Abstract
There is evidence that increased release of corticotropin-releasing factor (CRF) in the central nucleus of the amygdala (CeA) contributes to stress responsivity during cocaine withdrawal (WD). Recent studies suggest that tissue plasminogen activator (tPA) in the CeA is a downstream effector protein for CRF after acute “binge” cocaine administration. The purpose of this study was to determine if tPA modulates cocaine WD-induced stress responsivity. Wild-type (WT) and tPA-deficient (tPA − / − ) mice were subjected to chronic (14 days) “binge” cocaine (45 mg/kg per day) or its acute (1 day) WD. Extracellular tPA activity, CRF mRNA levels, and plasma corticosterone (CORT) levels were measured in tPA − / − and WT mice. Extracellular tPA activity was reduced by 50% in the CeA and medial amygdala of WT mice after chronic cocaine and returned to basal levels after acute WD. Unlike WT mice, tPA − / − mice did not display elevated amygdalar CRF mRNA levels during cocaine WD. In comparison to WT mice, tPA − / − mice showed a blunted plasma CORT response during acute WD. These results demonstrate that tPA activity in the amygdala (Amy) is altered by chronic cocaine exposure, and further suggest an involvement of tPA in modulating amygdalar CRF stress responsive system and hypothalamic–pituitary–adrenal axis in response to acute cocaine WD.
Keywords::
Introduction
It has been established that stress modulates the effects of drugs of abuse on the acquisition of drug self-administration, locomotor activity, and reinstatement of drug self-administration after extinction [reviewed in Piazza and Le Moal (Citation1997) and CitationKreek and Koob (1998)]. A growing body of evidence suggests that increased release of corticotropin-releasing factor (CRF) in the central nucleus of the amygdala (CeA) contributes to the anxiogenic and stress-like consequences of withdrawal (WD) common to all drugs of abuse (reviewed in CitationWeiss et al. (2001)]. Drug WD-induced anxiety and depression are considered to be important motivational factors for relapse to drug-seeking behavior (Basso et al. Citation1999; Smith and Aston-Jones Citation2008), and understanding the molecular underpinnings of this phenomenon could lead to better therapeutic strategies to combat addiction. A single dose of cocaine or acute (but not chronic) “binge” cocaine has been reported to induce CRF release from the rat CeA (Richter et al. Citation1995), and then an increase in CRF mRNA in the amygdala (Amy; Zhou et al. Citation1996). An increase in the levels of extracellular CRF peptide and mRNA (Richter and Weiss Citation1999; Zhou et al. Citation2003a; Rudoy et al. Citation2009) and a decrease in CRF1 receptor binding (Ambrosio et al. Citation1997) in the Amy are found in the rat undergoing WD from chronic cocaine exposure. The level of CRF-like immunoreactivity in the Amy is decreased during acute cocaine WD (Zorrilla et al. Citation2001). Furthermore, an immunoneutralization study implicates amygdalar CRF in the mediation of anxiogenic effects during acute cocaine WD (Sarnyai et al. Citation1995). These findings indicate that the interaction between CRF and its specific receptor may represent an important step in the neurobiological processes of anxiety associated with acute cocaine WD.
Cocaine and its acute WD stimulate the hypothalamic–pituitary–adrenal (HPA) axis in animal models of addiction (Moldow and Fischman Citation1987; Rivier and Vale Citation1987; Borowsky and Kuhn Citation1991; Zhou et al. Citation1996; Sarnyai et al. Citation1998; Mantsch et al. Citation2007). Although chronic “binge” cocaine treatment significantly enhances plasma corticosterone (CORT) levels, this effect was significantly blunted in comparison to the effects of acute cocaine on the HPA axis, indicating tolerance of HPA activity to chronic “binge” cocaine (Zhou et al. Citation1996). There is also evidence that cocaine abuse disrupts HPA axis function in humans. Cocaine abuse increases plasma adrenocorticotropic hormone (ACTH), beta-endorphin, and cortisol levels (Mendelson et al. Citation1992; Baumann et al. Citation1995). The effects of a challenge dose of cocaine or stress on ACTH secretion, however, are significantly lower in cocaine-dependent men than in occasional cocaine users or normal subjects (Mendelson et al. Citation1998; Back et al. Citation2010), showing tolerance as found in the animal model after chronic “binge” cocaine (Zhou et al. Citation1996). In an earlier study, higher basal plasma ACTH and cortisol levels were observed in cocaine addicts, 1 day after the cessation of cocaine use (Vescovi et al. Citation1992). In contrast, other clinical studies have reported that after a brief period of abstinence, basal and CRF-stimulated ACTH and cortisol levels in cocaine-dependent patients do not differ from or are even lower than those of healthy subjects (Mendelson et al. Citation1992, Citation1998; Baumann et al. Citation1995; Jacobsen et al. Citation2001; Aouizerate et al. Citation2006). Furthermore, recent human studies have found that psychological stressors elevate cocaine craving and HPA activity, and stress-induced HPA responses predict subsequent cocaine relapse (Sinha et al. Citation2003, Citation2006; Brady et al. Citation2009).
The serine protease tissue plasminogen activator (tPA) has recently been implicated in modulating the biochemical and behavioral effects of various drugs of abuse (Yamada et al. Citation2005). tPA is highly expressed in brain regions implicated in drug-induced anxiety and reward, including the medial amygdala (MeA), CeA, and nucleus accumbens (NAc), where it is thought to regulate a cascade of proteolytic events involved in neurite outgrowth and long-term potentiation (Samson and Medcalf Citation2006). tPA is released from neurons upon excitation (Gualandris et al. Citation1996; Baranes et al. Citation1998) and facilitates synaptic plasticity and the formation of new synapses (Centonze et al. Citation2002; Samson and Medcalf Citation2006). tPA can also directly modulate synaptic plasticity events by regulating N-methyl d-aspartate receptor function (Nicole et al. Citation2001; Norris and Strickland Citation2007).
Recent studies implicate a role for tPA in cocaine addiction. Cocaine induces tPA mRNA expression in the prefrontal cortex and NAc (Hashimoto et al. Citation1998; Bahi and Dreyer Citation2008). Acute “binge” cocaine enhances tPA activity in the central and medial nucleus of the Amy by a mechanism that requires activation of CRF1 receptors (Maiya et al. Citation2009). Further, cocaine-induced neuronal signaling and immediate early gene expression in the Amy and NAc, as well as its rewarding effects (as measured by conditioned place preference) are attenuated in tPA-deficient (tPA − / − ) mice (Maiya et al. Citation2009). It has also been shown that tPA is involved in modulation of cocaine-induced locomotor stimulation and sensitization (Ripley et al. Citation1999; Bahi and Dreyer Citation2008; Maiya et al. Citation2009). tPA has been shown to modulate morphine-induced dopamine release in the NAc and is required for morphine reward and locomotor sensitization (Nagai et al. Citation2004; Yan et al. Citation2007). In studies of stress, tPA also regulates stress-induced anxiety-like behavior (Pawlak et al. Citation2003; Norris and Strickland Citation2007). Finally, tPA activity in the Amy is regulated by CRF, a critical component of the behavioral response to stress (Matys et al. Citation2004).
Since the stress responsive CRF system is critically involved in cocaine WD, we examined the role of tPA in regulating the CRF system in the Amy and HPA axis after chronic cocaine and its acute WD in both wild-type (WT) and tPA − / − mice. First, our results show that chronic “binge” cocaine and 1-day acute WD dynamically regulate tPA activity, possibly by altering the expression of its cognate inhibitor plasminogen activator inhibitor-1 (PAI-1) in the Amy. Second, cocaine WD-induced increase in amygdalar CRF mRNA expression as well as activation of the HPA axis is impaired in tPA − / − mice. Together, these results implicate a novel role for tPA in modulating the stress-responsive CRF system and HPA axis in response to acute cocaine WD.
Methods
WT and tPA-deficient (tPA−/−) mice and “binge” cocaine administration
tPA − / − mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) where the mutation was backcrossed onto a C57B6 background for eight generations. These mice were then bred at The Rockefeller University's animal facility, where they were backcrossed to C57B6 mice (WT) for an additional 10 generations. Genome-wide microsatellite marker analysis (performed by Charles River Laboratories, Wimington, MA, USA) revealed that tPA − / − mice bred in The Rockefeller University facility are 99.1% identical to and hence congenic to C57B6 mice. WT mice were obtained from The Jackson Laboratory at 6 weeks-of-age and housed individually in a stress-minimized animal facility for 2–4 weeks before use and were provided with free access to food and water. In the present study, male WT and tPA − / − mice were used and housed in the same room under identical conditions. We followed the Principles of Laboratory Animal Care (NIH publication no 86–23, 1996). The specific protocol was approved by Animal Care and Use Committee of Rockefeller University. During all experimental procedures, the number of mice and potential suffering by treatments were minimized.
After adaptation to a standard 12-h light/dark cycle (lights on from 9:00 h to 21:00 h) for 3–4 weeks, animals were randomly assigned to treatment groups. On the experimental day, animals received three intraperitoneal (i.p.) injections of cocaine 45 mg/kg per day (3 × 15 mg/kg per injection) or an equal volume of saline in their home cages in a “binge” pattern: three times daily at 1-h intervals (9:30, 10:30, and 11:30). The cocaine dose and dosing schedule were chosen to mimic the pattern often observed in human cocaine abusers which is repeated administration over several hours in the evening, and with relation to the circadian rhythm of rest and activity (thus, in the early light time in the rat; Koob and Kreek Citation2007). Experiments were performed as follows: (1) Chronic cocaine: WT mice (n = 7–8/group) or tPA − / − mice (n = 6/group) received chronic “binge” cocaine or saline for 14 days and were then sacrificed 30 min after the last injection on day 14 and (2) 1-day WD: WT mice (n = 8/group) or tPA − / − mice (n = 5–6/group) received chronic “binge” cocaine or saline for 14 days and were then sacrificed 24 h after the last injection for 1-day WD. Each experiment had four groups: (1) WT or (2) tPA − / − mice treated with saline; (3) WT or (4) tPA − / − mice treated with “binge” cocaine. Mice were decapitated after brief exposure to CO2 (within 15 s), and specific brain and pituitary regions and plasma were collected for subsequent mRNA and hormonal analysis.
In situ zymography
In situ zymographies were performed as described before (Pawlak et al. Citation2003; Maiya et al. Citation2009). Briefly after chronic cocaine or 1-day WD, WT mice (n = 3–4/group) were anesthetized and perfused transcardially with ice-cold phosphate-buffered saline. Brains were removed, frozen, and embedded in optimal cutting temperature compound (Tissue-Tek, Sakura USA, Torrance, CA, USA). Zymographies were performed on coronal brain sections (20 μm) collected on silane-coated slides. Sections were overlaid with an agarose mixture containing 2.5% commercial instant nonfat milk and 25 μg/ml purified human plasminogen. The zymograms were developed in a humidified chamber at 37°C for 2.5 h and photographed in grayscale under dark field illumination. Lytic zone areas were measured using IMAGEJ 1.30 software. The Zymograms used for comparisons were processed and photographed at the same time.
Pai-1 Elisa
The ELISA was performed using a PAI-1 total antigen kit (Molecular Innovations, Southfield, MI, USA), and PAI-1 standards were prepared according to manufacturer's instructions. After chronic cocaine or 1-day WD, WT mice (n = 3–4/group) were sacrificed and the Amy was dissected bilaterally and homogenized, and the resulting homogenate was used to determine PAI-1 levels. ELISA was performed with duplicate samples using 200 μg protein per well according to manufacturer's instructions.
Brain dissection and preparation of RNA extracts
Each mouse brain was removed from the skull and placed in a chilled mouse brain matrix (ASI Instruments, Houston, TX, USA). Coronal slices containing the brain regions of interest were removed from the matrix and dissected on a chilled petri dish under a dissecting microscope, according to the mouse brain stereotaxic coordinates. Three regions were dissected as follows: the Amy, hypothalamus (Hypo), and anterior pituitary (AP). Then, the tissue was homogenized in guanidinium thiocyanate buffer and extracted with acidic phenol and chloroform (Chomczynski and Sacchi Citation1987). After the final ethanol precipitation step, each extract was re-suspended in diethylpyrocarbonate (DEPC)-treated H2O and stored at − 80°C.
Solution hybridization ribonuclease (RNase) protection assays
A 760-bp fragment of mouse CRF cDNA, a 2500-bp fragment of mouse CRF1 receptor cDNA, and a 476-bp fragment from the rat proopiomelanocortin (POMC) cDNA were subcloned into the polylinker region of the pSP64, pSP65, or pcDNA plasmid (Promega, Madison, WI, USA). The 33P-labeled cRNA antisense probes and unlabeled cRNA sense standards were synthesized using SP6 or T7 transcription system. A denaturing agarose gel containing 1.0 M formaldehyde showed that a single full-length transcript had been synthesized from each plasmid. As described previously (Zhou et al. Citation1996), RNA extracts were re-suspended in 30 μl buffer that contained 150 to 300 K cpm of a probe in 2 × TESS (10 mM Tris–HCl, pH 7.4; 10 mM EDTA; 0.3 M NaCl; 0.5% SDS) and hybridized overnight at 75°C. For RNase treatment, 250 μl of buffer containing 0.3 M NaCl, 5 mM EDTA, 10 mM Tris–HCl (pH 7.5), 40 μg/ml RNase A (Worthington Biochemicals, Freehold, NJ, USA), and 2 μg/ml RNase T1 (Calbiochem, San Diego, CA, USA) was added, and each sample was incubated at 30°C for 1 h. Trichloroacetic acid (TCA) precipitation was effected by 1 ml of 5% TCA and 0.75% sodium pyrophosphate. Precipitates were collected onto a filter by using a cell harvester (Brandel, Gaithersburg, MD, USA) and were measured in a scintillation counter with liquid scintillant (Beckman Instruments, Palo Alto, CA, USA). The procedure to measure mRNA levels involved comparison of values obtained from brain extracts to those obtained for a set of calibration standards. The calibration standards had known amounts of an in vitro sense transcript (1.25–80 pg). A new standard curve was generated each time experimental samples were analyzed, and all extracts of a particular tissue were assayed for each mRNA as a group on a single day. Total cellular RNA concentrations were measured by hybridization to a 33P-labeled probe complementary to 18S rRNA. The calibration standards for this curve contained 2.5–40 ng of total RNA from mouse brain. Selected samples of mouse pituitaries were subjected to solution hybridization and RNase treatment followed by gel electrophoresis (see Zhou et al. Citation1996, for validation and details).
Plasma CORT radioimmunoassay
At the time of decapitation, blood from each mouse was collected, and plasma was separated using refrigerated centrifuges and then stored at − 80°C. CORT levels were assayed by using a mouse CORT 125I kit (MP Biomedicals, Costa Mesa, CA, USA). All CORT values were determined in duplicate in a single assay, and the intra-assay coefficient of variation was 2.2%.
Data analysis
Multiple group comparisons were analyzed by two-way analysis of variance (ANOVA). Within genotype (WT or tPA − / − ), group differences in tPA, PAI-I, and mRNA levels of each gene in each brain region and in plasma CORT levels were analyzed using two-way ANOVA for treatment (cocaine/saline) and for treatment day (14 days cocaine, 1-day WD), followed by Newman–Keuls post-hoc tests. In some cases, a Student's t-test was used as indicated. The accepted level of significance for all tests was p < 0.05. All statistical analyses were performed using Statistica (version 5.5, StatSoft, Inc., Tulsa, OK, USA).
Results
Effects of chronic “binge” cocaine exposure and its acute WD on tPA and PAI-1 levels in the Amy and hippocampus of WT mice
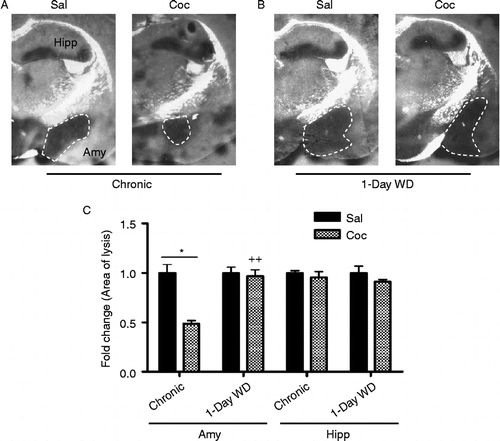
WT mice were injected with cocaine in chronic “binge” paradigm and were sacrificed 30 min or 24 h after the final cocaine injection. Extracellular tPA activity in the Amy and hippocampus (Hipp) was determined by in situ zymography. In the CeA and the MeA, tPA activity was decreased significantly by approximately 50% after chronic cocaine (). Two-way ANOVA showed a significant main effect of chronic cocaine treatment [F(1,10) = 5.64, p < 0.05]. Newman–Keuls post-hoc tests revealed that tPA activity was significantly decreased in the CeA and MeA of WT mice after 14 days chronic “binge” cocaine in comparison to that of saline-injected mice (p < 0.05). Significant differences were also found between chronic “binge” cocaine and acute 1-day WD groups (p < 0.01). After 1 day WD from chronic cocaine, however, amygdalar tPA activity in cocaine-injected animals was not different from that of saline-injected mice (). tPA activity in the Hipp remained unchanged at both time points (). These results suggest that chronic cocaine and its acute WD result in dynamic changes in tPA activity in the CeA and MeA, but not the Hipp.
Figure 1. Extracellular tPA activity in the Amy and Hipp of WT mice was measured by in situ zymography 30 min after 14 days of chronic “binge” pattern cocaine administration (Chronic Coc) and 1 day after 14 days of Chronic Coc as 1-day WD (A,B). tPA activity (indicated by dark lytic zones) in the CeA and MeA (A) was quantified by measuring the area of lysis outlined by dashed white line (C). Data in graph C are presented as mean ± SEM. tPA activity was significantly decreased in the CeA and MeA of WT mice 30 min after 14 days of chronic “binge” cocaine (Chronic Coc), compared to those of saline-injected mice (*p < 0.05). Significant differences are also found between Chronic Coc and 1-day WD groups (++p < 0.01) (n = 3–4/group). In the hippocampus, tPA activity remained unchanged at both time points examined (C).
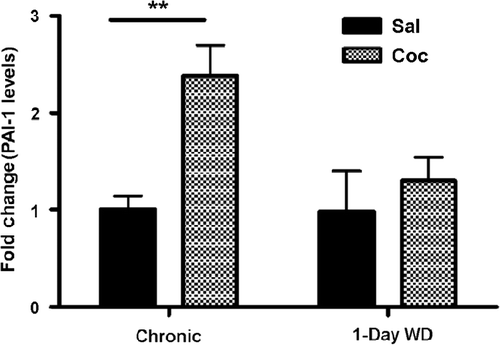
To explore the mechanism behind this dynamic regulation of tPA activity, we examined levels of PAI-1, an inhibitor of tPA activity, in the Amy. Chronic cocaine administration significantly increased PAI-1 levels in the Amy by two fold in comparison to saline administration (). Two-way ANOVA revealed a significant main effect of chronic cocaine treatment [F(1,14) = 9.10, p < 0.01]. Newman–Keuls post-hoc tests revealed that PAI-1 levels were significantly increased in cocaine-treated animals after 14 days chronic “binge” cocaine, compared to those of their saline-injected counterparts (p < 0.01). However, there was no statistical difference in PAI-1 levels between saline- and cocaine-injected WT mice during acute 1-day WD (). Together, our data suggest that cocaine-induced changes in PAI-1 may underlie alterations in tPA activity observed during chronic cocaine and acute WD.
Figure 2. PAI-1 levels in the Amy of WT mice were examined by ELISA 30 min after 14 days of chronic “binge” pattern cocaine administration (Chronic Coc) and 1-day after 14 days of Chronic Coc as 1-day WD. Data in the graph are presented as mean ± SEM. PAI-1 levels were significantly enhanced in cocaine-treated animals 30 min after 14 days of chronic “binge” cocaine (Chronic Coc), compared to those of their saline- injected counterparts (**p < 0.01) (n = 3–4/group).
tPA−/− mice display impaired amygdalar CRF mRNA response upon cocaine WD
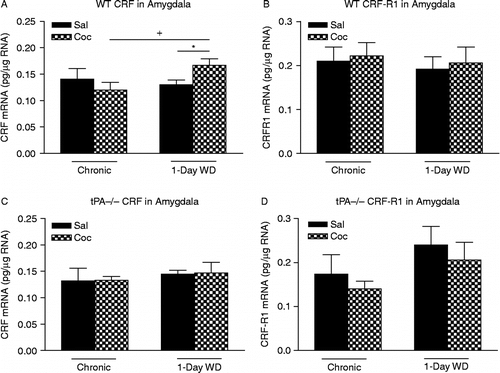
Acute WD from chronic cocaine exposure activates the stress responsive CRF system in rodents (reviewed in Weiss et al. Citation2001). Recent studies have also shown that acute “binge” cocaine enhances tPA activity in the Amy in a CRF/CRF1 receptor-dependent manner (Maiya et al. Citation2009). Hence, we examined CRF and CRF1 receptor mRNA levels in the Amy after chronic “binge” cocaine and its acute WD in both WT and tPA − / − mice. In the Amy of WT mice, two-way ANOVA revealed a significant effect of cocaine treatment × treatment day interaction [F(1,18) = 4.88, p < 0.05] for CRF mRNA levels. Newman–Keuls post-hoc tests revealed that there was a significant increase in CRF mRNA levels after acute 1-day WD, compared to those after 14 days chronic “binge” cocaine (p < 0.05), suggesting that acute cocaine WD enhanced CRF gene expression when compared to chronic cocaine exposure (). Consistent with previous studies carried out in rats (Zhou et al. Citation2003a), acute cocaine WD-induced increases in CRF mRNA levels in mice displayed a trend toward significance in comparison to their saline-injected counterparts (Student's t-test F(1,9) = 4.34, p = 0.06). In contrast, chronic cocaine and its acute WD did not alter CRF mRNA levels in the Amy of tPA − / − mice (). Since CRF mediates most of its biochemical and behavioral effects on anxiety through the CRF1 receptor, we examined CRF1 receptor mRNA levels in both WT and tPA − / − mice after chronic cocaine and acute WD. CRF1 receptor mRNA levels were unaltered in the Amy at either time points in WT mice (). Although two-way ANOVA revealed a significant effect of treatment day in tPA − / − mice [F(1,16) = 6.08, p < 0.05], Newman–Keuls post-hoc tests did not reveal any significant changes of CRF1 receptor mRNA levels at either time point ().
Figure 3. mRNA levels of corticotropin-releasing factor (CRF) (A,C) and CRF1 receptor (B,D) in the Amy of WT and tPA-deficient (tPA − / − ) mice were examined 30 min after 14 days of chronic “binge” pattern cocaine administration (Chronic Coc) and 1 day after 14 days of Chronic Coc as 1-day WD. Data in the graph are presented as mean ± SEM. (A) In the Amy of WT mice, significant differences are indicated: +p < 0.05 between 1-day WD vs. chronic cocaine (Chronic Coc) groups; *p = 0.06 between 1-day WD from cocaine (1-day WD Coc) and 1-day WD from saline (1-day WD Sal) groups. (B) No significant differences in CRF1 receptor mRNA levels in WT mice were observed at either of the time points examined. (C,D) No significant differences in mRNA levels of either CRF or CRF1 receptor in tPA − / − mice were observed at either of the time points examined (n = 4–6/group).
tPA−/− mice display impaired plasma CORT response upon cocaine WD
WD from chronic “binge” cocaine activates the HPA axis in the rat (Zhou et al. Citation2003a). Therefore, we measured plasma CORT levels (a marker of HPA axis activation) in both WT and tPA − / − mice.
In WT mice, two-way ANOVA showed a significant effect of cocaine treatment [F(1,27) = 26.3, p < 0.001] (). Newman–Keuls post-hoc tests further revealed that plasma CORT levels 30 min after the last injection on day 14 following chronic “binge” cocaine were significantly higher than those in saline control (p < 0.01). After 1-day WD, plasma CORT levels were also significantly higher than those in saline control (p < 0.01). Plasma CORT levels after 1-day acute WD were not different from those after 14 days chronic cocaine.
Figure 4. Plasma CORT levels in WT (A) and tPA-deficient (tPA − / − ) mice (B) were examined 30 min after 14 days of chronic “binge” pattern cocaine administration (Chronic Coc) and 1 day after 14 days of Chronic Coc (1-day WD). Data in the graph are presented as mean ± SEM. (A) In WT mice, significant differences are indicated: **p < 0.01 between chronic cocaine (Chronic Coc) vs. saline (Chronic Sal); ##p < 0.01 between 1-day acute WD (1-day WD) from cocaine vs. saline control (n = 5–8/group). (B) In tPA − / − mice, significant differences are indicated: *p < 0.05 between chronic cocaine (Chronic Coc) vs. saline (Chronic Sal).
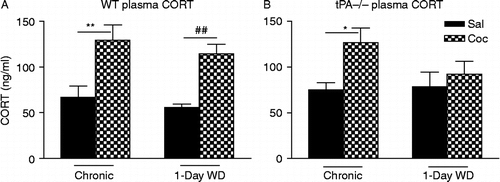
In tPA − / − mice, two-way ANOVA showed a significant effect of cocaine treatment [F(1,19) = 7.54, p < 0.05] (). Similar to WT mice, plasma CORT levels 30 min after the last injection on day 14 following chronic “binge” cocaine were significantly higher than those in saline control (Newman–Keuls post-hoc tests, p < 0.05). However, in contrast to WT mice, plasma CORT levels after 1-day acute WD were not higher than those in saline control. These results suggest that tPA − / − mice display impaired activation of the HPA axis during acute cocaine WD, and hence tPA may be necessary for this mechanism.
tPA−/− mice display decreased POMC mRNA levels in the AP during cocaine WD
To determine whether chronic “binge” cocaine or its WD induced changes in HPA activity is associated with changes in CRF, CRF1 receptor, and POMC mRNA expression in the Hypo or AP, we examined the above mRNA levels in both WT and tPA − / − mice. CRF and CRF1 receptor mRNA levels were unchanged in the Hypo or AP after either chronic cocaine or acute WD in both genotypes (). POMC, a precursor of ACTH and beta-endorphin, was not altered in the AP of WT mice. In tPA − / − mice, however, two-way ANOVA revealed a significant effect of cocaine treatment [F(1,21) = 4.36, p < 0.05]. Newman–Keuls post-hoc tests revealed that cocaine-injected tPA − / − mice undergoing acute WD showed significantly lower POMC mRNA levels in the AP compared to saline-injected tPA − / − mice ().
Table I. mRNA levels of CRF, CRF type 1 receptor (CRF-R1) or POMC (pg/μg RNA) in the Hypo or AP in the wildtype (WT) (A) and tPA-deficient (tPA − / − ) (B) mice were examined 30 min after 14 days of chronic “binge” pattern cocaine administration (Chronic Coc) or saline (Chronic Sal) and 1 day after 14 days of Chronic Coc or Chronic Sal as 1-day WD Coc or 1-day WD Sal.
Discussion
It has been recently found that acute (1 day) “binge” cocaine enhances tPA activity in the CeA and MeA of WT mice through a CRF1 receptor-mediated mechanism (Maiya et al. Citation2009). This increase in tPA activity by acute “binge” cocaine seems region-specific, because the alteration of tPA activity was not found in other brain regions of WT mice, including the NAc, frontal cortex, and Hipp (Maiya et al. Citation2009). The present study further investigated the effects of chronic “binge” cocaine and its acute WD on tPA activity. In sharp contrast to the acute effect, chronic “binge” cocaine dramatically decreased tPA activity by 50% in the CeA and MeA of WT mice in comparison to saline. tPA activity returned to basal levels 24 h after the final injection of cocaine. A two fold increase in amygdalar PAI-1, an inhibitor of tPA activity, was observed in WT mice after chronic “binge” cocaine, temporally coinciding with the decrease in the tPA activity. This increase in PAI-1 levels in the Amy was not observed in WT mice undergoing acute cocaine WD. These results suggest that chronic cocaine and acute WD can dynamically regulate tPA and PAI-1 activity in the Amy, and that the tPA/plasmin system may play a role in the behavioral effects of chronic cocaine and WD. For example, one could suggest that such cocaine-induced decrease in tPA activity is an adaptive change that may function to limit the activation of CRF mechanisms that contribute to WD-related HPA axis (see below).
It is hypothesized that drug WD-induced increases in amygdalar CRF activity play an important role in drug WD-related anxiety (Weiss et al. Citation2001). In fact, administration of CRF antagonists has been shown to reduce cocaine- and alcohol-induced anxiety-like behaviors (Basso et al. Citation1999; Valdez et al. Citation2002). In rats, acute cocaine WD stimulates CRF release and gene expression in the CeA (Richter and Weiss Citation1999; Zhou et al. Citation2003a; Rudoy et al. Citation2009). Acute morphine WD also increases extracellular levels of CRF peptide (Weiss et al. Citation2001) and CRF mRNA (McNally and Akil Citation2002; Befort et al. Citation2008) and decreases CRF1 receptor mRNA (Iredale et al. Citation2000) in the Amy of both rats and mice. An increase in extracellular levels of CRF in the CeA is also found in rats during acute WD from chronic ethanol (Merlo Pich et al. Citation1995; Olive et al. Citation2002) or cannabinoid (Rodriguez de Fonseca et al. Citation1997) exposure. The results from the present study demonstrate that chronic “binge” cocaine had no effect on CRF mRNA levels in the Amy of WT mice; however, acute WD from chronic “binge” cocaine significantly increased CRF mRNA levels in the Amy. This finding is consistent with most previously published reports (e.g., Zhou et al. Citation2003a; Rudoy et al. Citation2009).
Recent studies suggest that tPA is a downstream effector protein for CRF in the Amy. Central injections of CRF (Matys et al. Citation2004) as well as acute “binge” cocaine exposure (Maiya et al. Citation2009) increase tPA activity in the CeA and MeA in a CRF1 receptor-dependent manner. Hence, we expected amygdalar tPA activity after chronic “binge” cocaine and its acute WD to parallel the changes of CRF mRNA levels. Surprisingly, we did not observe a strong temporal association between the changes in CRF mRNA levels and tPA activity at these two time points. It is possible that a temporal connection exists between tPA activity and CRF peptide levels, rather than CRF mRNA levels. Alternatively, our results suggest that chronic “binge” cocaine-induced changes in tPA activity in WT mice might occur in a CRF-independent manner. In contrast to the WT mice, amygdalar CRF mRNA levels were unchanged during acute cocaine WD in tPA − / − mice. Although the mechanism is unclear, it is possible that tPA is involved in the alterations of CRF gene expression in cocaine WD.
We also compared HPA axis activation in WT and tPA − / − mice after chronic “binge” cocaine and its acute WD. Consistent with previous observations in rats (Zhou et al. Citation2003b), there was a significant elevation of plasma CORT levels in WT mice after acute WD from chronic “binge” cocaine administration. Another study found that rats trained to self-administer a moderate dose of cocaine (0.5 mg/kg per infusion) show an increase in plasma CORT levels after 1 day cessation from cocaine self-administration (Peltier et al. Citation2001). Our findings also agree with other observations of increased plasma CORT levels after acute cocaine WD (Borowsky and Kuhn Citation1991; Levy et al. Citation1992; Sarnyai et al. Citation1998; Carrasco et al. Citation2006; Cleck et al. Citation2008). Interestingly, plasma CORT levels were not elevated in tPA − / − mice undergoing WD, suggesting that cocaine WD induced-activation of the HPA axis is attenuated in these mice. This is not likely due to a general hyporesponsivity of the HPA axis in the tPA − / − mice as deficits were not observed after acute (1 day) cocaine-induced HPA activation in tPA − / − mice (Maiya et al. Citation2009). In fact, chronic (14 days) “binge” cocaine increased plasma CORT levels in both the WT (92%) and tPA − / − mice (71%) in comparison with saline, and the magnitude of CORT elevation did not differ significantly between the two genotypes in the present study. One possibility that arises from this result is that tPA − / − mice seem deficit in cocaine WD-induced HPA axis activation.
To determine whether the changes in HPA responsivity after chronic “binge” cocaine and during acute WD are associated with alterations in HPA gene expression, we measured mRNA levels of several components of the HPA axis: CRF mRNA levels in the Hypo, as well as CRF1 receptor and POMC mRNA levels in the AP. Consistent with our previous study in rats, we found that acute cocaine WD did not alter the above mRNA levels in WT mice. However, a significant decrease in pituitary POMC mRNA levels was observed after acute cocaine WD in tPA − / − mice. The mechanism involved in the interactions between POMC gene expression in the corticotropes and tPA during acute cocaine WD is not clear. However, since POMC is the precursor of ACTH and beta-endorphin, one would expect decreased ACTH biosynthesis and release from the AP, and a consequent impairment in pituitary–adrenal responses during cocaine WD in tPA − / − mice.
In summary, our results here indicate that chronic “binge” cocaine and its acute WD can dynamically modulate tPA activity in the Amy of WT mice. Although the biochemical measures in WT mice did not show strong temporal correlations between amygdalar tPA and CRF gene expression after chronic “binge” cocaine and during acute WD, our study in tPA − / − mice clearly suggests a potential novel role for tPA in modulating the stress responsive CRF system in the Amy and the HPA axis in response to acute cocaine WD. Since activation of the stress responsive CRF system during cocaine WD is thought to be involved in cocaine-seeking behavior in animals (Markou et al. Citation1998; Miczek et al. Citation2004; Koob and Kreek Citation2007; Koob Citation2008) and cocaine relapse in humans (Sinha et al. Citation2006; Back et al. Citation2010; Brady et al. Citation2009), one interesting hypothesis that stems from our study is that tPA may play a role in modulating cocaine-seeking behaviors. Therefore, it may be worthwhile to explore the usefulness of tPA inhibitors for the management of cocaine abuse. Our studies indicate that tPA may be an attractive therapeutic target for treating anxiety associated with cocaine addiction.
Acknowledgements
We greatly appreciate the use of Dr Bruce McEwen's facility. This work was funded by NlNDS Grant NS035704, NIAAA Grant AA014630 (to S.S.), and NIDA Research Center Grant DA-P60-05130 (to M.J.K.).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ambrosio E, Sharpe LG, Pilotte NS. 1997. Regional binding to corticotropin releasing factor receptors in brain of rats exposed to chronic cocaine and cocaine withdrawal. Synapse. 25:272–276.
- Aouizerate B, Ho A, Schluger JH, Perret G, Borg L, Le Moal M, Piazza PV, Kreek MJ. 2006. Glucocroticoid negative feedback in methadone-maintained former heroin addicts with ongoing cocaine dependence: Dose-response to dexamethasone suppression. Addict Biol. 11:84–96.
- Back SE, Hartwell K, Desantis SM, Saladin M, McRae-Clark AL, Price KL, Moran-Santa Maria MM, Baker NL, Spratt E, Kreek MJ, Brady KT. 2010. Reactivity to laboratory stress provocation predicts relapse to cocaine. Drug Alcohol Depend. 106:21–27.
- Bahi A, Dreyer JL. 2008. Overexpression of plasminogen activators in the nucleus accumbens enhances cocaine-, amphetamine- and morphine-induced reward and behavioral sensitization. Genes Brain Behav. 7:244–256.
- Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. 1998. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 21:813–825.
- Basso AM, Spina M, Rivier J, Vale W, Koob GF. 1999. Corticotropin-releasing factor antagonist attenuates the “anxiogenic-like” effect in the defensive burying paradigm but not in elevated plus-maze following chronic cocaine in rats. Psychopharmacology. 145:21–30.
- Baumann MH, Gendron TM, Becketts KM, Henningfield JE, Gorelick DA, Rothman RB. 1995. Effects of intravenous cocaine on plasma cortisol and prolactin in human cocaine abusers. Biol Psychiatry. 38:751–755.
- Befort K, Filliol D, Ghate A, Darcq E, Matifas A, Muller J, Lardenois A, Thibault C, Dembele D, Le Merrer J, Becker J, Poch O, Kieffer B. 2008. Mu-opioid receptor activation induces transcriptional plasticity in the central extended amygdala. Eur J Neurosci. 27:2973–2984.
- Borowsky B, Kuhn CM. 1991. Chronic cocaine administration sensitizes behavioral but not neuroendocrine responses. Brain Res. 543:301–306.
- Brady KT, McRae AL, Moran-Santa Maria MM, DeSantis SM, Simpson AN, Waldrop AE, Back SE, Kreek MJ. 2009. Response to corticotropin-releasing hormone infusion in cocaine-dependent individuals. Arch Gen Psychiatry. 66:422–430.
- Carrasco GA, Van de Kar LD, Sullivan NR, Landry M, Garcia F, Muma NA, Battaglia G. 2006. Cocaine-mediated supersensitivity of 5-HT2A receptors in hypothalamic paraventricular nucleus is a withdrawal-induced phenomenon. Neuroscience. 143:7–13.
- Centonze D, Napolitano M, Saulle E, Gubellini P, Picconi B, Martorana A, Pisani A, Gulino A, Bernardi G, Calabresi P. 2002. Tissue plasminogen activator is required for corticostriatal long-term potentiation. Eur J Neurosci. 16:713–721.
- Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 162:156–159.
- Cleck JN, Ecke LE, Blendy JA. 2008. Endocrine and gene expression changes following forced swim stress exposure during cocaine abstinence in mice. Psychopharmacology. 201:15–28.
- Gualandris A, Jones TE, Strickland S, Tsirka SE. 1996. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci. 16:2220–2225.
- Hashimoto T, Kajii Y, Nishikawa T. 1998. Psychotomimetic-induction of tissue plasminogen activator mRNA in corticostriatal neurons in rat brain. Eur J Neurosci. 10:3387–3399.
- Iredale PA, Alvaro JD, Lee Y, Terwilliger R, Chen YL, Duman RS. 2000. Role of corticotropin-releasing factor receptor-1 in opiate withdrawal. J Neurochem. 74:199–208.
- Jacobsen LK, Giedd JN, Kreek MJ, Gottschalk C, Kosten TR. 2001. Quantitative medial temporal lobe brain morphology and hypothalamic-pituitary-adrenal axis function in cocaine dependence: A preliminary report. Drug Alcohol Depend. 62:49–56.
- Koob GF. 2008. A role for brain stress systems in addiction. Neuron. 59:11–34.
- Koob GF, Kreek MJ. 2007. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 164:1149–1159.
- Kreek MJ, Koob GF. 1998. Drug dependence: Stress and dysregulation of brain reward pathways. Drug Alcohol Depend. 51:23–47.
- Levy AD, Li Q, Alvarez Sanz MC, Rittenhouse PA, Kerr JE, Van de Kar LD. 1992. Neuroendocrine responses to cocaine do not exhibit sensitization following repeated cocaine exposure. Life Sci. 51:887–897.
- Maiya R, Zhou Y, Norris E, Kreek MJ, Strickland S. 2009. Tissue plasminogen activator modulates the cellular and behavioral response to cocaine. Proc Natl Acad Sci USA. 106:1983–1988.
- Mantsch JR, Taves S, Khan T, Katz ES, Sajan T, Tang LC, Cullinan WE, Ziegler DR. 2007. Restraint-induced corticosterone secretion and hypothalamic CRH mRNA expression are augmented during acute withdrawal from chronic cocaine administration. Neurosci Lett. 415:269–273.
- Markou A, Kosten TR, Koob GF. 1998. Neurobiological similarities in depression and drug dependence: A self-medication hypothesis. Neuropsychopharmacology. 18:135–174.
- Matys T, Pawlak R, Matys E, Pavlides C, McEwen BS, Strickland S. 2004. Tissue plasminogen activator promotes the effects of corticotropin-releasing factor on the amygdala and anxiety-like behavior. Proc Natl Acad Sci USA. 101:16345–16350.
- McNally GP, Akil H. 2002. Role of corticotropin-releasing hormone in the amygdala and bed nucleus of the stria terminalis in the behavioral, pain modulatory and endocrine consequences of opiate withdrawal. Neuroscience. 112:605–617.
- Mendelson JH, Teoh SK, Mello NK, Ellingboe J, Rhoades E. 1992. Acute effects of cocaine on plasma adrenocorticotropic hormone, luteinizing hormone and prolactin levels in cocaine-dependent men. J Pharmacol Exp Ther. 263:505–509.
- Mendelson JH, Sholar M, Mello NK, Teoh SK, Sholar JW. 1998. Cocaine tolerance: Behavioral, cardiovascular, and neuroendocrine function in men. Neuropsychopharmacology. 18:263–271.
- Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. 1995. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 15:5439–5447.
- Miczek KA, Covington HE3rd, Nikulina EMJr, Hammer RP. 2004. Aggression and defeat: Persistent effects on cocaine self-administration and gene expression in peptidergic and aminergic mesocorticolimbic circuits. Neurosci Biobehav Rev. 27:787–802.
- Moldow RL, Fischman AJ. 1987. Cocaine induced secretion of ACTH, beta-endorphin, and corticosterone. Peptides. 8:819–822.
- Nagai T, Yamada K, Yoshimura M, Ishikawa K, Miyamoto Y, Hashimoto K, Noda Y, Nitta A, Nabeshima T. 2004. The tissue plasminogen activator-plasmin system participates in the rewarding effect of morphine by regulating dopamine release. Proc Natl Acad Sci USA. 101:3650–3655.
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. 2001. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 7:59–64.
- Norris EH, Strickland S. 2007. Modulation of NR2B-regulated contextual fear in the hippocampus by the tissue plasminogen activator system. Proc Natl Acad Sci USA. 104:13473–13478.
- Olive MF, Koenig HN, Nannini MA, Hodge CW. 2002. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 72:213–220.
- Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. 2003. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 6:168–174.
- Peltier RL, Guerin GF, Dorairaj N, Goeders NE. 2001. Effects of saline substitution on responding and plasma corticosterone in rats trained to self-administer different doses of cocaine. J Pharmacol Exp Ther. 299:114–120.
- Piazza PV, Le Moal M. 1997. Glucocorticoids as a biological substrate of reward: Physiological and pathophysiological implications. Brain Res Rev. 25:359–372.
- Richter RM, Weiss F. 1999. In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse. 32:254–261.
- Richter RM, Pich EM, Koob GF, Weiss F. 1995. Sensitization of cocaine-stimulated increase in extracellular levels of corticotropin-releasing factor from rat amygdala after repeated administration as determined by intracranial microdialysis. Neurosci Lett. 187:169–172.
- Ripley TL, Rocha BA, Oglesby MW, Stephens DN. 1999. Increased sensitivity to cocaine, and over-responding during cocaine self-administration in tPA knockout mice. Brain Res. 826:117–127.
- Rivier C, Vale W. 1987. Cocaine stimulates adrenocorticotropin (ACTH) secretion through a corticotropin-releasing factor (CRF)-mediated mechanism. Brain Res. 422:403–406.
- Rodriguez de Fonseca F, Carrera MR, Navarro M, Koob GF, Weiss F. 1997. Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science. 276:2050–2054.
- Rudoy CA, Reyes AR, Van Bockstaele EJ. 2009. Evidence for beta (1)-adrenergic receptor involvement in amygdalar corticotropin-releasing factor gene expression: Implications for cocaine withdrawal. Neuropsychopharmacology. 34:1135–1148.
- Samson AL, Medcalf RL. 2006. Tissue-type plasminogen activator: A multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 50:673–678.
- Sarnyai Z, Biro E, Gardi J, Vecsernyes M, Julesz J, Telegdy G. 1995. Brain corticotropin-releasing factor mediates ‘anxiety-like’ behavior induced by cocaine withdrawal in rats. Brain Res. 675:89–97.
- Sarnyai Z, Dhabhar FS, McEwen BS, Kreek MJ. 1998. Neuroendocrine-related effects of long-term, “binge” cocaine administration: Diminished individual differences in stress-induced corticosterone response. Neuroendocrinology. 68:334–344.
- Sinha R, Talih M, Malison R, Cooney N, Anderson GM, Kreek MJ. 2003. Hypothalamic-pituitary-adrenal axis and sympatho-adreno-medullary responses during stress-induced and drug cue-induced cocaine craving states. Psychopharmacology. 170:62–72.
- Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ. 2006. Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry. 63:324–331.
- Smith RJ, Aston-Jones G. 2008. Noradrenergic transmission in the extended amygdala: Role in increased drug-seeking and relapse during protracted drug abstinence. Brain Struct Funct. 213:43–61.
- Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, Koob GF. 2002. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: Regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 26:1494–1501.
- Vescovi PP, Coiro V, Volpi R, Passeri M. 1992. Diurnal variations in plasma ACTH, cortisol and beta-endorphin levels in cocaine addicts. Horm Res. 37:221–224.
- Weiss F, Ciccocioppo R, Parsons LH, Katner S, Liu X, Zorrilla EP, Valdez GR, Ben-Shahar O, Angeletti S, Richter RR. 2001. Compulsive drug-seeking behavior and relapse. Neuroadaptation, stress, and conditioning factors. Ann NY Acad Sci. 937:1–26.
- Yamada K, Nagai T, Nabeshima T. 2005. Drug dependence, synaptic plasticity, and tissue plasminogen activator. J Pharmacol Sci. 97:157–161.
- Yan Y, Yamada K, Mizoguchi H, Noda Y, Nagai T, Nitta A, Nabeshima T. 2007. Reinforcing effects of morphine are reduced in tissue plasminogen activator-knockout mice. Neuroscience. 146:50–59.
- Zhou Y, Spangler R, LaForge KS, Maggos CE, Ho A, Kreek MJ. 1996. Corticotropin-releasing factor and type 1 corticotropin-releasing factor receptor messenger RNAs in rat brain and pituitary during “binge”-pattern cocaine administration and chronic withdrawal. J Pharmacol Exp Ther. 279:351–358.
- Zhou Y, Spangler R, Ho A, Kreek MJ. Increased CRH mRNA levels in the rat amygdala during short-term withdrawal from chronic “binge” cocaine. Mol Brain Res. 2003a; 114:73–79.
- Zhou Y, Spangler R, Schlussman SD, Ho A, Kreek MJ. Alterations in hypothalamic-pituitary-adrenal axis activity and in levels of proopiomelanocortin and corticotropin-releasing hormone-receptor 1 mRNAs in the pituitary and hypothalamus of the rat during chronic “binge” cocaine and withdrawal. Brain Res. 2003b; 964:187–199.
- Zorrilla EP, Valdez GR, Weiss F. 2001. Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology. 158:374–381.