-stimulated translocation of GLUT4 in the rat hippocampus. Neuroendocrinology. 85:71–80.
- Plaschke K, Hoyer S. 1993. Action of the diabetogenic drug streptozotocin on glycolytic and glycogenolytic metabolism in adult rat brain cortex and hippocampus. Int J Dev Neurosci. 11:477–483.
- Plaschke K, Yun SW, Martin E, Hoyer S, Bardenheuer HJ. 1999. Interrelation between cerebral energy metabolism and behaviour in a rat model of permanent brain vessel occlusion. Brain Res. 830:320–329.
- Plaschke K, Feindt J, Djuric Z, Heiland S, Autschbach F, Lewicka S, Martin E, Bardenheuer HJ, Nawroth PP, Bierhaus A. 2006. Chronic corticosterone-induced deterioration in rat behaviour is not paralleled by changes in hippocampal NF-kappaB-activation. Stress. 9:97–106.
- Plotsky PM, Owens MJ, Nemeroff CB. 1998. Psychoneuroendocrinology of depression. Hypothalamic–pituitary–adrenal axis. Psychiatr Clin North Am. 21:293–307.
- Roder HM, Ingram VM. 1991. Two novel kinases phosphorylate tau and the KSP site of heavy neurofilament subunits in high stoichiometric ratios. J Neurosci. 11:3325–3343.
- Salkovic-Petrisic M, Hoyer S. 2007. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: An experimental approach. J Neural Transm Suppl. 72:217–233.
- Salkovic-Petrisic M, Tribl F, Schmidt M, Hoyer S, Riederer P. 2006. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J Neurochem. 96:1005–1015.
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. 2005. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease-is this type 3 diabetes?. J Alzheimers Dis. 7:63–80.
- Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. 1994. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 14:5373–5380.
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. 2008. Diabetes impairs hippocampal function through glucocorticoid-mediated effect on new and mature neurons. Nat Neurosci. 11:309–317.
- Stratakis CA, Chrousos GP. 1995. Neuroendocrinology and pathophysiology of the stress system. Ann NY Acad Sci. 771:1–18.
- Svaren J, Ehrig T, Abdulkadir SA, Ehrengruber MU, Watson MA, Milbrandt J. 2000. EGR1 target genes in prostate carcinoma cells identified by microarray analysis. J Biol Chem. 275:38524–38531.
- van der Staay FJ, van Nies J, Raaijmakers W. 1990. The effects of aging in rats on working and reference memory performance in a spatial holeboard discrimination task. Behav Neural Biol. 53:356–370.
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034.
- Vanitallie TB. 2002. Stress: A risk factor for serious illness. Metabolism. 51:40–45.
- Vecsei P. 1979. Glucocorticoids: Cortisol, cortisone, corticosterone, compound S, and their metabolites. In: Jaffe BM, Behrmann HR, editors. Methods of hormone radioimmunoassays. New York, NY: Academic Press. 767–792.
- Virgin CEJr, Ha TP, Packan DR, Tombaugh GC, Yang SH, Horner HC, Sapolsky RM. 1991. Glucocorticoids inhibit glucose transport and glutamate uptake in hippocampal astrocytes: Implications for glucocorticoid neurotoxicity. J Neurochem. 57:1422–1428.
- Vollmayr B, Faust H, Lewicka S, Henn FA. 2001. Brain-derived-neurotrophic-factor (BDNF) stress response in rats bred for learned helplessness. Mol Psychiatry. 6:471–474.
- Weiland NG, Orikasa C, Hayashi S, McEwen BS. 1997. Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol. 388:603–612.
- Wolf OT. 2003. HPA axis and memory. Best Pract Res Clin Endocrinol Metab. 17:287–299.
- Woolley CS, Gould E, McEwen BS. 1990. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 531:225–231.
- Young AH, Sahakian BJ, Robbins TW, Cowen PJ. 1999. The effects of chronic administration of hydrocortisone on cognitive function in normal male volunteers. Psychopharmacology (Berl). 145:260–266.
- Zhao WQ, Chen H, Quon MJ, Alkon DL. 2004. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 490:71–81.