Abstract
Myelofibrosis (MF) is a BCR–ABL1-negative myeloproliferative neoplasm diagnosed de novo or developed from essential thrombocythemia (ET) or polycythemia vera (PV). Average survival of a patient with MF is 5–7 years. Disease complications include fatigue, early satiety, pruritus, painful splenic infarcts, infections and leukemic transformation. Allogeneic hematopoietic stem cell transplant (HSCT) is the only potentially curative option for MF, but carries a risk of treatment-related mortality and is reserved for the few high-risk patients fit enough to endure the procedure. Other traditional therapies are palliative and supported by few randomized, controlled trials; thus, novel treatment strategies are needed. Discovery of the Janus kinase 2 (JAK2) gain-of-function mutation, JAK2V617F, in the majority (50–60%) of patients with MF led to increased understanding of the biology underlying MF and the development of JAK2 inhibitors to treat MF. Recent Food and Drug Administration (FDA) approval of the first JAK2 inhibitor, ruxolitinib, signaled a new era for treatment of MF. Additional JAK2 inhibitors, such as SAR302503, may become commercially available in the near future, and their distinct pharmacologic and efficacy profiles will help determine their use across the patient population. Data on JAK2 inhibitors, their role in an evolving treatment paradigm, and future directions for treatment of MF are discussed.
Introduction
The BCR–ABL1-negative myeloproliferative neoplasms (MPNs) arise from aberrant hematopoietic stem cell proliferation, and include primary myelofibrosis (PMF), polycythemia vera (PV) and essential thrombocythemia (ET). Myelofibrosis (MF) may also evolve from a previous diagnosis of PV or ET, known as post-PV or post-ET MF, respectively [Citation1,Citation2]. Characteristics of MF include dysfunctional hematopoiesis with cytopenias, extramedullary hematopoiesis contributing to splenomegaly, and a hypercatabolic state [Citation2,Citation3]. Disease-related symptoms primarily manifest as fatigue, night sweats, bone pain, fever, weight loss and spleen pain [Citation3]. Other potential complications include infections, bleeding, portal hypertension and transformation to acute myeloid leukemia (AML) [Citation4,Citation5]. While some patients with PV and ET have a shortened life expectancy compared with the general population, patients with MF experience the greatest morbidity and mortality among the MPNs [Citation2–4]. Patients with MF face a highly variable prognosis for survival, ranging from several months to more than 10 years [Citation4,Citation6]. The average survival of a patient with MF is 5–7 years, with the disease course characterized by progressively worsening symptoms [Citation4,Citation5].
Prior to the Food and Drug Administration (FDA) approval of ruxolitinib in November 2011, there were no drugs approved for the treatment of MF, and hematopoietic stem cell transplant (HSCT) was the only potentially curative treatment option. Improved understanding of the disease biology has led to an increase in randomized, controlled trials evaluating potential therapies, which until recently were lacking. With 2–5 new cases emerging per 100 000 people annually in the United States, significant unmet needs remain [Citation7].
Traditional treatment approaches
Assessing MF prognosis
The traditional treatment paradigm for MF consists largely of palliative measures that carry a risk of toxicity; thus, treatment is based on risk-stratification and the presence and severity of symptoms [Citation8,Citation9]. The International Prognostic Scoring System (IPSS) uses five risk factors to predict prognosis and assign a patient to a risk group: (1) age > 65 years; (2) hemoglobin < 10 g/dL; (3) leukocyte count > 25 × 109/L; (4) circulating blood blasts > 1%; and (5) the presence of constitutional symptoms [Citation4]. The Dynamic IPSS (DIPSS) uses the same five risk factors and has been validated to predict prognosis at any time during the disease course [Citation8,Citation10,Citation11]. The DIPSS was recently modified (DIPSS Plus) with the incorporation of three additional risk factors: (1) red blood cell transfusion needed; (2) platelet count < 100 × 109/L; and (3) unfavorable karyotype [complex or sole or two abnormalities, including + 8, − 7/7q −, i(17q), inv(3), − 5/5q −, 12q − or 11q23 rearrangement] [Citation10].
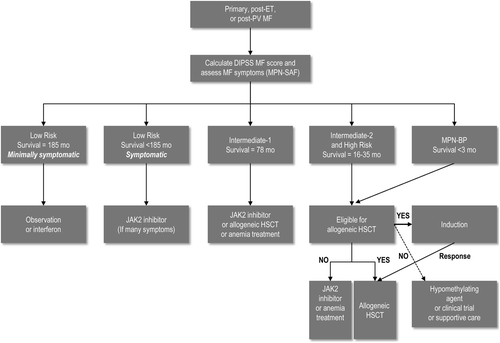
The presence of symptoms assessed with the MPN Symptom Assessment Form (SAF) and risk category can be used to determine appropriate treatment [Citation5,Citation12] (). Patients with low and intermediate-1 risk disease maintain a relatively good prognosis. Treatment is not always recommended in patients with these levels of risk, and often depends on the presence and severity of symptoms [Citation9]. Patients with intermediate-2 and high risk disease often have prominent symptoms and warrant treatment.
Figure 1. First-line therapy of myelofibrosis in 2012. Survival is expressed as the median. Low-risk patients described as “symptomatic” are patients with symptoms which, in the eye of the patient, are problematic and were not included in the IPSS/DIPSS calculations (i.e. severe fatigue, cachexia, pruritus, spleen symptoms). DIPSS, Dynamic International Prognostic Scoring System; ET, essential thrombocythemia; HSCT, hematopoietic stem cell transplant; JAK2, Janus kinase 2; MF, myelofibrosis; MPN-BP, myeloproliferative neoplasm – blast phase; MPN-SAF, Myeloproliferative Neoplasm Symptom Assessment Form; PV, polycythemia vera.
Allogeneic stem cell transplant
Allogeneic HSCT is the only potentially curative therapy for MPNs, but the procedure may be poorly tolerated in older patients and is associated with substantial treatment-related morbidity and mortality, thereby limiting its application in the typical MF patient population [Citation9,Citation13,Citation14]. A recent study demonstrated that while survival in intermediate-2 and high risk patients treated with HSCT was improved compared with patients treated without HSCT, risk of mortality was correlated with the DIPSS risk category [Citation14]. Thus, HSCT is generally considered only for patients with intermediate or high risk disease deemed fit enough to be eligible for the procedure.
Allogeneic HSCT results in grades 2–4 acute graft-versus-host disease (GVHD) in 20–40% of patients, creating significant liver dysfunction, skin rash and gastrointestinal disturbances. Chronic GVHD is a serious and common long-term complication reported in an additional 20–40% of patients [Citation15]. Analysis of data from a study of 289 patients with MF with a median age of 47 years treated with allogeneic HSCT between 1989 and 2002 showed that treatment-related mortality was 27% and overall survival (OS) at 5 years ranged from 30 to 40%. The most common causes of death post-transplant are relapse, infection and GVHD [Citation15]. Although the inclusion of a variety of conditioning regimens, GVHD prophylaxis regimens, and varying degrees of human leukocyte antigen (HLA)-matching complicate interpretation of these data, they indicate that allogeneic HSCT is associated with significant risk of morbidity or mortality and unpredictable benefit [Citation15].
The development of reduced intensity conditioning (RIC) regimens has the potential to help broaden the treatment population, particularly with older patients [Citation16]. A prospective trial (ClinicalTrials.gov ID NCT00599547) of RIC allogeneic HSCT in 103 patients with MF and a median age of 55 years reported an estimated 51% event-free survival rate at 5 years and 16% treatment-related mortality at 1 year, which compares favorably to the prognosis for higher-risk MF [Citation17]. Factors predicting poorer outcome included age over 55 years and HLA-mismatched donor. In another multicenter study evaluating RIC allogeneic HSCT in 100 patients, treatment-related mortality and OS at 3 years were 43% and 42%, respectively [Citation18]. Similar positive results were recently reported in well-selected older patients [Citation19]. While allogeneic HSCT remains a potentially curative treatment for individuals deemed fit enough to endure the procedure and its related toxicities, it remains of unproven benefit when applied to a wide population of patients [Citation20]. Because of the high risk of treatment-related mortality in older patients, most intermediate and high risk patients are instead treated medically with a goal of palliation [Citation20].
Available medical and surgical management options
Hydroxyurea, which inhibits ribonucleotide reductase and is an S-phase cell cycle-specific nucleotide-depleting agent [Citation21], is one of the most commonly used medical therapies for patients with appreciably symptomatic MPNs [Citation22]. In a retrospective series of 40 patients treated with hydroxyurea at a starting dose of 500 mg/day, clinical improvement by International Working Group (IWG) criteria was observed in approximately 40% of patients and persisted for a median of 13 months. Hydroxyurea also resulted in improvement in splenomegaly, bone pain, constitutional symptoms and pruritus. Worsening anemia and pancytopenia were the most frequently observed adverse events (AEs) [Citation23]. While generally well tolerated, the modest improvement in symptoms with hydroxyurea is temporary, and exacerbation of cytopenias frequently limits treatment.
The antiangiogenic and immunomodulatory properties of thalidomide and lenalidomide make them potentially effective medical therapies for MF. The combination of thalidomide and prednisone was evaluated in 21 patients with MF and myeloid metaplasia [Citation24]. Sustained objective responses in anemia occurred in 62% of patients; however, the high incidence of neuropathy observed with thalidomide limits its utility. A long-term investigation of 50 patients with MF demonstrated an overall response rate of 28% by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria and a median duration of response of 8.5 months with three different thalidomide–prednisone-based regimens (alone or in combination with either oral cyclophosphamide for 3 months or continuous etanercept) [Citation25]. Fourteen patients (28%) had expired after a median follow-up of 3 years, with a median survival of 36 months. Major toxicities included grade ≥ 3 neuropathy (6%) and grade ≥ 3 cytopenia (20%). With respect to lenalidomide, a phase 2 clinical trial (ClinicalTrials.gov ID NCT00227591) assessed the therapeutic efficacy of lenalidomide in combination with prednisone in 42 patients with MF. Clinical improvement in anemia and splenomegaly was observed in 19% and 10% of subjects, respectively. Similar to thalidomide, toxicity characterized by cytopenias (88% having at least one grade 3–4 event) and non-hematologic toxicity (45% having at least one grade 3–4 event) with lenalidomide was reported [Citation26]. A second report of lenalidomide plus prednisone in 40 patients with intermediate or high risk MF yielded an overall response rate by IWG criteria of 30% for anemia and 42% for splenomegaly. The median time to response was 12 weeks. Grade 3 and 4 AEs included anemia (42%), neutropenia (58%) and thrombocytopenia (13%) [Citation27]. An analysis combining results from three phase 2 trials indicated that lenalidomide-based therapy may be more effective than thalidomide-based therapy. Furthermore, fewer patients treated with lenalidomide plus prednisone discontinued therapy due to toxicity compared with patients receiving thalidomide-based therapy [Citation28].
Interferon alpha (IFN-α) increases the expression of tumor-associated antigens and major histocompatibility complex antigens, as well as increases the activity of T cells, macrophages and natural killer cells [Citation29]. The efficacy and safety of pegylated IFN-α2a has been investigated in the treatment of MF. In one investigation with a median starting dose of 45 µg/week (peak starting doses ranged from 30 to 300 µg/week), non-hematologic toxicities included grade 1–3 fatigue (23%), grade 1 liver function test elevation (5%), grade 2–3 skin/allergic reaction (5%), grade 1–2 nausea (4%) and also grade 2 mood disorder (4%), with hematologic toxicities grade 3 or lower of anemia (5%), thrombocytopenia (9%) and leukopenia (7%) [Citation30]. Responses meeting IWG criteria occurred in fewer than 20% of patients with MF, but despite low response rates (RRs), IFN-α2a used clinically as treatment is fairly well tolerated [Citation30]. A second study involved 18 adult patients, 14 with the JAK2V617F mutation and 15 with secondary MF, treated with pegylated IFN-α2a. All but two patients achieved a response, and six had a complete response; two experienced complete disappearance of splenomegaly. Two of the three transfusion-dependent patients became transfusion-independent [Citation31]. A third study reported results in 17 patients with grade 1/2 MF treated with IFN-α. Fourteen patients received IFN-α2b while three received pegylated IFN-α2a. Fourteen patients derived benefit, with a complete response in two, a partial response in seven, clinical improvement in one and stable disease in four patients [Citation32]. Treatment with interferon is likely most appropriate in minimally symptomatic patients with low risk disease ().
Hypomethylating agents such as azacitidine and decitabine have been studied in MF, but play a limited role in the treatment of MF. A phase 2 trial of azacitidine in 34 patients with MF resulted in a complete response in one patient and clinical improvement in seven patients. The median duration of response was short-lived for those only attaining clinical improvement (4 months). The primary AEs were hematologic, with grade 3–4 neutropenia in 29% of patients [Citation33].
Other palliative medications that provide benefit in patients with MPNs include oral alkylating agents, corticosteroids, androgenic steroids such as danazol, and erythropoiesis-stimulating agents (ESAs). The oral alkylating agents, melphalan and busulfan, have been shown to improve splenomegaly and other symptoms of disease, but may also exacerbate cytopenias. Busulfan may also increase the risk of blastic transformation, which is of particular concern in younger patients [Citation8,Citation22,Citation34]. Corticosteroids (e.g. prednisone 0.5 mg/kg/day) may be temporarily effective for treatment of symptoms and are commonly used in combination with other therapies [Citation20]. ESAs can also benefit patients with MPNs, but clinical utility is limited as anemia responses that do occur (in < 60% of patients) are of approximately 1 year in duration [Citation35,Citation36]. Treatment of anemia is most common for patients with intermediate and high risk disease ().
Splenectomy is a palliative measure used for debulking in patients with MPNs. Splenectomy improves splenomegaly-related symptoms (early satiety, splenic infarcts and splenic exacerbation of cytopenias), but has no clear effect on survival or disease course. Splenectomy is associated with increased risk of several complications; one study of 314 patients reported fatal complications in 6.7% of patients undergoing the surgery [Citation37]. When performed prior to allogeneic transplant, splenectomy may facilitate disease eradication. Some reports have also shown faster engraftment in splenectomized patients; however, the use of splenectomy prior to transplant remains controversial [Citation8,Citation38,Citation39]. Given the high complication rate and limited benefit of splenectomy, appropriate patient selection is crucial [Citation8].
Splenic radiotherapy can be used to treat MPNs, as extramedullary hematopoiesis has demonstrated considerable sensitivity to external beam radiotherapy in patients with MF. However, splenic radiotherapy demonstrates transient benefit in patients with MPNs and may exacerbate cytopenias, particularly thrombocytopenia [Citation8]. A perioperative mortality rate of 11% was reported in a series of patients treated with splenic irradiation followed by splenectomy, indicating that irradiation may complicate future splenectomy if attempted [Citation40]. Overall, traditional treatment options are limited and inadequate to address the morbidity and mortality associated with MF. Novel therapeutic strategies are needed.
Janus kinase 2 mutations and MF
The discovery of a Janus kinase 2 (JAK2) gain-of-function mutation, JAK2V617F, has led to significant improvements in the understanding of the biology underlying MPNs, as well as the development of ruxolitinib, a JAK2 inhibitor and the first drug approved by the FDA for the treatment of MF [Citation41–44]. The JAK2 inhibitors SAR302503 (TG101348), CYT387 and pacritinib (SB1518) show promise in the treatment of patients with MPNs and are currently in clinical development (). While all of these compounds inhibit JAK2 by competing with adenosine triphosphate (ATP) for the ATP-binding catalytic site in the kinase domain, there are significant differences in their inhibition of other JAK family kinases, which may influence efficacy and AE profiles. Familiarity with their pharmacologic properties and clinical effects will be critical in the management of MF as these agents become approved for use.
Table I. Janus kinase 2 (JAK2) inhibitors for myelofibrosis.
The Janus kinase family of receptor tyrosine kinases includes four different proteins: JAK1, JAK2, JAK3 and TYK2. The JAK family proteins play a crucial role in myeloid and lymphoid cell proliferation and differentiation; their reactions are essential for the intracellular interactions of cytokine receptors, resulting in activation of signal transducer activator of transcription (STAT) factors and downstream promotion of genes that regulate cellular proliferation and differentiation [Citation42,Citation45]. The JAK2V617F mutation results in constitutive activation of JAK2, driving myeloid cell proliferation and differentiation.
JAK2V617F is present in the majority of patients with MF (50–60%), ET (∼50%) and PV (95%) [Citation41–45]. Additional mutations relevant to the JAK–STAT pathway have been identified in patients with MPNs, including MPL [Citation46], LNK [Citation47], TET2 [Citation48] and ASXL1 [Citation49]. JAK2V617F and other mutations can occur in the same patient at the same time, and multiple clones with different mutational profiles can occur in a single patient. The presence of JAK2V617F is related to increasing symptoms and stage of disease, although the precise correlation remains unclear [Citation50,Citation51]. For example, patients with a JAK2V617F mutation appear to have a higher risk of infections [Citation52]; however, the relationship between the JAK2V617F mutation and survival has not been consistent across studies [Citation50].
Allele burden is defined as the ratio of JAK2V617F to total JAK2 in a given patient (JAK2V617F/[JAK2V617F + wild-type (WT) JAK2]). The correlation between allele burden at diagnosis and pathogenesis and prognosis also remains unresolved. In some studies, patients with lower allele burden have demonstrated a better prognosis than those with higher allele burdens [Citation52,Citation53]. One study in patients with MF demonstrates that a low JAK2V617F allele burden at diagnosis represents an independent factor associated with shortened survival in patients with PMF [Citation52]. A similar study demonstrates that low JAK2V617F allele burden is associated with both inferior overall and leukemia-free survival [Citation53]. However, a separate study of patients with MF linked high allele burden to progression of disease, as the majority of patients with leukemia in the study were homozygous for the JAK2V617F mutation. A second study in patients with PV also reported higher allele burden present in patients progressing to AML [Citation50,Citation54,Citation55]. Studies have consistently demonstrated a link between increasing allele burden and increasing spleen size and enhanced myelopoiesis [Citation54].
JAK2 inhibitors
Ruxolitinib
Ruxolitinib is an orally available inhibitor of both JAK1 and JAK2, with a 50% inhibitory concentration (IC50) of 3.3 nM and 2.8 nM, respectively [Citation56]. For comparison, the IC50 for JAK3 is 428 nM, and for TYK2, 19 nM [Citation56]. The phase 1/2 trial (ClinicalTrials.gov ID NCT00509899) of ruxolitinib included patients with relapsed and/or refractory disease or intolerance to previous therapy, and a Lille score of intermediate or high risk [Citation57]. The phase 1 portion of the trial identified the maximum tolerated dose (MTD) of ruxolitinib as 25 mg twice daily, or 100 mg daily with thrombocytopenia as the dose-limiting toxicity. Subsequently, a starting dose of 15 mg twice daily with escalation up to 25 mg twice daily if no response and no toxicity observed was recommended. Of the 153 patients enrolled with a median age of 65 years, 82% were JAK2V617F positive and 92% had splenomegaly.
RRs were 52% and 49% in the 15 and 25 mg twice-daily cohorts, with no difference in response between JAK2V617F-positive and -negative patients. Significant and rapid improvements in constitutional symptoms and exercise ability were observed, which correlated with decreases in cytokines interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α). However, although improvements in cytokine levels and constitutional symptoms correlate in clinical trials, and increased cytokine expression can lead to symptoms in other disorders, the existence of a causal relationship between cytokine levels and constitutional symptoms in MPNs remains theoretical. Fourteen percent of transfusion-dependent patients became transfusion-independent, and a lower-than-expected rate of leukemic transformation was also observed. Overall, treatment was well tolerated, with non-hematologic toxicity occurring in fewer than 10% of patients. The most common AE was myelosuppression, specifically, anemia and thrombocytopenia [Citation57].
The results of COMFORT-I (ClinicalTrials.gov ID NCT00952289) and COMFORT-II (ClinicalTrials.gov ID NCT00934544), two randomized, controlled phase 3 trials of ruxolitinib, have recently been reported. COMFORT-I enrolled patients with MF in the United States, Canada and Australia and compared two doses of ruxolitinib with placebo, with a primary endpoint of a ≥ 35% spleen volume reduction at 24 weeks [Citation58]. Eligibility criteria included measurable splenomegaly, no prior splenectomy and intermediate-2 or high risk MF. Patients were randomized to placebo or ruxolitinib 15 mg (if platelet count 100–200 × 109/L) or 20 mg (if platelet count > 200 × 109/L) orally twice daily.
The primary endpoint of spleen RR was observed in 41.9% of patients treated with ruxolitinib and 0.7% of patients treated with placebo [Citation58]. A greater than 50% improvement in symptoms was reported in 45.9% and 5.3% of patients, respectively. Compared with placebo, ruxolitinib resulted in a survival advantage (hazard ratio [HR], 0.5; 95% confidence interval [CI], 0.25–0.98) [Citation58]. Using IWG criteria, 41.2% of patients in the ruxolitinib group and 46.9% in the placebo group dependent on transfusions at baseline became transfusion-independent. Symptomatic patients demonstrated a benefit from ruxolitinib therapy in both spleen volume and total symptoms score [Citation58]. Of particular interest, reductions in spleen volume of as little as 10% were associated with significant improvement in quality of life (QoL), with larger spleen volume reductions resulting in greater improvements in QoL [Citation59]. A reduction in allele burden of 10.9% and 21.5% was reported at 24 and 48 weeks, respectively. Common AEs associated with ruxolitinib are thrombocytopenia (all grades, 69.7%), anemia (all grades, 96.1%), neutropenia (all grades, 18.7%) and bruising (all grades, 18.7%) [Citation58].
The COMFORT-II trial enrolled patients in Europe and compared ruxolitinib to best available therapy (BAT) with the similar primary endpoint of spleen reduction by 35%, but measured at 48 weeks [Citation60]. Patients with intermediate and high risk MF with palpable splenomegaly were randomized (2:1) to ruxolitinib or BAT; the doses of ruxolitinib were the same as COMFORT-I. The spleen RRs for ruxolitinib and BAT were 32% vs. 0%, respectively, at 24 weeks, and 28% vs. 0% at 48 weeks, respectively. Anemia and thrombocytopenia were more frequent in patients receiving ruxolitinib than in patients receiving BAT [Citation60].
On the basis of the COMFORT-I and COMFORT-II trials, ruxolitinib became the first JAK2 inhibitor approved for MF, having received approval by the FDA in November 2011. Ruxolitinib was approved for patients with intermediate and high risk disease, and to date, phase 3 data with ruxolitinib are limited to patients with intermediate-2 and high risk disease and platelet counts ≥ 100 × 109/L. Preliminary safety and efficacy data were recently presented from a phase 2 study of ruxolitinib in patients with intermediate-1 to high risk MF and platelet counts of 50–100 × 109/L [Citation61]. A post hoc analysis of both COMFORT studies demonstrated similar symptom and QoL responses from baseline to week 24, as well as similar increases in median spleen volume from baseline to week 24, for patients who received placebo in COMFORT-I compared with patients who received BAT in COMFORT-II. Neither patient group experienced clinically meaningful improvements in either symptoms or QoL, which suggests that BAT for patients with MF provides little improvement in symptoms, QoL or spleen size compared with placebo, and provides strong rationale for the use of JAK2 inhibitors for the treatment of MF [Citation62]. Based on available safety and efficacy data, treatment with JAK2 inhibitors is most appropriate for symptomatic patients with intermediate or high risk disease who are ineligible for allogeneic HSCT ().
SAR302503 (TG101348)
SAR302503 is a JAK2 inhibitor currently under investigation in patients with MF. As compared with ruxolitinib, SAR302503 more selectively inhibits JAK2 than JAK1 or JAK3 with IC50 values of 3, 105 and 996 nM, respectively. In addition, SAR302503 also inhibits Fms-like tyrosine kinase 3 (FLT3) [Citation7]. FLT3 is known to play a significant role in the development of AML, but the potential relevance of MPNs to pathogenesis remains unclear [Citation63,Citation64]. A phase 1 trial of SAR302503 with eligibility criteria of symptomatic splenomegaly and intermediate/high risk disease enrolled 59 patients; 31 were in the dose-confirmation stage [Citation65]. Subjects with platelet count above 50 × 109/L were included, with data available about tolerance and activity. The MTD of SAR302503 was determined to be 680 mg daily with dose-limiting toxicity of hyperamylasemia (with or without hyperlipasemia).
The phase 1 trial (ClinicalTrials.gov ID NCT00631462) of SAR302503 demonstrated rapid and durable responses in symptoms, despite little effect on cytokine levels [Citation65]. Using IWG criteria, 39% and 47% of patients achieved a spleen response by six and 12 cycles of treatment, respectively. More than half of patients with complaints of night sweats, fatigue, early satiety, pruritus and cough exhibited durable improvement. The 23 patients with an allele burden greater than 20% at baseline (median 60%) had significant (p = 0.002) improvement in allele burden after six and 12 cycles of treatment (31% and 32%, respectively). The most common grade 3/4 AEs were anemia (35%), thrombocytopenia (24%), diarrhea (10%), neutropenia (10%), hyperlipasemia (10%), nausea (3%) and vomiting (3%). Long-term data from the phase 1 study demonstrated safety results consistent with earlier analysis, as well as durable effects on spleen size and JAK2V617F allele burden [Citation66]. Greater than 50% of patients treated at a dose of more than 240 mg/day obtained a spleen response of more than 50%. The median spleen size at the start of the study was 18 cm, decreasing to 8 cm in those patients completing 30 months of therapy. A durable and significant (p = 0.03) reduction in allele burden from baseline (n = 51, median reduction 20%) remained at 24 months of treatment (n = 21, median reduction 9%).
Phase 2 (ClinicalTrials.gov ID NCT01420770) and 3 (ClinicalTrials.gov ID NCT01437787) clinical trials measuring the safety and efficacy of SAR302503 in patients with MF are ongoing. JAKARTA, the phase 3 trial of SAR302503 in intermediate-2 and high risk patients with MF, is an international, multicenter study currently recruiting subjects. This three-arm, randomized, crossover study will compare 400 mg and 500 mg once-daily doses of SAR302503 with placebo in the reduction of spleen volume, with secondary endpoints including symptom improvement (MFSAF score), overall and progression-free survival, and safety. Other study endpoints include changes in JAK2 allele burden as well as histological, cytogenetic and molecular effects of SAR302503 on the bone marrow. Subjects previously treated with a JAK2 inhibitor are excluded from JAKARTA, but may be eligible for other ongoing clinical trials such as a phase 2 study of SAR302503 in patients with MF previously treated with ruxolitinib (NCT01523171).
CYT387
CYT387 is a JAK1/2 inhibitor under investigation for treatment of MF. CYT387 inhibits JAK1, JAK2 and tyrosine kinase 2 (TYK2) receptor tyrosine kinase subtypes at clinically achievable concentrations with IC50 values of 11, 18 and 17 nM, respectively. Additionally, CYT387 inhibits JNK1 and cyclin-dependent kinase 2 (CDK2) [Citation67]. Preliminary safety and efficacy results from a multicenter phase 1/2 trial of 60 patients with intermediate and high risk MF treated with CYT387 orally once daily were recently reported [Citation68]. Among 42 (33 transfusion-dependent) patients that could be evaluated for an anemia response, the overall RR was 50% (58% in transfusion-dependent patients), with a median duration of anemia response of 20 weeks (range 12–54 weeks). Nineteen patients (32%) achieved transfusion-independency, with only two requiring single episodes of packed red blood cell transfusions. Transfusion-dependent patients were required to be transfusion-free for 12 or more weeks. The rate of 32% observed in this study is lower than the 46.9% observed in the placebo arm of the COMFORT-I study; both studies used the IWG response criteria [Citation58,Citation68]. Responses were independent of JAK2V617F status, karyotype or blast count. A spleen response was observed in 45% of patients, and a majority of patients experienced resolution of pruritus, night sweats and bone pain.
The MTD for CYT387 was established at 300 mg daily, with dose-limiting toxicities of grade 3 hyperamylasemia and grade 3 headache [Citation68,Citation69]. The most common AE due to CYT387 was transient lightheadedness and hypotension after the first dose occurring in 50% of patients, while grade 3/4 thrombocytopenia occurred in 25% of patients. The RR for anemia is unique among the JAK2 inhibitors reported thus far. Safety data from the expanded study in 163 patients revealed an 85% patient retention for the 9-month core study. Toxicity data were updated, with grades 3 and 4 thrombocytopenia occurring with a combined incidence of 16%, and new reports of grade 1 peripheral sensory neuropathy. Benefits to spleen size and constitutional symptoms, as well as transfusion-independence, were observed in the expanded patient population [Citation69].
Pacritinib (SB1518)
Pacritinib is a selective JAK2 and FLT3 inhibitor with IC50 values of 23, 19 and 22 nM for JAK2WT, JAK2V617F and FLT3, respectively [Citation70]. The IC50 values for JAK1, JAK3 and TYK2 are 1280, 520 and 50 nM, respectively. A phase 2 trial evaluated pacritinib 400 mg daily in patients with MF and palpable splenomegaly [Citation71]. Eleven patients (32%) showed ≥ 35% reduction in spleen volume at week 24 as assessed by magnetic resonance imaging (MRI); 15 (44%) showed decreases ≥ 50% in palpable splenomegaly; and six (18%) achieved clinical resolution of splenomegaly. Treatment was discontinued in 24% of patients due to AEs, including elevated bilirubin, thrombocytopenia, allergic reaction and gastrointestinal bleed. The most common treatment-related AEs were gastrointestinal in nature and manageable.
A second phase 2 trial reported results in 33 patients with MF and splenomegaly ≥ 5 cm below the left costal margin. Among 30 patients assessed by MRI, 29 had spleen volume reduction, 17 (57%) with reduction ≥ 25%. The most commonly reported AEs included diarrhea (81%, 6% grade 3), nausea (41%) and vomiting (22%). There were no grade 3 or 4 neutropenia or thrombocytopenia events reported [Citation72].
Other JAK2 inhibitors
Several other JAK2 inhibitors have been or are being investigated for the treatment of patients with MF, including AZD-1480, BMS-911543, NS-018, LY2784544, CEP-701 (lestaurtinib) and AT-9283. Some, such as CEP-701, are no longer under investigation for MF, while others are in early-stage development with no clinical data available at this time. Results of a phase 1 study involving 19 patients (17 with MF, one with post-essential thrombocythemia and one with PV) treated with LY2784544 were recently announced [Citation73]. A palpable spleen reduction of at least 35% was observed in 13 of 17 evaluable patients (76%). Seven patients with MF had a ≥ 50% reduction in spleen size, four classified as clinical improvement by IWG-MRT criteria. A drug-related serious AE was reported in four patients, including increased serum creatinine (n = 4, grade 2), hyperuricemia (n = 2, grade 4) and hyperkalemia (n = 1, grade 1). The MTD was determined to be 120 mg.
Emerging therapies
In addition to JAK2 inhibition, several other treatment strategies are under investigation for the treatment of MF and MPNs, including immunomodulating drugs (IMiDs), inhibition of the mammalian target of rapamycin (mTOR) pathway, and targeting epigenetic factors (). Pomalidomide is a second-generation immunomodulatory drug under evaluation in a range of doses for treatment of MPNs. A phase 2 trial evaluating low-dose pomalidomide (0.5 mg/day) and prednisone in 58 patients reported anemia responses in 24% of the JAK2V617F-positive patients [Citation74,Citation75]. Anemia responses were absent in those without the mutation, and predicted by pomalidomide-induced basophilia and the absence of marked splenomegaly [Citation74,Citation75]. An analysis of 82 evaluable patients with MF enrolled in three consecutive phase 1 and 2 clinical trials from 2007 to 2010 demonstrated an anemia response in 27% of patients according to IWG-MRT criteria. Anemia response occurred most often in the first 6 months of treatment, in the presence of JAK2V617F, and in the absence of marked splenomegaly. Grade 1 sensory neuropathy was observed in 16% of patients given pomalidomide for at least 12 months [Citation76].
Table II. Other investigational therapies for myelofibrosis (MF).
The mTOR inhibitor, everolimus, demonstrated favorable activity in a phase 1/2 trial of intermediate and high risk MF [Citation77]. A total of 69% and 80% of patients experienced complete resolution of systemic symptoms and pruritus, respectively [Citation77]. Responses in leukocytosis, anemia and thrombocytosis occurred in 15–25% of patients. Clinical responses were not associated with a reduction in JAK2V617F burden or cytokine levels. Everolimus was well tolerated, with grades 1 and 2 stomatitis the most frequent AE, occurring in 70% of patients.
Histone deacetylase (HDAC) inhibition has an inhibitory effect on the JAK–STAT pathway, and HDAC inhibitors givinostat (ITF2357), panobinostat (LBH589) and pracinostat (SB939) are all currently under investigation for MF. In a recent study, treatment with pracinostat resulted in a reduction in spleen size in 27% of patients, and IWG criteria anemia responses in approximately 10% of patients. The most common AEs were grades 1 and 2 fatigue (91%), peripheral edema, pain and diarrhea [Citation78]. Panobinostat in doses of 20, 25 and 30 mg three times daily also demonstrated encouraging activity in results from a phase 1/2 trial [Citation79]. Durable (> 6 months) responses in splenomegaly, decreases in leukoerythroblastosis, and responses in anemia were reported by IWG criteria. Givinostat also shows promise based on results of a randomized phase 2 trial in patients with PV that did not respond to hydroxyurea. A complete or partial response was observed in approximately 50% of patients, and the combination was generally well tolerated [Citation80].
Future directions
The approval of ruxolitinib by the FDA signifies a new era in the treatment of MF. The degree and duration of symptomatic responses reported with ruxolitinib and other investigational JAK2 inhibitors suggest that they will provide significant benefit to patients compared with traditional therapies. Improvements in symptoms have been closely correlated with decreases in splenomegaly [Citation59]. Available data suggest that treatment with JAK2 inhibitors is a safe and efficacious therapy for symptomatic patients and patients with intermediate or high risk disease. While the COMFORT-I and COMFORT-II trials of ruxolitinib enrolled symptomatic patients with intermediate and high risk disease, data are limited in patients with lower risk disease, and questions remain regarding whether treatment can slow progression of disease and decrease the risk for leukemic transformation. In addition, the malignant clone persists, and disease resistance occurs frequently either de novo or after an initial response to treatment with JAK2 inhibitors. Additional strategies may be needed to optimize QoL and improve OS.
Additional JAK2 inhibitors, such as SAR302503, are in late-stage clinical trials for treatment of MF. Understanding the differences in pharmacology, RRs and safety/tolerability profiles among JAK2 inhibitors will be critical for optimizing therapy and defining alternatives of treatment for intolerant or relapse/resistant patients. Such studies are already under way, for example a phase 2 trial (NCT01523171) of SAR302503 in patients previously treated with ruxolitinib. The differences among the JAK2 inhibitors provide an opportunity to further define the contribution to clinical efficacy and toxicity of other JAK proteins, related pathways and off-target effects of JAK2 inhibitors. The additional specificity of various JAK2 inhibitors for JAK1, FLT3 and other kinases will help to increase the understanding of the role of these kinases in the pathogenesis of disease.
Differences in pharmacology among the JAK2 inhibitors are a likely cause of the different outcomes observed in clinical trials. Significant decreases in allele burden have been achieved with SAR302503; however, the clinical relevance of this reduction remains unclear [Citation57,Citation65]. Treatment with ruxolitinib results in a reduction of both cytokine levels and constitutional symptoms; however, treatment with SAR302503 results in similar IWG spleen RRs and symptom reduction, but cytokine levels are not reduced upon treatment with SAR302503. This suggests that cytokine levels may not correlate with all outcomes and are not causally related to other outcomes. Patients treated with CYT387 demonstrate low rates of anemia and may attain transfusion-independence [Citation68].
While JAK2 inhibition marks a breakthrough in the treatment of MF, responses are variable and not always durable. Clinical outcomes in patients with MF may be enhanced by targeting other pathways relevant to the pathophysiology of MF in addition to JAK2 inhibition. Preclinical data suggest that the combination of JAK2 and HDAC inhibition may be more effective than either agent alone [Citation81], and a phase 1 clinical trial evaluating the combination of ruxolitinib and panobinostat is under way (ClinicalTrials.gov ID NCT01433445). Combining JAK2 inhibition with immunomodulation and antiangiogenesis also holds therapeutic promise, and is currently being investigated in a phase 2 trial using the combination of ruxolitinib and lenalidomide (ClinicalTrials.gov ID NCT01375140). Research in preclinical models may help to identify additional combinations of treatment, optimize pharmacodynamic interactions, and guide further clinical development of the JAK2 inhibitors to improve clinical outcomes in MF.
Conclusions
MPNs affect 2–5 per 100 000 people in the United States each year, with significant morbidity and mortality. Traditional treatments are primarily palliative and have proven inadequate to address the morbidity and mortality associated with MF. Ruxolitinib, which was recently approved by the FDA, as well as investigational JAK2 inhibitors, present new hope for patients with MF. JAK2 inhibitors have the potential to provide significant reductions in splenomegaly, palliation of disease-related symptoms, and improvements in hematologic parameters. Additionally, SAR302503 demonstrates a reduction in allele burden in clinical trials, CYT387 demonstrates low rates of anemia, and ruxolitinib has been shown to prolong OS. In light of the differences between inhibitors, the treatment paradigm is likely to evolve rapidly in the coming years. For example, SAR302503 exhibits potential due to its highly selective inhibition for JAK2 and potential to reduce allele burden. Drugs based on alternative treatment strategies are also entering clinical trials, and in the future, combinations of drugs targeting multiple pathways known to be disrupted in MF are likely to be required, to increase disease responses and prolong response duration. Increased research regarding the mechanisms of disease sensitivity and resistance can help design better treatments and optimize patient selection.
Supplementary Material
Download Zip (488.9 KB)Potential conflict of interest
A disclosure form provided by the author is available with the full text of this article at www.informahealthcare.com/lal.
Editorial support was provided by Eric Block, PhD, at Phase Five Communications Inc., and funded by sanofi-aventis US.
References
- Barosi G, Mesa RA, Thiele J, . Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008;22:437–438.
- Hoffman R, Rondelli D. Biology and treatment of primary myelofibrosis. Hematology Am Soc Hematol Educ Program 2007:346–354.
- Mesa RA, Niblack J, Wadleigh M, . The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international internet-based survey of 1179 MPD patients. Cancer 2007;109:68–76.
- Cervantes F, Dupriez B, Pereira A, . New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009;113:2895–2901.
- Scherber R, Dueck AC, Johansson P, . The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood 2011;118:401–408.
- Mesa RA, Li CY, Ketterling RP, . Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood 2005;105:973–977.
- Wernig G, Kharas MG, Okabe R, . Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617f-induced polycythemia vera. Cancer Cell 2008;13:311–320.
- Mesa RA. How I treat symptomatic splenomegaly in patients with myelofibrosis. Blood 2009;113:5394–5400.
- Tefferi A. How I treat myelofibrosis. Blood 2011;117:3494–3504.
- Gangat N, Caramazza D, Vaidya R, . DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011;29:392–397.
- Passamonti F, Cervantes F, Vannucchi AM, . A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010;115:1703–1708.
- Mesa RA, Schwager S, Radia D, . The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res 2009;33:1199–1203.
- Cherington C, Slack JL, Leis JF, . Induction therapy followed by allogeneic stem cell transplant for MPN blast phase: outcomes from Mayo Clinic in Arizona. Blood 2011;118(Suppl. 1): Abstract 1749.
- Scott BL, Gooley TA, Sorror ML, . The Dynamic International Prognostic Scoring System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood 2012;119:2657–2664.
- Ballen KK, Shrestha S, Sobocinski KA, . Outcome of transplantation for myelofibrosis. Biol Blood Marrow Transplant 2010;16:358–367.
- Zang DY, Deeg HJ. Allogeneic hematopoietic cell transplantation for patients with myelofibrosis. Curr Opin Hematol 2009;16:140–146.
- Kroger N, Holler E, Kobbe G, . Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 2009;114:5264–5270.
- Patriarca F, Bacigalupo A, Sperotto A, . Allogeneic hematopoietic stem cell transplantation in myelofibrosis: the 20-year experience of the Gruppo Italiano Trapianto di Midollo Osseo (GITMO). Haematologica 2008;93:1514–1522.
- Samuelson S, Sandmaier BM, Heslop HE, . Allogeneic haematopoietic cell transplantation for myelofibrosis in 30 patients 60–78 years of age. Br J Haematol 2011;153:76–82.
- Tefferi A. Primary myelofibrosis: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2011;86:1017–1026.
- Madaan K, Kaushik D, Verma T. Hydroxyurea: a key player in cancer chemotherapy. Expert Rev Anticancer Ther 2012;12:19–29.
- Cervantes F, Pereira A. Advances in the understanding and management of primary myelofibrosis. Curr Opin Oncol 2011;23:665–671.
- Martinez-Trillos A, Gaya A, Maffioli M, . Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patients. Ann Hematol 2010;89:1233–1237.
- Mesa RA, Steensma DP, Pardanani A, . A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003;101:2534–2541.
- Thapaliya P, Tefferi A, Pardanani A, . International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol 2011;86:96–98.
- Mesa RA, Yao X, Cripe LD, . Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase 2 trial E4903. Blood 2010;116:4436–4438.
- Quintas-Cardama A, Kantarjian HM, Manshouri T, . Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol 2009;27:4760–4766.
- Jabbour E, Thomas D, Kantarjian H, . Comparison of thalidomide and lenalidomide as therapy for myelofibrosis. Blood 2011;118:899–902.
- Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011;117:4706–4715.
- Gowin K, Thapaliya P, Samuelsson J, . Experience with pegylated interferon alpha-2a in advanced myeloproliferative neoplasms in an international cohort of 118 patients. Haematologica 2012 Mar 14. [Epub ahead of print]
- Ianotto JC, Kiladjian JJ, Demory JL, . PEG-IFN-α-2a therapy in patients with myelofibrosis. A study of the French Group d’Etudes des Myelofibroses (GEM) and France Intergroupe des syndromes Myeloproliferatifs (FIM). Br J Haematol 2009;146:223–225.
- Silver RT, Vandris K, Goldman JJ. Recombinant interferon-α may retard progression of early primary myelofibrosis: a preliminary report. Blood 2011;117:6669–6672.
- Quintas-Cardama A, Tong W, Kantarjian H, . A phase II study of 5-azacytidine for patients with primary and post-essential thrombocythemia/polycythemia vera myelofibrosis. Leukemia 2008; 22:965–970.
- Nielsen I, Hasselbalch HC. Acute leukemia and myelodysplasia in patients with a Philadelphia chromosome negative chronic myeloproliferative disorder treated with hydroxyurea alone or with hydroxyurea after busulphan. Am J Hematol 2003;74:26–31.
- Cervantes F, Alvarez-Larran A, Hernandez-Boluda JC, . Erythropoietin treatment of the anaemia of myelofibrosis with myeloid metaplasia: results in 20 patients and review of the literature. Br J Haematol 2004;127:399–403.
- Huang J, Tefferi A. Erythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin level. Eur J Haematol 2009;83:154–155.
- Mesa RA, Nagorney DS, Schwager S, . Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer 2006;107:361–370.
- Deeg HJ, Gooley TA, Flowers ME, . Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood 2003;102:3912–3918.
- Guardiola P, Anderson JE, Bandini G, . Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Societe Francaise de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood 1999;93:2831–2838.
- Elliott MA, Chen MG, Silverstein MN, . Splenic irradiation for symptomatic splenomegaly associated with myelofibrosis with myeloid metaplasia. Br J Haematol 1998;103:505–511.
- Baxter EJ, Scott LM, Campbell PJ, . Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365:1054–1061.
- James C, Ugo V, Le Couedic JP, . A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434:1144–1148.
- Kralovics R, Passamonti F, Buser AS, . A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005;352:1779–1790.
- Levine RL, Wadleigh M, Cools J, . Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7:387–397.
- Quintas-Cardama A, Kantarjian H, Cortes J, . Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov 2011;10:127–140.
- Pikman Y, Lee BH, Mercher T, . MPLW515l is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006;3:e270.
- Oh ST, Simonds EF, Jones C, . Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 2010;116:988–992.
- Delhommeau F, Dupont S, Della Valle V, . Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360:2289–2301.
- Carbuccia N, Murati A, Trouplin V, . Mutations of ASXl1 gene in myeloproliferative neoplasms. Leukemia 2009;23:2183–2186.
- Barosi G, Bergamaschi G, Marchetti M, . JAK2 V617F mutational status predicts progression to large splenomegaly and leukemic transformation in primary myelofibrosis. Blood 2007;110:4030–4036.
- Tefferi A, Lasho TL, Schwager SM, . The JAK2(V617F) tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specificity and clinical correlates. Br J Haematol 2005;131:320–328.
- Guglielmelli P, Barosi G, Specchia G, . Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009;114:1477–1483.
- Tefferi A, Lasho TL, Huang J, . Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia 2008;22:756–761.
- Passamonti F, Rumi E. Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica 2009;94:7–10.
- Passamonti F, Rumi E, Pietra D, . A prospective study of 338 patients with polycythemia vera: the impact of Jak2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 2010;24:1574–1579.
- Quintas-Cardama A, Vaddi K, Liu P, . Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010;115:3109–3117.
- Verstovsek S, Kantarjian H, Mesa RA, . Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010;363:1117–1127.
- Verstovsek S, Mesa RA, Gotlib J, . A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012;366:799–807.
- Mesa RA, Gotlib J, Gupta V, . Associations between improvements in myelofibrosis (MF) symptoms and quality of life measures with splenomegaly reduction in COMFORT-1: A randomized, double-blind, phase III trial of the JAK1 and JAK2 inhibitor ruxolitinib versus placebo in patients with MF. Blood 2011;118(Suppl. 1): Abstract 3842.
- Harrison C, Kiladjian JJ, Al-Ali HK, . JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012;366:787–798.
- Talpaz M, Hamburg SI, Jamieson K, . Preliminary safety and efficacy of ruxolitinib in patients (pts) with primary and secondary myelofibrosis (MF) with platelet counts (PC) of 50–100x109/L. J Clin Oncol 2012;30(Suppl.): Abstract 6630.
- Mesa RA, Verstovsek S, Cervantes F, . Comparison of the efficacy of placebo and best available therapy for the treatment of myelofibrosis. Blood 2011;18(Suppl. 1): Abstract 1753.
- Estey EH. Acute myeloid leukemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012;87:89–99.
- Li L, Bailey E, Greenblatt S, . Loss of the wild-type allele contributes to myeloid expansion and disease aggressiveness in FLT3/ITD knockin mice. Blood 2011;118:4935–4945.
- Pardanani A, Gotlib JR, Jamieson C, . Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol 2011;29:789–796.
- Pardanani A, Gotlib J, Jamieson C, . SAR302503: interim safety, efficacy and long-term impact of JAK2 V617F allele burden in a phase I/II study in patients with myelofibrosis. Blood 2011;118(Suppl. 1): Abstract 3838.
- Tyner JW, Bumm TG, Deininger J, . CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010;115:5232–5240.
- Pardanani AD, Caramazza D, George G, . Safety and efficacy of CYT387, a JAK-1/2 inhibitor, for the treatment of myelofibrosis. J Clin Oncol 2011;29(Suppl.): Abstract 6514.
- Pardanani A, Gotlib J, Gupta V, . An expanded multicenter phase I/II study of CYT387, a JAK-1/2 inhibitor for the treatment of myelofibrosis. Blood 2011;118(Suppl. 1): Abstract 3849.
- Hart S, Goh KC, Novotny-Diermayr V, . SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011;25:1751–1759.
- Komrokji RS, Wadleigh M, Seymour JF, . Results of a phase 2 study of pacritinib (SB1518), a novel oral JAK2 inhibitor, in patients with primary, post-polycythemia vera, and post-essential thrombocythemia myelofibrosis. Blood 2011;118(Suppl. 1): Abstract 282.
- Deeg HJ, Odenike O, Scott BL, . Phase II study of SB1518, an orally available novel JAK2 inhibitor, in patients with myelofibrosis. J Clin Oncol 2011;29(Suppl.): Abstract 6515.
- Verstovsek S, Mesa RA, Rhoades SK, . Phase I study of the JAK2 V617F inhibitor, LY2784544, in patients with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET). Blood 2011;118(Suppl.1): Abstract 2814.
- Begna KH, Mesa RA, Pardanani A, . A phase-2 trial of low-dose pomalidomide in myelofibrosis. Leukemia 2011;25:301–304.
- Mesa RA, Pardanani AD, Hussein K, . Phase1/-2 study of pomalidomide in myelofibrosis. Am J Hematol 2010;85:129–130.
- Begna K, Pardanani A, Mesa RA, . Pomalidomide therapy for myelofibrosis: analysis of results from three consecutive clinical trials. Blood 2011;118(Suppl. 1): Abstract 1759.
- Guglielmelli P, Barosi G, Rambaldi A, . Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood 2011;118:2069–2076.
- Quintas-Cardama A, Kantarjian HM, Borthakur G, . Therapy with histone deacetylase inhibitor SB939 for patients with myelofibrosis. Blood 2011;118(Suppl. 1): Abstract 3863.
- Mascarenhas J, Mercado A, Rodriguez A, . Prolonged low dose therapy with pan-deacetylase inhibitor, panobinostat (LBH589), in patients with myelofibrosis. Blood 2011;118(Suppl. 1): Abstract 794.
- Rambaldi A, Finazzi G, Vannucchi AM, . A phase II study of the HDAC inhibitor givinostat in combination with hydroxyurea in patients with polycythemia vera resistant to hydroxyurea monotherapy. Blood 2011;118(Suppl. 1): Abstract 1748.
- Baffert F, Evrot E, Ebel N, . Improved efficacy upon combined JAK1/2 and pan-deacetylase inhibition using ruxolitinib (INC424) and panobinostat (LBH589) in preclinical mouse models of JAK2V617F-driven disease. Blood 2011;118(Suppl. 1): Abstract 798.