Abstract
There is considerable evidence that inhaled toxicants such as cigarette smoke can cause both irreversible changes to the genetic material (DNA mutations) and putatively reversible changes to the epigenetic landscape (changes in the DNA methylation and chromatin modification state). The diseases that are believed to involve genetic and epigenetic perturbations include lung cancer, chronic obstructive pulmonary disease (COPD), and cardiovascular disease (CVD), all of which are strongly linked epidemiologically to cigarette smoking. In this review, we highlight the significance of genomics and epigenomics in these major smoking-related diseases. We also summarize the in vitro and in vivo findings on the specific perturbations that smoke and its constituent compounds can inflict upon the genome, particularly on the pulmonary system. Finally, we review state-of-the-art genomics and new techniques such as high-throughput sequencing and genome-wide chromatin assays, rapidly evolving techniques which have allowed epigenetic changes to be characterized at the genome level. These techniques have the potential to significantly improve our understanding of the specific mechanisms by which exposure to environmental chemicals causes disease. Such mechanistic knowledge provides a variety of opportunities for enhanced product safety assessment and the discovery of novel therapeutic interventions.
| Abbreviations: | ||
| BALF, | = | bronchoalveolar lavage fluid |
| COPD, | = | chronic obstructive pulmonary disease |
| CVD, | = | cardiovascular disease |
| EGFR, | = | epidermal growth factor receptor |
| HDAC, | = | histone deacetylase |
| K-RAS, | = | Kirsten rat sarcoma viral oncogene homolog |
| LOH, | = | loss of heterozygosity |
| MI, | = | microsatellite instability |
| MLA, | = | mouse lymphoma assay |
| NNK, | = | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone |
| NSCLC, | = | non-small cell lung cancer |
| PBMC, | = | peripheral blood mononuclear cells |
| SCLC, | = | small cell lung cancer |
| TSNA, | = | tobacco specific N-nitrosamines |
| TSP, | = | Tumor Sequencing Project |
Table of Contents
Abstract 877
Keywords 877
Key abbreviations used 877
Genomic and epigenomic perturbations produced by cigarette smoke 878
Lung cancer 879
Chronic obstructive pulmonary disease 881
Cardiovascular disease 882
DNA methylation signatures 882
Next generation genomics and epigenomics platforms 883
Discussion 883
Conclusion 884
Declaration of interest 885
References 885
Genomic and epigenomic perturbations produced by cigarette smoke
Exposure to mutagens
In man, the process of detoxification of tobacco carcinogens has two phases. Phase I xenobiotic metabolizing enzymes transform oxidizable substrates into electrophiles, and phase II-related enzymes attack oxidized substrates via nucleophilic reactions, providing an efficient process for xenobiotic metabolism and excretion. Accordingly, the genes encoding the enzymes involved in xenobiotic metabolism are up-regulated in the bronchial epithelium of smokers (CitationSpira et al., 2004; CitationBeane et al., 2011) and in smoke-exposed rodent lung tissue (CitationGebel et al., 2004; CitationGebel et al., 2006). Some metabolites formed during the detoxification process can be highly reactive and may create covalent adducts at guanidine and adenine bases. Adducts are normally eliminated through the DNA excision repair pathway, but if an adduct is still present during replication, the DNA polymerase may bypass such an altered base leading to a mutation (CitationPfeifer et al., 2002; CitationHang, 2010).
The mutagenicity of tobacco smoke has been demonstrated in various assays including the mouse lymphoma assay (MLA) (CitationOECD, 1997). In a recent study, Guo et al. tested smoke condensate from 11 different cigarette brands and found that they all demonstrated dose-dependent mutagenic effects (CitationGuo et al., 2011). Moreover, the tobacco-specific N-nitrosamine (TSNA), nicotine-derived 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces lung tumors in all tested species (mice, rat, hamster and mink) regardless of the route of administration.
Some of these agents or their combinations may also induce genomic aberrations in smokers. Genetic lesions, such as loss-of-heterozygosity (LOH) and other chromosomal aberrations are found in dysplastic, hyperplastic but also even histologically normal bronchial epithelium in clinically cancer-free smokers (CitationFranklin et al., 1997; CitationMao et al., 1997; CitationWistuba et al., 1997; CitationNelson et al., 1998). There is a higher frequency of overall LOH in smoker lungs as compared to non-smokers, and among former smokers the LOH at 3p14 (containing the tumor suppressor gene FHIT) is less frequent than in active smokers.
Epigenomic changes induced by tobacco carcinogens
While most compounds that are present in cigarette smoke and in various other environmental contaminants are non-mutagenic, some apparent non-mutagenic compounds can interfere with gene expression by means of epigenetic mechanisms. There is ample evidence for smoking-induced epigenetic events in the clinical context. Hypermethylated promoters have been observed in clinically cancer-free and cancer-bearing smokers. These promoters can be detected in cell samples derived from sources as diverse as bronchoscopy, sputum, broncho-alveolar lavage fluid (BALF), alveolar macrophages, or lymphoblasts (CitationBelinsky et al., 1998; CitationPalmisano et al., 2000; CitationLamy et al., 2002; CitationKim et al., 2004; CitationRusso et al., 2005; CitationBelinsky et al., 2005; CitationBelinsky, 2005; CitationBelinsky et al., 2006; CitationKerr et al., 2007; CitationBaryshnikova et al., 2008; CitationMonick et al., 2012). Even though promoter hypermethylation can persist for many years after smoking cessation (CitationZöchbauer-Müller et al., 2003; CitationBhutani et al., 2008), current smokers in general, have a higher mean methylation index than former smokers, supporting the view that hypermethylation, unlike somatic mutations, is reversible (CitationYanagawa et al., 2011). While the methylation of Retinoic Acid Receptor Beta (RARB), p16, fragile histidine triad (FHIT), and RASSF1A (RAS association domain family 1A) correlate weakly with smoking status, the degree of methylation increases with intensity of smoking.
The smoking effect has even been detected in cell-free DNA present in plasma. When methylation of at least one of Kif1a, DCC, RARB, or NISCH was considered, methylation frequency was smoking-dependent; while none of the light- or non-smoker controls showed plasma DNA methylation, the cumulative smoking dose (pack-years) correlated well with the methylation frequency in cancer-free heavy smokers (CitationOstrow et al., 2010).
This frequent hypermethylation in smoker tissues has been explained by the high levels of DNA methyltransferase 1 (DNMT1) that correlated with smoking status in lung tumor samples (CitationLin et al., 2010). In vitro and murine in vivo experiments have further shown that NNK acts through Akt signaling and inhibits DNMT1 protein degradation. Subsequently, DNMT1 protein accumulation leads to increased tumor suppressor gene hypermethylation (CitationDamiani et al., 2008; CitationLin et al., 2010; CitationLiu et al., 2010).
In an attempt to develop biomarkers of exposure, recent work has identified regions of altered DNA methylation in the lungs of SENCAR (SENsitive to CARcinogens) mice exposed to a single dose of 7,12-dimethylbenz[α]anthracene (DMBA) with or without cigarette smoke (CitationPhillips and Goodman, 2009). The aberrant methylation was detected at very early time points, before any obvious lung histopathology. Based on their results, CitationPhillips and Goodman (2009) suggested that regions of altered DNA methylation could serve as both biomarkers of exposure and effect.
Numerous in vitro studies have attempted to reproduce smoke-induced epigenomic changes observed in the lung of a smoker (CitationMass and Wang, 1997; CitationLiu et al., 2007; CitationLiu et al., 2010). A recent study has shown that a gene-specific promoter methylation is induced in immortalized lung epithelial cells after prolonged (several months) exposure to cigarette smoke condensate (CitationLiu et al., 2010). Currently, it is not known whether the methylation is reversible and what would be the time frame for demethylation events after removal from smoke exposure. Reversible smoke effects on DNA methylation have been documented in cultured lung cancer cells. In A549 cells, the pro-metastatic oncogene synuclein-γ (SNCG) is silenced by CpG island (genomic region that contains a high frequency of CG-dimer) methylation. Cigarette smoke extract (CSE)-induced SNCG demethylation accompanied by gene overexpression in just 3 days of treatment. The demethylation was associated with a twofold decrease of DNMT3B mRNA. Withdrawal of the treatment resulted in the recovery of DNMT3B expression and concomitant re-establishment of SNCG CpG methylation (CitationLiu et al., 2007).
Lung cancer
The various forms of lung cancer display phenotypically diverse cell types and according to the clonal evolution model, cancer arises from a single cell, the extent of the cumulative genomic instability dictating tumor progression (CitationNowell, 1976). More recent evidence suggests that within a tumor, only a small population of cells has a self-renewing capacity making them able to drive malignant growth (CitationEramo et al., 2008). According to this cancer stem cell theory (reviewed in CitationRivera et al., 2011), either restricted progenitors or more differentiated lung cells could convert to cancer stem cells with self-renewing capacity. Such cell populations have already been identified for both small cell (SCLC) and non-small cell lung cancer (NSCLC) (CitationEramo et al., 2008).
Lung cancer development involves various genomic perturbations, such as point mutations, deletions, and gene amplifications. The short arm of chromosome 3 contains several candidate tumor suppressor genes, and deletions within this region are detected in nearly 100% of lung cancers (CitationZabarovsky et al., 2002). The appearance of activating mutations in dominant oncogenes, or inactivating mutations in recessive tumor suppressor genes, as well as chromosomal aberrations are generally regarded as late events in tumor development. However, in support of the field cancerization theory (CitationSteiling et al., 2008; CitationGazdar and Minna, 2009), some mutations have been described in non-malignant lung tissue adjacent to resected lung tumors (CitationNelson et al., 1998; CitationZabarovsky et al., 2002).
A recent study has confirmed that cigarette smoking is linked to specific copy number alterations in NSCLC. Analyses of early stage tumors showed that heavy smokers (>60 pack-years) have more copy number gains than light and non-smokers. These gains were found to be predominantly affecting oncogenes and genes associated with tumor growth. Interestingly, copy number losses located in intergenic regions were found in light and non-smoker tumors, a phenomenon not seen in tumors from heavy smokers (CitationHuang et al., 2011).
Even though adenocarcinoma is becoming the dominant lung cancer type in smokers today, it presents a variable, distinct subtype that is also found in non-smokers (CitationSubramanian and Govindan, 2010). However, lung tumors found in non-smokers are genetically different from those found in smokers (CitationSun et al., 2007). While mutations in the GTPase gene encoding KRAS and the tumor suppressor gene encoding p53 are more specific to smoking-related lung adenocarcinoma, the sequence encoding epidermal growth factor receptor (EGFR) kinase domain often becomes mutated very early in the course of smoking-independent lung adenocarcinoma development (CitationHerbst et al., 2008; CitationLantuéjoul et al., 2009; CitationDacic et al., 2010; CitationBroët et al., 2011). KRAS and EGFR mutations have been found to be mutually exclusive, and there was an inverse correlation between smoking status (number of pack-years and duration of smoking) and the frequency of EGFR mutations (CitationYanagawa et al., 2011). It has been proposed, supported by the positive correlation between smoke-free years and the occurrence of EGFR mutations, that tobacco smoking suppresses EGFR mutation (CitationGarinis et al., 2001; CitationDacic et al., 2010; CitationLee et al., 2010; CitationBroët et al., 2011; CitationYanagawa et al., 2011). An inverse correlation has been shown between the methylation of RASSF1A, and FHIT and runt-related transcription factor 3 (RUNX3) versus EGFR mutation (CitationYanagawa et al., 2011).
The gene encoding tumor suppressor p53 is commonly mutated in cancers, and contrary to the EGFR mutation, p53 mutations occur more frequently in smokers than in non-smokers (CitationRyberg et al., 1994; CitationGarinis et al., 2001; CitationPfeifer et al., 2002). Anna et al. showed that among lung cancer patients, prolonged duration of smoking increased the frequency of p53 mutations. While the mutation frequency was 14.3% in smokers of less than 20 years, half of those who had smoked for more than 20 years carried the mutation (CitationGarinis et al., 2001; CitationAnna et al., 2009). The occurrence of p53 mutations are not random and several ‘hot-spots’ were observed within the region that encodes the DNA-binding domain of p53 (CitationDenissenko et al., 1996). In the subsequent study, it was demonstrated that the locations of the mutation “hot-spots” correlated with the methylation status of the proximal CpG dinucleotides (CitationDenissenko et al., 1997).
The Tumor Sequencing Project (TSP) studied 188 lung adenocarcinomas and identified a group of somatic mutations that might affect the key pathways in adenocarcinoma development. The mutated genes that were found included formerly identified tumor suppressors, such as p53, CDKN2A, and STK11 and oncogenes such as KRAS, EGFR, and NRAS. Additionally, several additional mutated genes were discovered, including putative tumor suppressor genes, such as ATM, NF1, RB1, and APC as well as putative proto-oncogenes, such as ERBB4, KDR, FGFR4 and NTRK. Many of these genes were not only mutated, but the identified loci had also undergone copy number and/or gene expression changes. It was concluded that even though lung adenocarcinomas are highly heterogeneous, the pathways that are affected are most likely the same among the majority of the different subtypes. The dominant pathways that were affected included those of MAPK, p53, Wnt, cell cycle and mTOR (CitationDing et al., 2008).
In cancer biology, the most relevant, or at least the best understood, epigenetic mechanism refers to DNA hypermethylation of specific loci (CpG islands) in the promoter region of known and presumptive tumor suppressor genes which is a common hallmark of human tumors in general (CitationEsteller, 2007), and of lung cancer in particular (CitationZochbauer-Muller et al., 2002; CitationBelinsky, 2005). Importantly, there is now strong evidence that aberrant promoter methylation and the consequent silencing of tumor suppressor genes is a critical step during tumor development, and is frequently detected in so-called precursor (benign) lesions (CitationBelinsky et al., 1998). Thus, aberrant promoter methylation is regarded as an early event in the multistep process of carcinogenesis.
Genes ‘targeted’ by promoter hypermethylation encompass a broad functional spectrum including some involved in cell cycle arrest such as p16 (major target) and p53, DNA repair such as MGMT (O6-methylguanine-DNA methyltransferase), or growth arrest such as RASSF1A. Importantly, although it requires at least 5 years of follow-up, there is a strong indication that clinically cancer-free chronic smokers with hypermethylated promoters in actual and presumptive tumor suppressor genes are at an elevated risk of developing lung cancer (CitationBelinsky et al., 1998; CitationPalmisano et al., 2000; CitationJarmalaite et al., 2003; CitationBaryshnikova et al., 2008; CitationFeng et al., 2008). The frequency of p16 hypermethylation, which is considered one of the prime aberrations in lung cancer development, increases with disease progression (CitationBelinsky et al., 1998; CitationBelinsky, 2005). In fact, p16 is frequently fully inactivated in lung cancer via allele-loss and inactivation of the remaining allele by homozygous deletion, hypermethylation, or mutation (CitationCairns et al., 1995; CitationMerlo et al., 1995; CitationRusin et al., 1996). The consequence of p16 inactivation is an increased cyclin D expression, which leads to persistent hyper-phosphorylation of retinoblastoma protein (Rb) with resultant evasion of cell cycle arrest (CitationGautschi et al., 2007).
Though ‘targeted’ hypermethylation is more recognized in cancer development, global hypo-methylation is almost always linked to malignancy; in normal human cells, more than twice as many cytosine residues are methylated compared to tumor cell DNA. Nearly half of the genome consists of highly repeated DNA sequences that are accountable for the global hypomethylation, a common trait in cancers, which can result in increased possibility of chromosomal rearrangements (CitationDunn, 2003). Activation of proto-oncogenes, including c-myc, c-fos, and c-Ha-ras, which upon demethylation become abnormally expressed, set off uncontrolled cellular proliferation. Finally, it is possible that inserted pro-viral sequences (silenced by methylation) become demethylated, which can lead to reactivation and increased infectivity (CitationDunn, 2003; CitationSchär and Fritsch, 2011).
Tobacco smoke-induced methylation and resulting loss of gene expression has been detected in cell lines derived from cigarette smoke- or tobacco carcinogen-induced mouse lung tumors that showed reduced expression of death associated protein (DAP) kinase. The loss of expression correlated with the promoter methylation state and could be reversed by treatment with 5-aza-2′-deoxycytidine, a compound that inhibits DNA methyl transferase. The aberrant DAP kinase methylation was linked to the earliest pre-neoplastic state of lung adenocarcinoma, and the authors hypothesized that silencing of a crucial component of several apoptotic pathways could allow the expansion of the malignant cell colony leading to lung carcinogenesis (CitationPulling et al., 2004).
The methylated-CpG island recovery assay has identified genes that are hypermethylated in lung cancer cells. The data were used to compare the methylation status of A549 lung cancer cells relative to normal human bronchial epithelial cells. Judged by the location of the differentially methylated CpG islands (close to the ends of known or predicted genes or to the exons/introns within genes), it was hypothesized that the elements have a regulatory function. Notably, one fifth of the top 50 genes that were identified in the screen belong to the homeobox gene family (LHX2, LHX4, PAX7,HOXB13, LBX1, SIX2, HOXD3, DLX1, HOXD1, ONECUT2, and PAX9) (CitationRauch et al., 2006). In addition to gene specific methylation studies, a genome-wide screen was carried out to identify new cancer-specific methylation markers. The study included normal and malignant cell lines and relied on 5-aza-2′-deoxycytidine treatment as the readout for demethylation and increased gene expression. The candidates for cancer cell-specific promoter methylation sites were compared to those found in primary epithelial tumors to establish novel malignancy-associated methylation markers (CitationShames et al., 2006). An extensive depiction of methylation events and cancer is provided by the MethyCancer database, which documents the interplay between DNA methylation, gene expression, and cancer (CitationHe et al., 2008).
DNA methylation has also been detected in plasma. A panel of six genes (APC, CDH1, MGMT, DCC, RASSF1A, and AIM1) was used to compare serum and tumor DNA methylation profiles: if a given gene was methylated in serum, it was always also methylated in the tumor tissue. By contrast, tumor methylation was not always mirrored in the serum sample (CitationBegum et al., 2011). A progressive rise in plasma DNA methylation has also been detected in samples from patients with malignant lung tumors as compared to subjects with normal computer tomography (CT) scan (CitationOstrow et al., 2010). These studies are encouraging non-invasive approaches to advance the clinical management of lung cancer to distinguish between cancerous and noncancerous abnormal CT findings. Such methylation markers could be followed early on in heavy smokers and the individuals harboring aberrant methylation could be subjected to more intensive screening, resulting in earlier tumor detection and improved prognosis. Moreover, as Dolinoy et al. have proposed, novel therapeutic intervention strategies targeting reversible unfavorable epigenetic modifications could be developed (CitationDolinoy and Jirtle, 2008).
It has been shown that there is increased methylation for certain individual genes as well as the overall number of methylated genes with the progression of pre-neoplastic lesion towards invasive adenocarcinoma (CitationLicchesi et al., 2008; CitationChung et al., 2011). Selamat et al. have further mapped early, intermediate, and late methylation changes occurring throughout the development from histologically normal lung tissue (adjacent to the tumor) to atypical adenomatous hyperplasia, further to adenocarcinoma in situ and invasive lung adenocarcinoma. Unfortunately, the number of samples with smoking information was small and no correlation could be drawn between smoking status and DNA methylation levels in the tissue samples examined. Aberrant hypomethylation was evident only in the invasive carcinoma samples, indicating that unlike promoter hypermethylation, hypomethylation is a late event during lung adenocarcinoma development (CitationSelamat et al., 2011). This is in line with a study demonstrating that global DNA hypomethylation is highly associated with tumor progression (CitationAnisowicz et al., 2008). While global hypomethylation, being a late event in the course of disease, may not directly bring value to disease prediction, this association is important for understanding the tumor biology and may have implications in therapeutic intervention.
Clearly, epigenetics plays a major role in tumor progression; however, it might also be involved in the earlier stages of cancer development. According to the previously described cancer stem cell theory, the capacity to self-renew could be the result of abnormal activation of developmental pathways in cells destined for differentiation (CitationRivera et al., 2011). It is possible that such pathway activation involves epigenetic gene regulation, which evidently contributes to the cell’s ability to express or silence diverse genes during development.
Both in vitro and genetic studies have documented the involvement of the histone code in lung carcinogenesis (CitationPeters et al., 2001; CitationShan et al., 2008; CitationHaberland et al., 2009; CitationChi et al., 2010). Histone deacetylase 6 (HDAC6) is required for the epithelial-mesenchymal transition of TGF-α SMAD3 signaling-mediated lung cancer cell metastasis (CitationShan et al., 2008). Tumor cell lines engineered to have conditional HDAC alleles have shown that HDAC1 and HDAC2 have redundant functions, and while they are expendable in post-mitotic cells, at least one of them is required for tumor cell survival (CitationHaberland et al., 2009). Mutations and deregulation in histone lysine demethylases (HDMs) have been found in many types of cancers (CitationChi et al., 2010). The perturbed patterns of histone tail methylations are associated with increased chromosomal instabilities and tumor risk, and this applies to both ‘writing’ and ‘erasing’ of the methylation code. For example, the lack of histone H3 lysine 9 methylation in histone methyltransferase (HMTase)-deficient mice leads to impaired genomic stability (CitationPeters et al., 2001).
COPD
As lung cancer and COPD share many characteristics in the early stages of disease development, and since each condition increases the susceptibility to the other, it is reasonable to assume that they may be affected by similar genomic perturbations (CitationSundar et al., 2011; CitationYang et al., 2011).
In a recent study, white blood cell DNA isolated from subjects with well-defined COPD (using spirometry) was used for an array-based DNA methylation screen. The association of 349 CpG sites with the severity of COPD was reproduced in two family-based cohorts, and several of the 330 genes affected were related to immune and inflammatory processes. A significant association was found between SERPINA1 (the gene encoding α1-antitrypsin), hypomethylation, and low lung function (CitationQiu et al., 2012).
Cigarette smoke has considerable oxidative stress potential, leading to an imbalance between histone acetylation and deacetylation. This may account for the enhanced expression of inflammatory mediators, which in turn leads to amplified pulmonary inflammation (CitationRahman et al., 2002; CitationRahman, 2002). Cigarette smokers have increased acetylation of histones H3 and H4 (CitationSzulakowski et al., 2006), and in C57BL/6J mice cigarette smoke exposure increases the levels of acetylated histones H3 and H4 on lysine 9 after just 3 days of exposure (CitationYang et al., 2008). In rat lung, cigarette smoke results in altered histone acetylation, and in analogy to COPD, the resulting excessive release of pro-inflammatory cytokines was insensitive to glucocorticoid treatment (CitationMarwick et al., 2004). Mechanistically, the abnormal histone acetylation seems to be due to cigarette smoke-induced phosphorylation and subsequent ubiquitination and proteosomal degradation of HDAC2 as documented by both in vitro (macrophages, human bronchial and primary small airway epithelial cells) and in vivo (mouse lung) smoke exposure models (CitationAdenuga et al., 2009; CitationAdenuga and Rahman, 2010). COPD patients have reduced HDAC activity measured in the peripheral lung tissue, alveolar macrophages, and bronchial biopsy specimens (CitationIto et al., 2005). Lung tissue and peripheral blood mononuclear cells (PBMC) from COPD patients were also found to have reduced levels of Sirtuin 1, a type III HDAC that is also classified as an anti-aging molecule (CitationRajendrasozhan et al., 2008; CitationIto and Barnes, 2009). In agreement with the reduced HDAC levels and activity, it has been shown that the acetylation of histones H2A, H2B, H3, and H4 is increased in the lungs and alveolar macrophages of COPD patients (CitationChen et al., 2008).
Szulakowski et al. reported a correlation between COPD severity and decreased HDAC2 levels in the cytoplasm, and between both cytoplasmic and nuclear HDAC2 with the reduced lung function (CitationSzulakowski et al., 2006). Oxidative stress is one of the major causative factors for COPD and most likely leads to the observed degradation of HDAC2 with consequent increase in the expression of pro-inflammatory cytokines. It has been proposed that the inhibition of HDAC2 activity also contributes to the glucocorticoid resistance seen in COPD inflammation (CitationBarnes et al., 2005; CitationBarnes, 2006; CitationBarnes, 2009).
CVD
From the many risk factors for cardiovascular disease (CVD), smoking is causally linked to both disease onset and progression. Vascular remodeling, characteristic of CVD, involves chronic inflammation and the release of various cytokines and chemokines, and largely establish the initiation and progress of an atherosclerotic lesion (CitationWierda et al., 2010). Lung inflammation, the major driver of both lung cancer and COPD, has also been linked to CVD. It is believed that when lung inflammation, or even acute lung injury, turns systemic, it further stimulates events that lead to the activation of the vascular endothelium, heart attack, and stroke (CitationVan Eeden et al., 2012).
Direct DNA damage has been linked to cardiovascular disease; the increase of micronuclei, which is indicative of DNA damage, correlates with the severity of atherosclerosis. Genomic instability, in the forms of LOH and microsatellite instability (MI), has been documented in smooth muscle cells of human plaques. The loci involved include the TGF-1 receptor (MI), mismatch repair genes (LOH), and nitric oxide synthase (LOH). Currently, it is not clear what the source of DNA damage is in the disease, or whether the damage is a cause or a consequence of disease progression (CitationAndreassi and Botto, 2003).
Several independent studies have emerged showing that DNA methylation is an important aspect in CVD pathology. Analyses of peripheral lymphocytes have indicated that DNA from subjects with angiographically confirmed coronary artery disease (CAD) had undergone more extensive genomic methylation than that of healthy controls. There was also a significant positive correlation of global DNA methylation with plasma homocysteine levels, which is an independent risk factor in CAD patients (CitationSharma et al., 2008). Kim et al. have probed global genomic DNA methylation within ALU and Satellite 2 (AS) repetitive elements in peripheral blood lymphocytes (PBL) as a population based CVD risk assessment. Their study showed a positive correlation between PBL DNA methylation and prevalence of CVD or its risk factors, more pronounced in men than women (CitationKim et al., 2010). Finally, in the apolipoprotein E (apoE) mutant mouse, an animal model for CVD, the epigenetic changes in PBMCs DNA were found within coding sequences as well as repeated interspersed sequences. Strikingly, the altered methylation pattern could be detected before any noticeable atherosclerotic lesions were present, suggesting that DNA methylation plays a causative role during the course of CVD development (CitationLund et al., 2004). Epigenetic gene regulation has been reported to occur in platelets from smokers, in a study of monoamine oxidase-B (MAOB) (CitationLaunay et al., 2009). These authors reported that “the methylation frequency of the MAOB gene promoter was markedly lower in smokers than in non-smokers, due to cigarette smoke-induced increase of nucleic acid demethylases activity”.
In addition to DNA methylation, vascular homeostasis and atherosclerosis biology depend on the histone code. Whilst some functions mediated by HDACs are cytoplasmic (i.e. not associated with chromatin), chromatin immunoprecipitation has shown that the FGF2 promoter is bound by HDAC5. The promoter is thus presumed to be repressed by HDAC5 leading to transcriptional inactivity (CitationZhou et al., 2011). Post-translational histone modifications have also been involved in endothelial nitric oxide synthase 4 (eNOS4) regulation in endothelial cells, an important feature of vascular biology. It has been proposed that histone modifications are essential in maintaining the eNOS4 expression and that erasing the histone mark results in the hypoxic repression of the eNOS4 gene (CitationFish et al., 2010).
DNA methylation signatures
As DNA methylation plays a critical role in many different cellular processes including chronic inflammation, which lays a foundation to many smoking-related diseases, such as COPD, lung cancer, and CVD, the whole genome methylation signatures could provide valuable information on possible perturbations as a result of cigarette smoking. DNA methylation signatures, and the changes thereof, complement the conventional gene expression profiling in assessing the extent of damage that smoking causes to different organs. Li et al. have carried out a whole-genome DNA methylation analysis on human PBMCs and demonstrated 20 genomic features of regulatory, coding, non-coding, RNA-coding, and repeat sequences with a distinct methylation signature. There was a considerable inverse correlation between the allele-specific methylation (ASM) and allele-specific gene expression (ASE). As PBMCs represent a significant, non-invasive sample source, risk assessment could benefit considerably from analysis of the PBMC DNA methylome (CitationLi et al., 2010). Similar to gene expression analyses, the challenge is to categorize the individual methylated genes in a meaningful way to build a predictive classifier. Assessment of network perturbation amplitude by applying systems biology data to causal biological networks could identify specific signatures beyond single genes for each of the many smoking-related diseases (CitationMartin et al., 2012).
The methylation analyses should be explored as a method to improve stratification of subjects in addition to gene expression. Most likely the gene expression will be inversely correlated to the methylation levels. Furthermore, this work may pinpoint “interchromosomal networks” (chromosomal regions) that are subject to joint epigenetic mechanisms regulating gene expression (CitationZhao et al., 2006). A variety of DNA methylation biomarkers, have been developed in the field of tumor classification as well as disease and therapy prognosis (CitationSandoval and Esteller, 2012).
Next generation genomics and epigenomics platforms
As reviewed above, a large body of information exists on different genomic and epigenetic changes that are caused by smoking and other xenobiotic compounds. It is, however, possible to obtain an even more comprehensive picture on these perturbations by employing new and advanced technologies. CitationPleasance et al. (2010) have assessed the genome-wide mutational load extracted from an SCLC cell line by a massively parallel sequencing technology. The authors showed a comprehensive, somatically acquired, mutational profile including base substitutions, insertions, and deletions, as well as copy number changes and genomic rearrangements. The 23,000 SCLC mutations could be classified into distinct mutation signatures. Overall, these remarkable mutational patterns observed in the SCLC genome highlight the utility of genome-wide analyses to obtain true DNA signatures associated with lung cancer and carcinogen exposure. The patterns would not have been identified by simply sequencing limited genomic regions (CitationPleasance et al., 2010).
Several parallel sequencing technologies including pyro-sequencing, fluorescence-based sequencing-by-synthesis and sequencing-by-ligation, ion semiconductor and single molecule real time (SMRT) sequencing, have been developed in the last decade, offering read lengths and throughputs ranging from about 75,000 single-end long reads (~1 kb) to 6 billion 2 × 100 bp paired-end reads (Supplementary Material; CitationNiedringhaus et al.,2011).
High-throughput sequencing application in genomics
Regardless of the technology used, high-throughput sequencing has applications in genomics and epigenomics. Apart from its use in de-novo and re-sequencing of whole genomes, deep sequencing of a genome (DNA-seq) enables a wide range of analysis. Structural variations, copy number variations, single nucleotide polymorphisms (SNPs) and small insertions or deletions can be identified based on the comparison of a re-sequenced genome to a reference genome. If genomic regions of interest are known, targeted re-sequencing can be used to reduce costs, complexity and time.
High-throughput sequencing application in epigenomics
Whole genome DNA methylation can be investigated either at a single nucleotide level using shotgun bisulfite sequencing (MethylC-seq) and reduced representation bisulfite sequencing (RRBS), or at the level of a few tens of nucleotides with methylated DNA immunoprecipitation (MeDIP-seq), methylated DNA capture by affinity purification (MethylCap-seq), methylated DNA binding domain sequencing (MBD-seq) or methylation-sensitive restriction enzyme sequencing (MRE-seq) (CitationBock et al.,2010; CitationHarris et al.,2010). MethylC-seq and RRBS require the sequencing of both the untreated and the bisulfite treated genomes in order to compare them and identify methylated cytosines. With MeDIP-seq, MethylCap-seq and MBD-seq, methylated genomic DNA fragments are enriched before sequencing. On the contrary, MRE-seq enriches unmethylated genomic DNA fragments. These methods reduce the amount of sequencing necessary, but do not provide single nucleotide accuracy.
Histone modifications can be studied using chromatin immunoprecipitation (ChIP) with antibodies specific for a given histone modification. After precipitation, the DNA bound to the precipitated chromatin can be hybridized on a microarray chip (ChIP-chip) or sequenced (ChIP-seq) on a high-throughput platform. ChIP-chip will require a large number of arrays to cover whole mammalian genomes, while ChIP-seq provides the whole genome coverage even at a low sequencing depth (CitationBarski et al., 2007).
With the development of platforms based on high-throughput sequencing, genomics and epigenomics analyses are freed from the requirement for prior knowledge inherent to array-based platforms. The platforms thus move closer to measuring what is in the sample rather than what is on the chip. Furthermore, as high-throughput sequencing directly measures the DNA present in a sample, it can capture several modifications and alterations at once. The data generated by these platforms are therefore more flexible in the way they can be analysed and provide wider spectrum of genomic information. Computational resources and methodologies for the analysis of large-scale epigenomics datasets have been developed in recent year, however, as noted by Huss, “custom tools are needed to optimally analyze ChIP-seq data on histone modification and BS-seq data on DNA methylation” (CitationHuss, 2010). Furthermore, reaching the full potential of whole genome epigenomics studies is still hindered by the lack of guidelines and repositories for data submission. This would speed up research and allow for advanced applications such as therapeutic interventions (CitationOngenaert, 2010).
These next generation technology platformsshould be preferred methodologies to execute DNA and chromatin bound analyses. They have proven to be powerful tools, and are clearly the future means of identifying genetic and epigenetic aberrations triggered by compounds in tobacco smoke. In their recent review, Pfeifer et al. have given this assertion some perspective: “the analysis of a single cancer genome may generate almost twice as much mutation data as the whole literature on sequencing the p53 gene accumulated during 20 years” (CitationPfeifer and Hainaut, 2011).
Discussion
Better understanding of the mechanisms for the major smoking-related disease was the main aim of a recent report (CitationU.S. Department of Health and Human Services. 2010); however, the report was quite limited on how this understanding might actually be gained. Product safety assessment through clinical and epidemiological studies, generally decades after the initial exposure, rarely explains the mechanisms that link biological perturbations to a given exposure. In contrast, predictive systems biology approaches offer a valuable means for the assessment of biological perturbations before the phenotypic outcomes manifest as well as for pinning down the mechanisms involved in the body’s response to potentially harmful substances. Global measurements from in vitro cellular and in vivo animal experimental models can be causally linked to a biological network and this offers immense power to identify important pathways and targets for intervention (CitationHoeng et al., 2012).
A major component of systems biology is to identify the gene-related network perturbations caused by xenobiotics. For example, a constitutively increased growth pattern in gene expression profiling may stem from an activating mutation in a cell growth signaling gene such as KRAS or EGFR. While both effects may provide similar pictures at gene expression level, their impact on disease progression is clearly different. The integration of epigenetics and genomics to the systems biology approach will lead to the identification of a full panel of biomarkers and to a more reliable depiction of DNA damages caused by cigarette smoke. As some of the epigenetic and genomic changes, e.g. DNA methylation, can be reversible, their targeting has been considered for therapies. In this context, it is of interest to also study these effects upon smoking cessation to determine how many of the changes presented above reverse completely upon cessation, and how long does the reversal process take.
Given the very rapid increase in knowledge on genomic effects of cigarette smoke, our recommendation is that this research be continued to definitively determine pathways (i.e.mechanisms) by which smoke actually causes diseases such as those describe here. This genomic investigation will spur the formulation of new testable hypotheses and present mechanistic insight on how exposure to chemicals and mixtures are related to disease onset and progression, thus providing the basis for biomarkers selection and justification. It might then be possible to identify specific chemicals (or classes of chemicals) in the smoke that would be linked to specific diseases, a long-term goal (CitationWynder, 1980) which has so far received only minimal success. Research could then be pursued on reducing or eliminating those chemicals in smoke, which have been identified by systems biology as necessary to substantially perturb disease networks as opposed to the current assumption that ‘harmful and potentially harmful constituents’ (CitationU.S. Department of Health and Human Services, 2011) of the smoke matrix can be reliably identified by traditional toxicological or epidemiological studies.
Our approach may be more robust than the simplistic application of genetic toxicology testing of either whole smoke or of cigarette smoke condensate. Mutagenicity testing of highly complex mixtures seems scientifically irrational, but it is an approach that is often used when comparing cigarette types (CitationDemarini et al.,2008) and smokeless products (CitationJohnson et al.,2009). It might also be possible to take the now-established mechanisms for the different diseases in humans, and see whether these mechanisms also exist (or could be improved) in animal species that have been used as surrogates for humans (CitationSchleef, 2006), with the aim of building better animal models (CitationCoggins, 2010). Transgenic strains of laboratory animals are becoming increasingly more popular and seemingly limitless in scope.
It is very likely that multiple redundant pathways exist for each of the disease states we have reviewed. The adoption of a systems biology approach relying on results from high-throughput sequencing genomics studies may therefore be a significant contribution enabling the elucidation of these pathways and of their interplay. We propose that the use of systems biology can go beyond just toxicological assessment, and can be applied in other areas such as drug development, pharmacology, and personalized medicine. These measurements should be considered in order to gain better understanding of the mechanism of actions of cigarette smoke on disease development as also indicated by Hammons et al. in their recent review (CitationHammons and Lyn-Cook, 2011). Furthermore, the genomic and epigenetic mechanisms may serve as causal links between exposure, dose and duration. Recently, Hou et al. summarized how exposure to environmental chemicals can cause epigenetic changes in a dose-dependent manner (CitationHou et al., 2012). Moreover, the initiation of biological perturbations resulting in malignancy by certain chemical compounds can be explained by epigenetic mechanisms (CitationStein, 2012).
Conclusion
The limitations of the traditional risk assessments include the lack of mechanistic information, uncertainty about the toxicity pathways that might potentially be affected, relevance of the endpoints of toxicity, default assumptions for dose-response extrapolations, and uncertainty about intra- and inter-species “safety factors” (CitationBarlow et al.,2006; CitationEdwards and Preston, 2008; CitationHartung, 2009). In this review, we have focused on the genomic and epigenomic perturbations caused by tobacco smoke and how the latest technological developments enable the research community to investigate them. In general, toxicogenomics is not yet fully accepted in the toxicological sciences, due to regulatory concerns and the lack of published proof of concept studies (CitationMendrick, 2008). All these aspects are currently being addressed and the advances in molecular system biology unveils the multifaceted nature of disease processes. The toxicology community should incorporate this emerging knowledge, whereby it can take an iterative approach of examining proof of concepts, assessing the value of new information, and developing decision rules. Programs such as NexGen (http://www.epa.gov/risk/nexgen/index.htm) from the US Environmental Protection Agency guide the community in this direction.
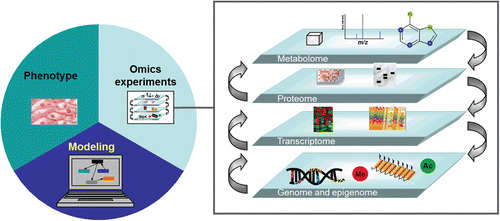
For a complete understanding of the effects of these perturbations, all components of network biology should be included (). This allows the mechanistic understanding of the biological responses that attempt to protect the organism from potentially harmful substances and the identification of predictive biomarkers for disease onset. Ultimately, the combined quantifiable genomic and epigenomic perturbations will allow us to measure the genomic impact of a substance on a biological system.
Figure 1. Predictive systems biology for toxicological assessment. The different levels of omics are needed to fully understand risks associated with cigarette smoking. High throughput genomic analyses are an essential part of this mechanistic approach.
Declaration of interest
The affiliation of the authors is shown on the cover page. The authors are all employees of Philip Morris International (PMI) with the exception of Christopher Coggins who served as a paid consultant to PMI for preparation of this review. PMI is one of the world’s largest producers and marketers of cigarettes. Christopher Coggins is an independent toxicology consultant, specializing primarily on issues concerned with the health impacts of airborne materials from environmental or occupational exposures or use of consumer products such as tobacco containing products. The views expressed in the paper are solely those of the authors and the paper was prepared exclusively by the authors.
Supplementary Material
Download PDF (84.5 KB)Related Research Data
References
- Adenuga D, Rahman I. (2010). Protein kinase CK2-mediated phosphorylation of HDAC2 regulates co-repressor formation, deacetylase activity and acetylation of HDAC2 by cigarette smoke and aldehydes. Arch Biochem Biophys 498:62–73.
- Adenuga D, Yao H, March TH, Seagrave J, Rahman I. (2009). Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol 40:464–473.
- Andreassi MG, Botto N. (2003). DNA damage as a new emerging risk factor in atherosclerosis. Trends Cardiovasc Med 13:270–275.
- Anisowicz A, Huang H, Braunschweiger KI, Liu Z, Giese H, Wang H, Mamaev S, Olejnik J, Massion PP, Del Mastro RG. (2008). A high-throughput and sensitive method to measure global DNA methylation: application in lung cancer. BMC Cancer 8:222.
- Anna L, Holmila R, Kovács K, Gyorffy E, Gyori Z, Segesdi J, Minárovits J, Soltész I, Kostic S, Csekeo A, Husgafvel-Pursiainen K, Schoket B. (2009). Relationship between TP53 tumour suppressor gene mutations and smoking-related bulky DNA adducts in a lung cancer study population from Hungary. Mutagenesis 24:475–480.
- Barlow S, Renwick AG, Kleiner J, Bridges JW, Busk L, Dybing E, Edler L, Eisenbrand G, Fink-Gremmels J, Knaap A, Kroes R, Liem D, Müller DJ, Page S, Rolland V, Schlatter J, Tritscher A, Tueting W, Würtzen G. (2006). Risk assessment of substances that are both genotoxic and carcinogenic report of an International Conference organized by EFSA and WHO with support of ILSI Europe. Food Chem Toxicol 44:1636–1650.
- Barnes PJ, Adcock IM, Ito K. (2005). Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J 25:552–563.
- Barnes PJ. (2006). Reduced histone deacetylase in COPD: clinical implications. Chest 129:151–155.
- Barnes PJ. (2009). Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 6:693–696.
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129:823–837.
- Baryshnikova E, Destro A, Infante MV, Cavuto S, Cariboni U, Alloisio M, Ceresoli GL, Lutman R, Brambilla G, Chiesa G, Ravasi G, Roncalli M. (2008). Molecular alterations in spontaneous sputum of cancer-free heavy smokers: results from a large screening program. Clin Cancer Res 14:1913–1919.
- Beane J, Vick J, Schembri F, Anderlind C, Gower A, Campbell J, Luo L, Zhang XH, Xiao J, Alekseyev YO, Wang S, Levy S, Massion PP, Lenburg M, Spira A. (2011). Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev Res (Phila) 4:803–817.
- Begum S, Brait M, Dasgupta S, Ostrow KL, Zahurak M, Carvalho AL, Califano JA, Goodman SN, Westra WH, Hoque MO, Sidransky D. (2011). An epigenetic marker panel for detection of lung cancer using cell-free serum DNA. Clin Cancer Res 17:4494–4503.
- Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, Baylin SB, Herman JG. (1998). Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci USA 95:11891–11896.
- Belinsky SA. (2005). Silencing of genes by promoter hypermethylation: key event in rodent and human lung cancer. Carcinogenesis 26:1481–1487.
- Belinsky SA, Klinge DM, Dekker JD, Smith MW, Bocklage TJ, Gilliland FD, Crowell RE, Karp DD, Stidley CA, Picchi MA. (2005). Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res 11:6505–6511.
- Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K, Haney J, Kennedy TC, Hirsch FR, Miller Y, Franklin WA, Herman JG, Baylin SB, Bunn PA, Byers T. (2006). Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res 66:3338–3344.
- Bhutani M, Pathak AK, Fan YH, Liu DD, Lee JJ, Tang H, Kurie JM, Morice RC, Kim ES, Hong WK, Mao L. (2008). Oral epithelium as a surrogate tissue for assessing smoking-induced molecular alterations in the lungs. Cancer Prev Res (Phila) 1:39–44.
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A. (2010). Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28:1106–1114.
- Broët P, Dalmasso C, Tan EH, Alifano M, Zhang S, Wu J, Lee MH, Régnard JF, Lim D, Koong HN, Agasthian T, Miller LD, Lim E, Camilleri-Broët S, Tan P. (2011). Genomic profiles specific to patient ethnicity in lung adenocarcinoma. Clin Cancer Res 17:3542–3550.
- Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W. (1995). Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet 11:210–212.
- Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM. (2008). Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 3:e3316.
- Chi P, Allis CD, Wang GG. (2010). Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10:457–469.
- Chung JH, Lee HJ, Kim BH, Cho NY, Kang GH. (2011). DNA methylation profile during multistage progression of pulmonary adenocarcinomas. Virchows Arch 459:201–211.
- Coggins CR. (2010). A further review of inhalation studies with cigarette smoke and lung cancer in experimental animals, including transgenic mice. Inhal Toxicol 22:974–983.
- Dacic S, Shuai Y, Yousem S, Ohori P, Nikiforova M. (2010). Clinicopathological predictors of EGFR/KRAS mutational status in primary lung adenocarcinomas. Mod Pathol 23:159–168.
- Damiani LA, Yingling CM, Leng S, Romo PE, Nakamura J, Belinsky SA. (2008). Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res 68:9005–9014.
- DeMarini DM, Gudi R, Szkudlinska A, Rao M, Recio L, Kehl M, Kirby PE, Polzin G, Richter PA. (2008). Genotoxicity of 10 cigarette smoke condensates in four test systems: comparisons between assays and condensates. Mutat Res 650:15–29.
- Denissenko MF, Chen JX, Tang MS, Pfeifer GP. (1997). Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc Natl Acad Sci USA 94:3893–3898.
- Denissenko MF, Pao A, Tang M, Pfeifer GP. (1996). Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 274:430–432.
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455:1069–1075.
- Dolinoy DC, Jirtle RL. (2008). Environmental epigenomics in human health and disease. Environ Mol Mutagen 49:4–8.
- Dunn BK. (2003). Hypomethylation: one side of a larger picture. Ann N Y Acad Sci 983:28–42.
- Edwards SW, Preston RJ. (2008). Systems biology and mode of action based risk assessment. Toxicol Sci 106:312–318.
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. (2008). Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15:504–514.
- Esteller M. (2007). Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet 16 Spec No 1:R50–R59.
- Feng Q, Hawes SE, Stern JE, Wiens L, Lu H, Dong ZM, Jordan CD, Kiviat NB, Vesselle H. (2008). DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol Biomarkers Prev 17:645–654.
- Fish JE, Yan MS, Matouk CC, St Bernard R, Ho JJ, Ho JJ Jr, Gavryushova A, Srivastava D, Marsden PA. (2010). Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem 285:810–826.
- Franklin WA, Gazdar AF, Haney J, Wistuba II, La Rosa FG, Kennedy T, Ritchey DM, Miller YE. (1997). Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J Clin Invest 100:2133–2137.
- Garinis GA, Gorgoulis VG, Mariatos G, Zacharatos P, Kotsinas A, Liloglou T, Foukas P, Kanavaros P, Kastrinakis NG, Vassilakopoulos T, Vogiatzi T, Field JK, Kittas C. (2001). Association of allelic loss at the FHIT locus and p53 alterations with tumour kinetics and chromosomal instability in non-small cell lung carcinomas (NSCLCs). J Pathol 193:55–65.
- Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. (2007). Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer 55:1–14.
- Gazdar AF, Minna JD. (2009). Multifocal lung cancers–clonality vs field cancerization and does it matter? J Natl Cancer Inst 101:541–543.
- Gebel S, Gerstmayer B, Bosio A, Haussmann HJ, Van Miert E, Müller T. (2004). Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis 25:169–178.
- Gebel S, Gerstmayer B, Kuhl P, Borlak J, Meurrens K, Müller T. (2006). The kinetics of transcriptomic changes induced by cigarette smoke in rat lungs reveals a specific program of defense, inflammation, and circadian clock gene expression. Toxicol Sci 93:422–431.
- Guo X, Verkler TL, Chen Y, Richter PA, Polzin GM, Moore MM, Mei N. (2011). Mutagenicity of 11 cigarette smoke condensates in two versions of the mouse lymphoma assay. Mutagenesis 26:273–281.
- Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN. (2009). Genetic dissection of histone deacetylase requirement in tumor cells. Proc Natl Acad Sci USA 106:7751–7755.
- Hammons G, Lyn-Cook B. (2011). Epigenetics in tobacco smoke exposure. Curr Top Toxicol 7:63–77.
- Hang B. (2010). Formation and repair of tobacco carcinogen-derived bulky DNA adducts. J Nucleic Acids 2010:709521.
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, Olshen A, Ballinger T, Zhou X, Forsberg KJ, Gu J, Echipare L, O’Geen H, Lister R, Pelizzola M, Xi Y, Epstein CB, Bernstein BE, Hawkins RD, Ren B, Chung WY, Gu H, Bock C, Gnirke A, Zhang MQ, Haussler D, Ecker JR, Li W, Farnham PJ, Waterland RA, Meissner A, Marra MA, Hirst M, Milosavljevic A, Costello JF. (2010). Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28:1097–1105.
- Hartung T. (2009). A toxicology for the 21st century–mapping the road ahead. Toxicol Sci 109:18–23.
- He X, Chang S, Zhang J, Zhao Q, Xiang H, Kusonmano K, Yang L, Sun ZS, Yang H, Wang J. (2008). MethyCancer: the database of human DNA methylation and cancer. Nucleic Acids Res 36:D836–D841.
- Herbst RS, Heymach JV, Lippman SM. (2008). Lung cancer. N Engl J Med 359:1367–1380.
- Hoeng J, Deehan R, Pratt D, Martin F, Sewer A, Thomson TM, Drubin DA, Waters CA, de Graaf D, Peitsch MC. (2012). A network-based approach to quantifying the impact of biologically active substances. Drug Discov Today 17:413–418.
- Hou L, Zhang X, Wang D, Baccarelli A. (2012). Environmental chemical exposures and human epigenetics. Int J Epidemiol 41:79–105.
- Huang YT, Lin X, Liu Y, Chirieac LR, McGovern R, Wain J, Heist R, Skaug V, Zienolddiny S, Haugen A, Su L, Fox EA, Wong KK, Christiani DC. (2011). Cigarette smoking increases copy number alterations in nonsmall-cell lung cancer. Proc Natl Acad Sci USA 108:16345–16350.
- Huss M. (2010). Introduction into the analysis of high-throughput-sequencing based epigenome data. Brief Bioinformatics 11:512–523.
- Ito K, Barnes PJ. (2009). COPD as a disease of accelerated lung aging. Chest 135:173–180.
- Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. (2005). Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 352:1967–1976.
- Jarmalaite S, Kannio A, Anttila S, Lazutka JR, Husgafvel-Pursiainen K. (2003). Aberrant p16 promoter methylation in smokers and former smokers with nonsmall cell lung cancer. Int J Cancer 106:913–918.
- Johnson MD, Schilz J, Djordjevic MV, Rice JR, Shields PG. (2009). Evaluation of in vitro assays for assessing the toxicity of cigarette smoke and smokeless tobacco. Cancer Epidemiol Biomarkers Prev 18:3263–3304.
- Kerr KM, Galler JS, Hagen JA, Laird PW, Laird-Offringa IA. (2007). The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers 23:5–30.
- Kim H, Kwon YM, Kim JS, Lee H, Park JH, Shim YM, Han J, Park J, Kim DH. (2004). Tumor-specific methylation in bronchial lavage for the early detection of non-small-cell lung cancer. J Clin Oncol 22:2363–2370.
- Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. (2010). DNA methylation as a biomarker for cardiovascular disease risk. PLoS ONE 5:e9692
- Lamy A, Sesboüé R, Bourguignon J, Dautréaux B, Métayer J, Frébourg T, Thiberville L. (2002). Aberrant methylation of the CDKN2a/p16INK4a gene promoter region in preinvasive bronchial lesions: a prospective study in high-risk patients without invasive cancer. Int J Cancer 100:189–193.
- Lantuéjoul S, Salameire D, Salon C, Brambilla E. (2009). Pulmonary preneoplasia–sequential molecular carcinogenetic events. Histopathology 54:43–54.
- Launay JM, Del Pino M, Chironi G, Callebert J, Peoc’h K, Mégnien JL, Mallet J, Simon A, Rendu F. (2009). Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS ONE 4:e7959.
- Lee YJ, Shim HS, Kang YA, Hong SJ, Kim HK, Kim H, Kim SK, Choi SH, Kim JH, Cho BC. (2010). Dose effect of cigarette smoking on frequency and spectrum of epidermal growth factor receptor gene mutations in Korean patients with non-small cell lung cancer. J Cancer Res Clin Oncol 136:1937–1944.
- Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, Zheng H, Yu J, Wu H, Sun J, Zhang H, Chen Q, Luo R, Chen M, He Y, Jin X, Zhang Q, Yu C, Zhou G, Sun J, Huang Y, Zheng H, Cao H, Zhou X, Guo S, Hu X, Li X, Kristiansen K, Bolund L, Xu J, Wang W, Yang H, Wang J, Li R, Beck S, Wang J, Zhang X. (2010). The DNA methylome of human peripheral blood mononuclear cells PLoS.Biol 8:e1000533.
- Licchesi JD, Westra WH, Hooker CM, Herman JG. (2008). Promoter hypermethylation of hallmark cancer genes in atypical adenomatous hyperplasia of the lung. Clin Cancer Res 14:2570–2578.
- Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, Wang YC. (2010). The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest 120:521–532.
- Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Davis S, Zhang Y, Hussain M, Xi S, Rao M, Meltzer PA, Schrump DS. (2010). Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 29:3650–3664.
- Liu H, Zhou Y, Boggs SE, Belinsky SA, Liu J. (2007). Cigarette smoke induces demethylation of prometastatic oncogene synuclein-gamma in lung cancer cells by downregulation of DNMT3B. Oncogene 26:5900–5910.
- Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar-Garea A, Ballestar E, Esteller M, Zaina S. (2004). DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 279:29147–29154.
- Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, Lee JJ, Ro JY, Broxson A, Yu R, Morice RC, Kemp BL, Khuri FR, Walsh GL, Hittelman WN, Hong WK. (1997). Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst 89:857–862.
- Martin F, Thomson TM, Sewer A, Drubin DA, Mathis C, Weisensee D, Pratt D, Hoeng J, Peitsch MC. (2012). Assessment of network perturbation amplitude by applying high-throughput data to causal biological networks. BMC Syst Biol 6:54.
- Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. (2004). Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 31:633–642.
- Mass MJ, Wang L. (1997). Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat Res 386:263–277.
- Mendrick DL. (2008). Toxicogenomics and classic toxicology: how to improve prediction and mechanistic understanding of human toxicity. Methods Mol Biol 460:1–22.
- Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. (1995). 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1:686–692.
- Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH, Philibert RA. (2012). Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet 159B:141–151.
- Nelson HH, Wiencke JK, Gunn L, Wain JC, Christiani DC, Kelsey KT. (1998). Chromosome 3p14 alterations in lung cancer: evidence that FHIT exon deletion is a target of tobacco carcinogens and asbestos. Cancer Res 58:1804–1807.
- Niedringhaus TP, Milanova D, Kerby MB, Snyder MP, Barron AE. (2011). Landscape of next-generation sequencing technologies. Anal Chem 83:4327–4341.
- Nowell PC. (1976). The clonal evolution of tumor cell populations. Science 194:23–28.
- OECD. (1997). Guideline for the Testing OF Chemicals. In Vitro Mammalian Cell Gene Mutation Test. Available at: http://www.oecd-ilibrary.org/docserver/download/fulltext/9747601e.pdf?expires=1332790058&id=id&accname=freeContent&checksum=01D755899A5653A11A1D2211DD51662D
- Ongenaert M. (2010). Epigenetic databases and computational methodologies in the analysis of epigenetic datasets. Adv. Genet. 71:259–295.
- Ostrow KL, Hoque MO, Loyo M, Brait M, Greenberg A, Siegfried JM, Grandis JR, Gaither Davis A, Bigbee WL, Rom W, Sidransky D. (2010). Molecular analysis of plasma DNA for the early detection of lung cancer by quantitative methylation-specific PCR. Clin Cancer Res 16:3463–3472.
- Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA. (2000). Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 60:5954–5958.
- Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, Schöfer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T. (2001). Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107:323–337.
- Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. (2002). Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21:7435–7451.
- Pfeifer GP, Hainaut P. (2011). Next-generation sequencing: emerging lessons on the origins of human cancer. Curr Opin Oncol 23:62–68.
- Phillips JM, Goodman JI. (2009). Inhalation of cigarette smoke induces regions of altered DNA methylation (RAMs) in SENCAR mouse lung. Toxicology 260:7–15.
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, Varela I, Nik-Zainal S, Davies HR, Ordoñez GR, Mudie LJ, Latimer C, Edkins S, Stebbings L, Chen L, Jia M, Leroy C, Marshall J, Menzies A, Butler A, Teague JW, Mangion J, Sun YA, McLaughlin SF, Peckham HE, Tsung EF, Costa GL, Lee CC, Minna JD, Gazdar A, Birney E, Rhodes MD, McKernan KJ, Stratton MR, Futreal PA, Campbell PJ. (2010). A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463:184–190.
- Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA. (2004). Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res 64:3844–3848.
- Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, Rennard S, Agusti A, Anderson W, Lomas DA, DeMeo DL. (2012). Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med 185:373–381.
- Rahman I. (2002). Oxidative stress, transcription factors and chromatin remodelling in lung inflammation. Biochem Pharmacol 64:935–942.
- Rahman I, Gilmour PS, Jimenez LA, MacNee W. (2002). Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem 234-235:239–248.
- Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. (2008). SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177:861–870.
- Rauch T, Li H, Wu X, Pfeifer GP. (2006). MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res 66:7939–7947
- Rivera C, Rivera S, Loriot Y, Vozenin MC, Deutsch E. (2011). Lung cancer stem cell: new insights on experimental models and preclinical data. J Oncol 2011:549181.
- Rusin MR, Okamoto A, Chorazy M, Czyzewski K, Harasim J, Spillare EA, Hagiwara K, Hussain SP, Xiong Y, Demetrick DJ, Harris CC. (1996). Intragenic mutations of the p16(INK4), p15(INK4B) and p18 genes in primary non-small-cell lung cancers. Int J Cancer 65:734–739.
- Russo AL, Thiagalingam A, Pan H, Califano J, Cheng KH, Ponte JF, Chinnappan D, Nemani P, Sidransky D, Thiagalingam S. (2005). Differential DNA hypermethylation of critical genes mediates the stage-specific tobacco smoke-induced neoplastic progression of lung cancer. Clin Cancer Res 11:2466–2470.
- Ryberg D, Kure E, Lystad S, Skaug V, Stangeland L, Mercy I, Børresen AL, Haugen A. (1994). p53 mutations in lung tumors: relationship to putative susceptibility markers for cancer. Cancer Res 54:1551–1555.
- Sandoval J, Esteller M. (2012). Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 22:50–55.
- Schär P, Fritsch O. (2011). DNA repair and the control of DNA methylation. Prog Drug Res 67:51–68.
- Schleef RR. (2006) Animal Models for Three Cigarette-Smoke-Induced Diseases.Boca Raton: CRC Press, Taylor & Francis.
- Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, Siegmund KD, Kerr KM, Laird-Offringa IA. (2011). DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS ONE 6:e21443.
- Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N, Jiang A, Perou CM, Kim YH, Pollack JR, Fong KM, Lam CL, Wong M, Shyr Y, Nanda R, Olopade OI, Gerald W, Euhus DM, Shay JW, Gazdar AF, Minna JD. (2006). A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med 3:e486.
- Shan B, Yao TP, Nguyen HT, Zhuo Y, Levy DR, Klingsberg RC, Tao H, Palmer ML, Holder KN, Lasky JA. (2008). Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J Biol Chem 283:21065–21073.
- Sharma P, Kumar J, Garg G, Kumar A, Patowary A, Karthikeyan G, Ramakrishnan L, Brahmachari V, Sengupta S. (2008). Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol 27:357–365.
- Spira A, Schembri F, Beane J, Shah V, Liu G, Brody JS. (2004). Impact of cigarette smoke on the normal airway transcriptome. Chest 125:115S.
- Steiling K, Ryan J, Brody JS, Spira A. (2008). The field of tissue injury in the lung and airway. Cancer Prev Res (Phila) 1:396–403.
- Stein RA. (2012). Epigenetics and environmental exposures. J Epidemiol Community Health 66:8–13.
- Subramanian J, Govindan R. (2010). Lung cancer in ‘Never-smokers’: a unique entity. Oncology (Williston Park, NY) 24:29–35.
- Sun S, Schiller JH, Gazdar AF. (2007). Lung cancer in never smokers–a different disease. Nat Rev Cancer 7:778–790.
- Sundar IK, Mullapudi N, Yao H, Spivack SD, Rahman I. (2011). Lung cancer and its association with chronic obstructive pulmonary disease: update on nexus of epigenetics. Curr Opin Pulm Med 17:279–285.
- Szulakowski P, Crowther AJ, Jiménez LA, Donaldson K, Mayer R, Leonard TB, MacNee W, Drost EM. (2006). The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 174:41–50.
- U.S. Department of Health and Human Services. (2010). How tobacco smoke causes disease: The biology and behavioral basis for smoking-attributable disease: A report of the Surgeon General., Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA.
- U.S. Department of Health and Human Services. (2011). Harmful and potentially harmful constituents in tobacco products and tobacco smoke; request for comments., Food and Drug Administration, Federal Register, Vol. 76. Washington, DC: Docket No. FDA-2011-N-0271.
- Van Eeden S, Leipsic J, Paul Man SF, Sin DD. (2012). The relationship between lung inflammation and cardiovascular disease. Am J Respir Crit Care Med 186:11–16.
- Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. (2010). Epigenetics in atherosclerosis and inflammation. J Cell Mol Med 14:1225–1240.
- Wistuba II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, Samet JM, Srivastava S, Minna JD, Gazdar AF. (1997). Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst 89:1366–1373.
- Wynder EL. (1980). Some concepts of the less harmful cigarette, in A safe cigarette? Banbury Report 3. Cold Spring Harbor Laboratory.
- Yanagawa N, Tamura G, Oizumi H, Endoh M, Sadahiro M, Motoyama T. (2011). Inverse correlation between EGFR mutation and FHIT, RASSF1A and RUNX3 methylation in lung adenocarcinoma: relation with smoking status. Anticancer Res 31:1211–1214.
- Yang IA, Relan V, Wright CM, Davidson MR, Sriram KB, Savarimuthu Francis SM, Clarke BE, Duhig EE, Bowman RV, Fong KM. (2011). Common pathogenic mechanisms and pathways in the development of COPD and lung cancer. Expert Opin Ther Targets 15:439–456.
- Yang SR, Valvo S, Yao H, Kode A, Rajendrasozhan S, Edirisinghe I, Caito S, Adenuga D, Henry R, Fromm G, Maggirwar S, Li JD, Bulger M, Rahman I. (2008). IKK alpha causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung. Am J Respir Cell Mol Biol 38:689–698.
- Zabarovsky ER, Lerman MI, Minna JD. (2002). Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 21:6915–6935.
- Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. (2006). Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet 38:1341–1347.
- Zhou B, Margariti A, Zeng L, Xu Q. (2011). Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res 90:413–420.
- Zochbauer-Muller S, Minna JD, Gazdar AF. (2002). Aberrant DNA methylation in lung cancer: biological and clinical implications. Oncologist 7:451–457.
- Zöchbauer-Müller S, Lam S, Toyooka S, Virmani AK, Toyooka KO, Seidl S, Minna JD, Gazdar AF. (2003). Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int J Cancer 107:612–616.