
Abstract
Acute myeloid leukemia (AML) blasts express high levels of interlekin-3 (IL-3) receptor-α (CD123). CSL360 is a recombinant, chimeric immunoglobulin G1 (IgG1), anti-CD123 monoclonal antibody (MoAb) that neutralizes IL-3 and demonstrates anti-leukemic activity in vitro. This phase 1 study assessed safety, pharmacokinetics and bioactivity of weekly intravenous CSL360 for 12 weeks in 40 patients with advanced AML across five dose levels (0.1–10.0 mg/kg). Other than mild infusion reactions, CSL360 was well tolerated. The maximal tolerated dose was not reached. The half-life was 4.9 days, and the area under the curve (AUC) and maximum concentration (Cmax) increased proportionally with dose. Doses ≥ 3.0 mg/kg resulted in complete saturation and down-regulation of CD123 and abolition of ex vivo proliferative responsiveness to IL-3, indicating adequate blockade of IL-3 signaling. Two patients responded, with one remaining in complete remission after 17 doses. CSL360 bound CD123 specifically, but did not induce anti-leukemic activity in most patients. While safe, MoAb blockade of CD123 function is insufficient as a therapeutic strategy.
Introduction
Treatment of acute myeloid leukemia (AML) has changed little over the last 30 years, and cure in younger patients is dependent upon induction and consolidation therapy with cytosine arabinoside- and anthracycline-containing regimens, and allogeneic stem cell transplant in selected patients. Relapse is common and salvage treatment with cytotoxic chemotherapy is rarely curative [Citation1]. The treatment of older adults (> 65 years of age) with AML remains very unsatisfactory, with median survival less than a year and little improvement in overall prognosis over the past three decades [Citation2,Citation3].
There is growing evidence to suggest that AML arises from leukemic stem cells (LSCs) [Citation4,Citation5]. LSCs exhibit many characteristics similar to those of normal hematopoietic stem cells (HSCs), such as surface CD34 + expression and absent or low level CD38 expression, and are thought to arise from normal stem cells through accumulation of oncogenic insults or from differentiated progenitor cells that have re-acquired the capacity for self-renewal [Citation4,Citation6]. LSCs have been shown to be quiescent and relatively resistant to DNA damaging chemotherapy such as cytarabine and anthracyclines [Citation7,Citation8]. It is hypothesized that the persistence of LSCs underpins relapse after initial remission, and thus the eradication of LSCs can be considered a requirement for cure, and an important therapeutic goal.
One potential therapeutic target in AML is the surface receptor CD123, the interleukin-3 receptor alpha chain (IL-3Rα). CD123 is constitutively expressed on normal committed hematopoietic progenitor cells. CD123 is expressed in a variety of hematological neoplasms, including AML, myelodysplasia, B-cell acute lymphoblastic leukemia, Hodgkin lymphoma, hairy cell leukemia and chronic myeloid leukemia [Citation9–11]. The majority of AML blasts express surface CD123. This expression is consistently at a higher density than observed for normal CD34 + cells [Citation12–14] and does not vary by subtype of AML [Citation14,Citation15]. CD123 also appears to be a distinguishing marker of AML LSCs. High levels of CD123 expression are found on CD34 + CD38− LSCs, in contrast to minimal or absent expression on HSCs in normal BM [Citation9,Citation16–18], and it is presumed that this heightened expression is of functional importance to the leukemic clone. Higher expression of CD123 on AML LSCs at diagnosis has been reported to be associated with a poorer prognosis [Citation19].
7G3 is a murine anti-CD123 monoclonal antibody directed specifically against the N-terminal domain of the human IL-3Rα [Citation20]. It binds to human surface CD123 and prevents IL-3 binding to its receptor in a dose-dependent manner [Citation20]. In vitro blockade of IL-3Rα by 7G3 in AML cell lines completely abolishes proliferation induced by IL-3 [Citation18], suggesting that deprivation of paracrine or autocrine cytokine stimulation may be a mechanism for anti-leukemic activity. Preclinical in vivo data demonstrated that CD123 plays a vital role in engraftment and establishment of AML disease in a repopulating xenograft model using NOD/SCID (non-obese diabetic/severe combined immune deficiency) mice. Anti-CD123 monoclonal antibody profoundly reduced engraftment of human AML cells, but not normal bone marrow (BM) cells, and in vivo treatment by 7G3 was shown to reduce not only disease burden in NOD/SCID mice with established AML, but also secondary transplantability [Citation18].
CSL360 is a 7G3-derived immnoglobuin G1 (IgG1) recombinant chimeric monoclonal antibody, and has identical biological activity against the receptor [ and ]. A toxicology study in non-human primates (cynomolgus macaques) using up to 100 mg/kg of CSL360 showed no significant effects on hematological parameters [Citation18]. We report the results of the first-in-human trial of CSL360 in patients with advanced AML.
Figure 1. Both CSL360 and 7G3 effectively neutralize IL-3-induced proliferation (A) and signaling (B) in primary AML cells. (A) Primary AML cells (1 × 105 cells), collected at diagnosis from non-CSL360 trial patients who provided informed consent for inclusion on another HREC-approved research study [Citation39], were cultured in RPMI/10% FCS and 10 ng/mL IL-3 in the presence of increasing concentrations of CSL360 or 7G3 for 48 h at 37°C. Cell proliferation was determined by measuring 3[H]-thymidine incorporation. The mean and SEM of proliferation as a percentage of control (IL-3 alone) from four different AML patient samples are shown. (B) Primary AML cells (1 × 105 cells) were incubated with 10 μg/mL CSL360 or 7G3 for 15 min prior to stimulation with 10 ng/mL IL-3 for a further 15 min at 37°C. The percentage of pSTAT5-positive blasts was determined by flow cytometry.
![Figure 1. Both CSL360 and 7G3 effectively neutralize IL-3-induced proliferation (A) and signaling (B) in primary AML cells. (A) Primary AML cells (1 × 105 cells), collected at diagnosis from non-CSL360 trial patients who provided informed consent for inclusion on another HREC-approved research study [Citation39], were cultured in RPMI/10% FCS and 10 ng/mL IL-3 in the presence of increasing concentrations of CSL360 or 7G3 for 48 h at 37°C. Cell proliferation was determined by measuring 3[H]-thymidine incorporation. The mean and SEM of proliferation as a percentage of control (IL-3 alone) from four different AML patient samples are shown. (B) Primary AML cells (1 × 105 cells) were incubated with 10 μg/mL CSL360 or 7G3 for 15 min prior to stimulation with 10 ng/mL IL-3 for a further 15 min at 37°C. The percentage of pSTAT5-positive blasts was determined by flow cytometry.](/cms/asset/ed05201b-5592-4a9e-97a7-147d76035971/ilal_a_956316_f0001_b.gif)
Materials and methods
Study design
This was an open-label, multi-center dose-escalation phase 1 study of CSL360 in patients with relapsed, refractory or high-risk AML (ClinicalTrial.gov Identifier: NCT00401739). The primary objectives were to investigate the safety and the pharmacokinetic profile of CSL360. The secondary objectives were to determine the maximum tolerated dose (MTD), assess its anti-leukemic activity, investigate its biological activity, determine the biologically effective dose(s) and investigate its immunogenicity.
The study was conducted according to the Declaration of Helsinki and relevant International Conference on Harmonisation Good Clinical Practice guidelines, and with approval from the local Human Research Ethics Committee (HREC) of all participating study sites. All participants provided written and dated informed consent before participating in this study.
Patients
Patients with relapsed AML (after at least one cycle of conventional chemotherapy), or refractory AML (after at least one cycle of induction chemotherapy), or newly diagnosed AML (if unfit for standard chemotherapy and, if ≤ 70 years old, with poor risk features: unfavorable cytogenetics or history of myelodysplasia) were included. Patients were considered eligible if they: (1) provided written informed consent, (2) were ≥ 18 years of age, (3) had an Eastern Cooperative Oncology Group (ECOG) performance score of 0, 1 or 2, (4) had resolved all clinically significant toxic effects of any prior surgery, radiotherapy, targeted therapy or chemotherapy, (5) were ≥ 4 weeks following intravenous (IV) immunoglobulin or non-myeloablative chemotherapy, or > 3 months post-stem cell transplant (SCT), (6) had adequate renal (serum creatinine ≤ 1.5 × upper limit of normal [ULN]) and hepatic function (total bilirubin ≤ 1.5 × ULN). Patients were excluded if any of the following applied: (1) peripheral blood (PB) blast count > 30 × 09/L, or rapidly progressive AML, (2) prior solid organ transplant, (3) active graft-versus-host disease (GVHD), (4) concurrent treatment with other anti-cancer therapy or any investigational agent within 30 days, (5) evidence of AML in the central nervous system, (6) active bacterial or fungal infection, (7) hepatitis B, hepatitis C or human immunodeficiency virus (HIV) infection, (8) active invasive malignancy other than adequately treated basal or squamous cell carcinoma of the skin or cervical carcinoma in situ or (9) previous allergic reaction to a Chinese hamster ovary (CHO) cell line derived product. The study was conducted at six sites in Australia commencing in June 2007 and was completed in September 2010.
Treatment
Initial treatment with CSL360 was administered as an IV infusion over 90 min on days 1, 8, 15 and 22. The weekly dosing regimen was based on preclinical pharmacokinetics (PK) data. All patients were admitted for administration of the first dose and observed overnight. If there was no evidence of disease progression after the first four doses, further weekly doses were administered up to a total of 12. Patients achieving a complete (CR) or partial response according to National Comprehensive Cancer Network (NCCN) response criteria [Citation21] were permitted to receive additional weekly doses beyond the 12 doses. Five dose levels of CSL360 were planned at 0.1, 0.3, 1.0, 3.0 and 10.0 mg/kg. No intra-patient dose escalation was allowed.
All patients received appropriate supportive care for the prevention of tumor lysis syndrome as per institutional practice. The use of prophylactic granulocyte colony stimulating factor (G-CSF) was not permitted during the study except for the treatment of febrile neutropenia.
In the event of an infusion reaction, the infusion was slowed or interrupted and medication with oral paracetamol and IV antihistamine and/or IV corticosteroids at the discretion of the investigators would be given immediately and as pre-medication for all subsequent doses.
Study assessments
Study assessments (history, physical examination, ECOG performance, hematology and biochemistry) were performed at screening, daily in the first week, then weekly during therapy and at 30 days after completion. All adverse events (AEs) and serious adverse events (SAEs) were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE version 3.0). Pre-existing conditions or laboratory abnormalities present at screening and which did not worsen during the study period were not considered as AEs in the study. All patients receiving a dose of CSL360 in the dose-escalation cohorts were evaluable for determination of dose-limiting toxicity (DLT) and MTD. DLTs were defined as CSL360-related (possibly, probably or definitely) AE ≥ grade 3 occurring during the first four doses of CSL360.
Dose escalation
Patients were assigned sequentially to dose-escalation cohorts of a minimum of three. Patients who did not complete the first four doses were replaced if they were discontinued due to disease progression. Dose escalation was allowed to the next higher level if no patient experienced a DLT. If one patient experienced a DLT at a particular dose, a total of at least six patients were to be enrolled. If no further DLT occurred, dose-escalation would recommence. If two or more patients enrolled at a dose level experienced a DLT, additional patients would be enrolled at the next lower dose level to a total of six patients, and no further dose-escalation was allowed. The MTD was defined as the highest dose level with no more than one DLT during the first four doses of CSL360. If ≤ 1 DLT was observed at 10.0 mg/kg in the first six patients, expansion up to 20 patients was allowable for this highest dose cohort.
Pharmacokinetics and immunogenicity testing
Serum samples were collected before dosing, at 45 min after starting the first infusion, then immediately after infusion, 1, 2, 3, 4, 6 and 24 h post dosing and on days 3, 4, 5 and 7. The same PK sampling schedule was repeated for dose 4. Additional samples were collected prior to, and immediately post-, doses 2 and 3. Serum samples (stored at − 70°C) were analyzed by non-competitive sandwich enzyme immuno-assay for unbound CSL360 concentrations: microplates (Nunc Maxisorp; Thermo Fisher Scientific, Roskilde, Denmark) were coated with soluble IL-3Rα (R&D Systems, Minneapolis, MN) and freeze dried; 10 mL of serum was serially diluted in assay diluent (phosphate buffered saline with 0.1% casein) in the pre-coated microplates; samples were incubated for 1 h, allowing the free CSL360 in the sample to bind to the immobilized receptor; plates were washed to remove any unbound CSL360 and non-specific serum components; bound CSL360 was detected by incubating with polyclonal goat anti-human IgG conjugated with horseradish peroxidase (HRP) (KPL, Gaithersburg, MD) for 1 h; after a further wash step, the substrate 3,3,5,5′-tetramethylbenzidine (TMB) (KPL) was added; TMB reacts in the presence of HRP to form a colored product, which was measured spectrophotometrically at wavelength 450 nm with a 620 nm reference using a Tecan Sunrise™ absorbance reader with Magellan™ V6.5 software (Tecan, Grödig, Austria); the amount of colored product was directly proportional to the amount of free CSL360 present in the sample. The following PK parameters were estimated: elimination half-life (t1/2), maximum concentration (Cmax), time to reach maximum concentration (Tmax), clearance, volume of distribution and area under the plasma concentration–time curve (AUC) from 0 to 24 h. Samples for immunogenicity testing were collected prior to first infusion, then on days 1, 15 and 29 (± 3), then monthly during treatment and at study exit, and assayed for the presence of anti-CSL360 antibody using a bridging enzyme immunoassay.
Pharmacodynamics study
Twenty milliliters of PB and 5 mL of BM were collected within 2 weeks of day 1, at day 16 and day 29 (± 3 days). A range of biological correlative studies were performed to determine: (1) the extent of binding of CSL360 to AML blasts and LSCs by flow cytometry; (2) whether there was evidence of CD123 down-regulation in the presence of CSL360; and (3) whether in vivo exposure to CSL360 affected the ex vivo proliferative response of AML cells to IL-3.
To compare the IL-3 neutralizing activity of 7G3 and CSL360, primary AML cells (1 × 105 cells) were cultured in RPMI/10% fetal calf serum (FCS) and 10 ng/mL IL-3 in the presence of increasing concentrations of 7G3 or CSL360 for 36 h at 37°C prior to pulsing with 0.5 μCi 3[H]-thymidine for 12 h and harvesting onto glass fiber filters. Radioactivity was quantitated by liquid scintillation counting. To compare the efficacy of these two antibodies in inhibiting IL-3 signaling pathways, primary AML cells (1 × 105 cells) were incubated in 10 μg/mL 7G3 or CSL360 for 15 min prior to stimulation with 10 ng/mL IL-3 for a further 15 min at 37°C. The percentage of blasts expressing phosphorylated signal transducer and activator of transcription 5 (pSTAT5) was determined by intracellular flow cytometry after gating on the CD45dim population.
The levels of CD123 expression and the extent of binding saturation of CSL360 with its intended target (IL-3Rα) on AML blast cells after infusion was assessed by flow cytometry using two anti-CD123 antibodies (clone 7G3-PE or 9F5-PE; BD Biosciences, Franklin Lakes, NJ). 9F5 recognizes a distinctly different epitope on CD123 from 7G3 and CSL360 [Citation20]. PB mononuclear cells (PBMCs) and BM mononuclear cells (BMCs) were separated by Ficoll (GE Healthcare, Uppsala, Sweden) density gradient centrifugation. Cells were incubated with a cocktail consisting of fluorochrome labeled antibodies to CD45, CD34, CD3 and CD123 (BD Biosciences). AML blasts were identified based on a low side scatter (SSC)/CD45dim profile. LSC-enriched cells were identified from the blast gate as CD34+/CD38−.
The number of anti-CD123 antibodies bound/cell was determined from the mean fluorescence intensity (MFI) observed for each antibody using Quantibrite-PE beads (BD Biosciences) as detailed by the manufacturer. CD123 receptor occupancy was calculated using the following formula: 100 − 100 × (number of CD123 molecules detected by 7G3/total number of CD123 molecules as detected by 9F5).
To assess the ability of a patient's mononuclear cells (MNCs) (from either PB or BM) exposed to CSL360 in vivo to respond to IL-3, MNCs were isolated by Ficoll gradient centrifugation and washed to remove bound CSL360. The washed MNCs were then cultured in RPMI/5% FCS with increasing concentrations of IL-3 in the presence or absence of 10% autologous plasma for 36 h at 37°C prior to pulsing with 0.5 μCi 3[H]-thymidine for 12 h and harvesting onto glass fiber filters. Radioactivity was quantitated by liquid scintillation counting.
Statistical analysis
Safety data were summarized for each dose level. The incidence of all AEs was tabulated by body system. PK parameters were summarized for each dose level using mean ± standard deviation, or geometric mean as appropriate. Disease response was defined according to the NCCN response criteria for AML [Citation21].
Role of the funding source
The trial was sponsored and fully funded by CSL Ltd. The sponsor was involved in study design and collection, compilation and preliminary analysis of clinical data. Employees of the sponsor conducted the biomarker and PK assays, and were involved in interpretation of the data and contributed to manuscript writing.
Results
Patient characteristics
Fifty-one patients were screened, with 11 screen failures. The key characteristics of the 40 eligible patients (male n = 27; female n = 13) of median age 63 years (range 22–85) are summarized in . At the time of initial diagnosis of AML, 27 patients had de novo disease, 12 patients had a prior history of myelodysplasia and one patient had therapy-related AML. The disease status at study entry was as follows: relapsed AML (n = 25), primary refractory disease (n = 8) and newly diagnosed high-risk AML ineligible for conventional chemotherapy (n = 7). Of the 25 patients with relapsed disease, seven had relapsed AML after allogeneic SCT and one after autologous SCT. Sixteen patients had cytogenetic analysis available at initial diagnosis, with 12 having unfavorable abnormalities according to standard criteria [Citation22]. The median number of prior therapies was two (range 0–4).
Table I. Patient characteristics.
Patient flow and dosing of CSL360
Twenty-eight patients entered the dose-escalation phase for safety evaluation across five dose levels (0.1 mg/kg n = 4; 0.3 mg/kg n = 4; 1.0 mg/kg n = 5; 3.0 mg/kg n = 9; and 10.0 mg/kg n = 6). Seven of the 28 patients received fewer than four infusions. Reasons for discontinuation prior to the fourth dose were: progressive disease (n = 2), consent withdrawal (n = 2) and fatal neutropenic sepsis (n = 3: two due to AML, and one considered a DLT as possibly related to CSL360). No other DLTs were observed among the 28 patients evaluable for determination of a MTD. An additional 12 patients were enrolled subsequently in an expanded 10.0 mg/kg cohort. Overall, a total of 304 infusions were administered to 40 patients. Thirty-three patients received four or more doses including all 12 patients in the expanded 10.0 mg/kg cohort. Twelve patients completed 12 weekly doses as planned, including one patient who received a total of 17 doses. The reasons for discontinuation before completion of the 12 planned doses were: progressive disease (n = 17); SAE (n = 3); investigator decision (n = 5); and consent withdrawal (n = 3).
Toxicities
CSL360 was generally well tolerated. The most common AEs of any grade observed during the study were headache (32.5%), diarrhea (27.5%), nausea (27.5%) and infection (17.3%). The most common AE considered related to CSL360 by investigators was infusion-related reaction, which was reported in seven patients (17.5%). lists grade ≥ 3 AEs observed in more than one patient in the entire trial, irrespective of possible causality. Overall, AEs and SAEs did not increase in frequency as the dose level increased. Six of 62 (10%) SAEs were considered possibly related to CSL360, and these included: grade 2 infusion reaction (n = 2), grade 3 gamma-glutamyltransferase increase (n = 1), grade 3 culture-negative sepsis (n = 1), grade 3 generalized arthromyalgia (n = 1) and grade 4 invasive fungal infection (n = 1; after third dose of 3.0 mg/kg and considered a DLT, although neutropenia due to AML was an alternative cause). No MTD for CSL360 was identified in this study.
Table II. Summary of grade ≥ 3 adverse events occurring in > 1 patient.
Pharmacokinetics
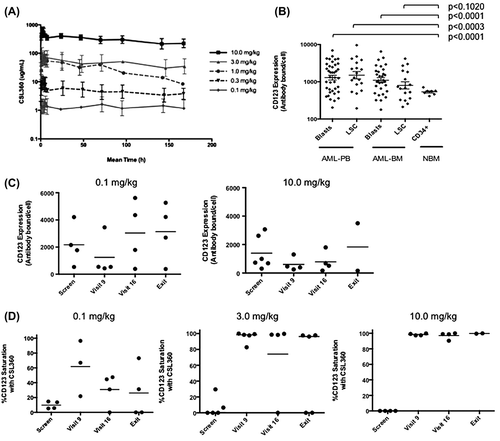
After administration as an IV infusion, CSL360 concentrations displayed a relatively flat concentration–time profile over a dosing interval of approximately 7 days. The mean CSL360 concentration–time profiles demonstrated that exposure to CSL360 increased in a dose-proportional manner over the dose range from 0.1 to 10.0 mg/kg. These data were supported by dose-proportional increases in AUC(0-∞) after dose 1, AUC(0-τ,ss) after dose 4 and the Cmax after the first and fourth doses. After the administration of four doses of CSL360, the serum concentration of CSL360 approximated steady-state []. The Cmax measured after the administration of the fourth dose of CSL360 at 10.0 mg/kg was 672.2 ± 203.6 mg/L (n = 4). The Tmax was approximately 3 h, irrespective of the dose of CSL360 administered. The t1/2, volume of distribution and clearance were also independent of the CSL360 dose level. The t1/2 following the first dose of CSL360 at 10.0 mg/kg was approximately 4.9 days (118.5 ± 49.3 h). Calculations indicated a small volume of distribution (47.0 ± 14.3 mL/kg) and clearance (0.30 ± 0.13 mL/h/kg) as expected for an IgG1 monoclonal antibody [Citation23].
Figure 2. Steady-state exposure of CSL360 and its effects on CD123 expression and saturation in vivo. (A) Mean serum CSL360 concentration–time profile following the fourth dose administration. Mean and SD are shown for all five dose cohorts. (B) The level of CD123 expression was quantitated on the blasts and LSC population of all patients at screening and in CD34 + cells from normal bone marrow (NBM). Ficoll-separated mononuclear cells from PB were stained with a cocktail consisting of CD45, CD34, CD38 and CD123. Blasts were identified based on a low SSC/CD45dim phenotype and the LSC enriched population was defined as CD34+/CD38−. (C) CD123 expression is down-regulated by exposure to CSL360 in vivo. The expression of CD123 on PB blasts at day 16 (visit 9), day 29 ± 3 (visit 16) and where available study exit, from selected patients was measured using 9F5 and is represented as antibodies bound/cell. Data from the 0.1 mg and 10 mg/kg cohorts are shown, and a reduction was observed in all cohorts (not shown). (D) CD123 saturation by CSL360 in PB blasts demonstrated by the absence of 7G3 binding in the presence of 9F5 binding in 0.1, 3.0 and 10.0 mg/kg cohorts is shown. Individual patient data and the mean are represented.
Immunogenicity and infusion-related reactions
In all pretreatment samples, anti-CSL360 antibody titers were negative. CSL360-specific binding antibodies were detected (titer ≥ 50) in 11 of 35 patients tested (31.4%). All 11 patients found to have anti-CSL360 antibody were amongst the 19 patients in the 0.1, 0.3, 1.0 and 3.0 mg/kg dose cohorts. High titers occurred predominantly in the lower dose cohorts (0.1, 0.3, 1.0 mg/kg). In the 3.0 mg/kg cohort, only low titers of anti-CSL360 antibody were found during treatment, and these had no significant effect on PK parameters, suggesting that this higher dose of CSL360 could achieve sufficient serum concentrations to neutralize any anti-CSL360 antibody. CSL360-specific binding antibodies were not detected in any of the 16 patients in the 10.0 mg/kg cohort.
Infusion-related reactions were observed in three of the 11 patients who had detectable CSL360-specific binding antibodies. A total of 11 infusion-related reactions occurred amongst seven patients in the study. Five patients had just one reaction, and two patients (one in the 3.0 mg/kg cohort, one in the 10.0 mg/kg cohort) each had three reactions. Ten of the reactions were grade 2 and one reaction was grade 1. All settled with protocol-prescribed slowing of the rate and standard medication. Three patients experienced a reaction during their first infusion (one patient in the 0.3 mg/kg cohort, two in the 10.0 mg/kg cohort). The other four patients experienced their first reaction after at least three doses of CSL360. Three of the four patients had detectable CSL360-specific binding antibodies at least 2 weeks before onset of the infusion reaction. The fourth patient experienced three infusion reactions but did not have detectable CSL360-specific binding antibodies. However, this patient was in the 10.0 mg/kg cohort, and therefore the possible effects of undetected CSL360-specific binding antibodies in this patient could not be discounted. All patients recovered satisfactorily within 3 h of the onset of the infusion reaction, without sequelae, and none of the grade 2 infusion reactions constituted a DLT. Routine premedication was given prophylactically for all subsequent infusions in patients who had experienced infusion-related toxicity, and thereafter reactions were not observed. Infusion reactions were not observed for eight of 11 patients who had detectable CSL360-specific binding antibodies.
Biological correlations
Specific binding of CD123 by CSL360
Quantitative flow cytometry was used to determine the level of CD123 expression on AML cells in PB and BM from all patients at screening []. These data demonstrated that CD123 was detected on all samples. A wide range of expression exhibiting a log-normal distribution was observed (range of 178–9600 anti-CD123 antibodies bound/cell). A significantly higher expression of CD123 was evident on blasts in both PB and BM compared to CD34 + progenitors from healthy donors.
Down-regulation of CD123 expression in AML blasts by CSL360
By using the alternative anti-CD123 specific antibody 9F5 to quantify the IL-3Rα density in AML samples post-CSL360 infusion, a demonstrable reduction of antibody bound per AML blast cell and LSC was seen in evaluable samples from patients at all dose levels when compared to that at screening, indicating that there was down-regulation of CD123 antigen surface expression in response to CSL360 []. At the lowest dose level, the down-regulation appeared transient and unsustained. However, at higher dose levels (≥ 3.0 mg/kg), the level of CD123 expressed on blast cells was significantly reduced from a median number of 2345 (antibodies bound/cell) at screening to 773 CD123 antigens per blast cell on day 16 (p = 0.0175) and 552 on day 29, suggesting that sustained down-regulation of CD123 was dose-dependent.
Saturation of CD123 on AML blasts by CSL360
As CSL360 recognized the same epitope as 7G3, specific binding of CSL360 to IL-3Rα on day 16 and day 29 was demonstrated indirectly by the absence of 7G3 binding in the presence of 9F5 binding. CSL360 was shown to bind to CD123 on both LSCs and blasts at all dose levels. This specific binding was first observed at the lowest dose cohort of 0.1 mg/kg on day 16 []. Furthermore, complete, sustained saturation of CD123 by CSL360 was seen following dosing at 3.0 and 10.0 mg/kg. The duration of CD123 saturation by CSL360 was dose-dependent on both AML blasts and LSCs in PB and BM: at lower dose cohorts (0.1 and 0.3 mg/kg), CD123 saturation was transient and significantly reduced by day 29 (7 days after the fourth infusion); however, at higher doses (3.0 and 10.0 mg/kg), the saturation of CD123 was maintained at 100% on day 29.
CSL360 in plasma abolishes ex vivo IL-3 mediated proliferation of MNCs
To determine whether the concentration of CSL360 achieved in vivo was sufficient to block IL-3-induced proliferation, MNCs from patients’ PB or BM samples were tested in proliferation assays. The addition of 10% of autologous serum collected on day 16 (after the third dose of CSL360) to a culture of cells from patients in the 3.0 or 10.0 mg/kg cohorts was sufficient to markedly inhibit IL-3 mediated ex vivo proliferation (), while GM-CSF-induced blast proliferation was unaffected (data not shown). This demonstrated that the excess of unbound CSL360 in the serum was able to effectively block IL-3 mediated signaling and proliferation of AML cells.
Figure 3. Plasma concentrations of CSL360 at higher dose cohorts are sufficient to inhibit IL-3-induced proliferation of patient mononuclear cells ex vivo. Ficoll-separated mononuclear cells from patient PB or BM taken at day 16 (1 day post-dose 3) were cultured for 48 h at 37°C in increasing concentrations of IL-3 in the presence (black circles) or absence (gray circles) of 10% autologous plasma collected on the same day. Proliferation was measured by incorporation of 3[H]-thymidine. Each plot represents a single patient from cohort 1 (A), 2 (B), 4 (C) or 5 (D). The mean of triplicate measurements is shown.
![Figure 3. Plasma concentrations of CSL360 at higher dose cohorts are sufficient to inhibit IL-3-induced proliferation of patient mononuclear cells ex vivo. Ficoll-separated mononuclear cells from patient PB or BM taken at day 16 (1 day post-dose 3) were cultured for 48 h at 37°C in increasing concentrations of IL-3 in the presence (black circles) or absence (gray circles) of 10% autologous plasma collected on the same day. Proliferation was measured by incorporation of 3[H]-thymidine. Each plot represents a single patient from cohort 1 (A), 2 (B), 4 (C) or 5 (D). The mean of triplicate measurements is shown.](/cms/asset/d6caa4c8-564d-41be-ba67-f82724c3d615/ilal_a_956316_f0003_b.gif)
Clinical anti-leukemic activity
The majority of patients showed no improvement in blast percentage in BM aspirates during the study. As per NCCN response criteria, 93.3% (28 of 30 patients with a BM biopsy adequate for response assessment) had persistent disease after four doses of CSL360. In the 18 patients who received 10.0 mg/kg, the median BM aspirate blast counts at screening and day 29 were 38% and 41%, respectively. Of the 16 patients with CD34 + CD38− AML LSC fractions identified at screening, only minor changes were observed during the entire treatment period, except for one patient who demonstrated a marked increase. Among 27 patients who received doses of CSL360 sufficient to achieve sustained CD123 blockade at ≥ 3.0 mg/kg, two patients (one in the 3.0 mg/kg cohort and the other in the 10.0 mg/kg cohort) achieved a complete morphologic remission (defined as BM blasts < 5%) after four doses of CSL360, giving an overall response rate of 7.4% at day 29. All other patients had persistent disease.
The responders were two of seven patients who had relapsed AML post-allogeneic SCT. At the time of study entry, neither had clinical evidence of GVHD. One of these two patients achieved a durable CR (i.e. maintained after 12 doses). That patient was a 22-year-old male with multiply relapsed AML who had experienced only brief CRs after two allografts. The second allograft, an umbilical cord blood transplant, was approximately 4 months prior to the start of study treatment with CSL360. He was on no immunosuppression and had no evidence of GVHD at study entry. The screening BM blast count of 52% was reduced to 2% after three doses of 3.0 mg/kg CSL360 (). CR with normalized PB counts was subsequently confirmed after 12 doses. This patient was withdrawn after a total of 17 doses following the development of a tension pneumothorax complicating pre-existing lung disease. One other allograft recipient in second relapse with 10% BM blasts at study entry achieved a morphologic remission with 1% BM blasts after four doses of CSL360 at 10.0 mg/kg. However, the response was not durable, with progressive disease documented after 11 doses (BM blasts of 40%).
Figure 4. Flow cytometry profile of PB and BM of subject AWM002 indicating sustained clearance of the blast population from day 16. Ficoll-separated mononuclear cells from patients taken at screening, day 16 (visit 9: 1 day post-dose 3), day 31 (visit 16: 10 days post-dose 4) and day 74 (post-dose 12) were analyzed by fluorescence activated cell sorting (FACS). AML blasts were identified by gating on the CD45dim/CD34 + population.

Discussion
Eradication of LSCs is an important goal in the treatment of AML. Strong evidence in animal models supports this hypothesis [Citation24,Citation25]. Recently, several cell-surface targets which are preferentially overexpressed by AML LSCs have been identified, including CD44, C-type lectin-like molecule 1 (CLL-1), CD47, T cell immunoglobulin mucin-3 (TIM-3), CD96, CD33 and CD123 [Citation20,Citation26–32]. The presence of the high IL-3Rα expression on the surface of AML blasts and CD34+/CD38 − AML LSC fraction, but not in normal HSCs, suggests that CD123 is an attractive cell-surface target amenable to immunotherapy. Various strategies of immunotherapy have been investigated targeting specific AML-associated surface antigens such as CD33 using gemtuzumab ozogamicin [Citation16,Citation33] and lintuzumab [Citation34]. An anti-CD123 immunotoxin targeting CD123-expressing AML cell lines showed in vitro cytotoxic activity [Citation35]. In vivo blockade using a specific anti-CD123 monoclonal antibody (7G3) in a mouse model of established AML demonstrated a reduction in leukemic burden [Citation18].
CSL360 was the first chimeric monoclonal antibody developed to selectively target cell surface CD123 on AML blasts and LSCs. The half-life of CSL360 in patients was approximately 4.9 days (dose-dependent range: 1.4–8.8 days). This was in agreement with the PK properties of other therapeutic monoclonal antibodies, and supported the use of a weekly dosing schedule in this trial. The development of the anti-CSL360 antibodies was detected only in suboptimal dose cohorts. These antibodies appeared to be non-neutralizing, with high and sustained CSL360 concentrations achieved with weekly dosing in the 3.0 and 10.0 mg/kg dose cohorts.
We and others [Citation6,Citation7] have confirmed the high expression of CD123 in AML blasts and LSC fraction. Specific binding of CSL360 to its intended target IL-3Rα on AML blasts and LSCs was demonstrated in all PB and BM samples. In patients receiving doses of ≥ 3.0 mg/kg, sustained binding of CSL360 to its target was demonstrated, and down-regulation of CD123 expression was also observed. Adequate concentrations of CSL360 were achieved and sustained in serum at 10.0 mg/kg, sufficient to saturate CD123 on leukemic cells in vivo and to abolish IL-3 mediated proliferation of blasts ex vivo.
CSL360 was safe and tolerable, and a MTD was not reached. Six of 62 (10%) SAEs were considered possibly related to CSL360, including one grade 4 invasive fungal infection. The incidence of sepsis was within the expected rate observed in patients with advanced AML in this trial. The incidence of infusion-related toxicity was low, reported in 11 of 304 infusions (3.6%), and the infusion reactions (all grade 1 and 2) were manageable or preventable with premedication.
While the study population represented patients with active leukemia and a very poor prognosis, only one durable CR was seen in 27 patients treated with doses that achieved sustained CD123 saturation. Of seven patients with relapsed AML post-allogeneic SCT, two achieved a complete morphologic remission in BM after four infusions. While a direct role cannot be proved for CSL360, the absence of GVHD at study entry and the lack of development of GVHD subsequently argue against this being an epiphenomenon. Although the mechanism of action of CSL360 remains unclear in these patients, antibody-dependent cell-mediated cytotoxicity (ADCC) is one possibility, as data in NOD/SCID mice suggest that residual murine immunity contributed to the action of 7G3 [Citation18]. Induction of the graft-versus-leukemia effect through an unknown mechanism is an alternative possibility.
These data indicate that blockade of CD123 and abolition of IL-3 signaling alone by CSL360 is an ineffective therapeutic strategy in AML. While functional inhibition of CD123 does not warrant further clinical investigation in this setting, using CD123 to target AML LSCs for clearance remains a rational and attractive experimental strategy. Recently, human CD123-specific chimeric antigen receptor (CAR)-transduced T-cells have been developed, demonstrating killing of CD123-positive AML cell lines in vitro [Citation36–38] and anti-leukemic activity in vivo in immunodeficient mouse models [Citation37,Citation38].
Elimination of CD123-expressing leukemia through ADCC also shows potential. While CSL360 was not designed to induce potent ADCC and the activity of CD123-targeted ADCC was not tested in this trial, the intriguing observation of rapid clearance of disease in one patient post-allograft is consistent with such a mechanism of action. Any potential ADCC effect of CSL360 was likely compromised in most patients by a high leukemic load and low effector cell numbers. It is noteworthy that the two responses were observed in patients who had prior allografts and may have had innate immune cell function less compromised by chemotherapy. When ADCC activity was optimized by enhancing Fc-mediated effector function in a second-generation anti-CD123 monoclonal antibody that targets the same epitope (CSL362), increased cell killing in vivo was observed in a murine xenograft model [Citation39]. Consequently, the safety and biological activity of an ADCC-mediated CD123 targeting approach is being investigated in a phase 1 study of CSL362 for patients with poor-prognosis AML. Unlike the study reported here, the CSL362 trial is being conducted in the post-consolidation setting, where low leukemic burden and adequate effector cell numbers and function should maximize the potential for ADCC to occur.
ilal_a_956316_sm4706.zip
Download Zip (23.3 MB)Acknowledgements
This trial was funded by CSL Ltd.
We thank all patients participating in the study, and the research nurses who worked with us during its conduct. We also thank Dr. David Gearing for his role in the preclinical development of CSL360, and for helpful discussions. We would like to thank Mark Biondo, Sarah Hosback and Rosyln Davis from CSL Ltd. for developing, validating and performing the pharmacodynamics and PK and immunogenicity assays. S.Z.H. was supported by a Webster scholarship (Melbourne Health) and Max Whiteside Clinical Fellowship (Leukaemia Foundation of Australia); A.W.R.'s work was supported by a fellowship from the Victorian Cancer Agency and grants (461219, 637309, 1016647) from the National Health and Medical Research Council (NHMRC) of Australia; A.F.L.'s work was supported by grants from the NHMRC and from Cancer Australia; H.S.R.'s work was supported by the Peter Nelson Leukaemia Research Fund.
Potential conflict of interest: SB, SB, MB, MD and RB are employees of CSL Ltd or were employees during the clinical trial; AFL is a paid consultant to CSL Ltd; AWR acted as a paid consultant to CSL Ltd during the pre-clinical development of CSL360, but not during the clinical trial, or subsequently; AJM received paid consultancy for the analysis and interpretation of pharmacokinetic data. SZH, DSR, MSH, SD, IDL, PM, IK, KFB, GK, AWB, TMY, HSR and HI have no conflicts relating to this manuscript to declare.
References
- Breems DA, Van Putten WL, Huijgens PC, et al. Prognostic index for adult patients with acute myeloid leukemia in first relapse. J Clin Oncol 2005;23:1969–1978.
- Kantarjian H, O’Brien S, Cortes J, et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer 2006;106:1090–1098.
- Burnett AK, Hills RK, Green C, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood 2010;115:948–956.
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–737.
- Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014;506: 328–333.
- Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol 2004;5:738–743.
- Guzman ML, Swiderski CF, Howard DS, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci USA 2002;99:16220–16225.
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275–284.
- Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000;14:1777–1784.
- Florian S, Sonneck K, Hauswirth AW, et al. Detection of molecular targets on the surface of CD34+/CD38- stem cells in various myeloid malignancies. Leuk Lymphoma 2006;47:207–222.
- Munoz L, Nomdedeu JF, Lopez O, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001;86:1261–1269.
- De Waele M, Renmans W, Vander Gucht K, et al. Growth factor receptor profile of CD34 + cells in AML and B-lineage ALL and in their normal bone marrow counterparts. Eur J Haematol 2001;66:178–187.
- Testa U, Riccioni R, Diverio D, et al. Interleukin-3 receptor in acute leukemia. Leukemia 2004;18:219–226.
- Testa U, Riccioni R, Militi S, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood 2002;100:2980–2988.
- Graf M, Hecht K, Reif S, et al. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. Eur J Haematol 2004;72:89–106.
- Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005;106: 4086–4092.
- van Rhenen A, Feller N, Kelder A, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res 2005;11:6520–6527.
- Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009;5:31–42.
- Vergez F, Green AS, Tamburini J, et al. High levels of CD34 + CD38low/-CD123 + blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica 2011;96: 1792–1798.
- Sun Q, Woodcock JM, Rapoport A, et al. Monoclonal antibody 7G3 recognizes the N-terminal domain of the human interleukin-3 (IL-3) receptor alpha-chain and functions as a specific IL-3 receptor antagonist. Blood 1996;87:83–92.
- Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003;21:4642–4649.
- Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000;96:4075–4083.
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004;93:2645–2668.
- Wang JC. Evaluating therapeutic efficacy against cancer stem cells: new challenges posed by a new paradigm. Cell Stem Cell 2007;1:497–501.
- Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med 2007;13:470–481.
- Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica 2012;97:966–974.
- Hauswirth AW, Florian S, Printz D, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123 + AML stem cells. Eur J Clin Invest 2007;37:73–82.
- Bakker AB, van den Oudenrijn S, Bakker AQ, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res 2004;64:8443–8450.
- Jan M, Chao MP, Cha AC, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci USA 2011;108:5009–5014.
- Hosen N, Park CY, Tatsumi N, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci USA 2007;104:11008–11013.
- Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009;138:286–299.
- Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009;138:271–285.
- Tsimberidou AM, Giles FJ, Estey E, et al. The role of gemtuzumab ozogamicin in acute leukaemia therapy. Br J Haematol 2006;132: 398–409.
- Raza A, Jurcic JG, Roboz GJ, et al. Complete remissions observed in acute myeloid leukemia following prolonged exposure to lintuzumab: a phase 1 trial. Leuk Lymphoma 2009;50:1336–1344.
- Du X, Ho M, Pastan I. New immunotoxins targeting CD123, a stem cell antigen on acute myeloid leukemia cells. J Immunother 2007;30:607–613.
- Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol 2013;161: 389–401.
- Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013;122:3138–3148.
- Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014;123:2343–2354.
- Busfield SJ, Biondo M, Wong M, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia 2014 Apr 7. [Epub ahead of print]