Abstract
Cystic fibrosis (CF) is an autosomal recessive monogenetic disease that afflicts nearly 70 000 patients worldwide. The mutation results in the accumulation of viscous mucus in multiple organs especially in the lungs, liver and pancreas. High associated morbidity and mortality is caused by CF due to the lack of effective therapies. It is widely accepted that morbidity and mortality caused by CF is primarily due to the respiratory manifestations of the disease. Consequently, several approaches were recently developed for treatment of lung complications of CF. However, the lack of effective methods for delivery and especially targeted delivery of therapeutics specifically to lung tissues and cells limits the efficiency of the therapy. Local pulmonary delivery of therapeutics has two major advantages over systemic application. First, it enhances the accumulation of therapeutics specifically in the lungs and therefore increases the efficiency of the treatment. Second, local lung delivery substantially prevents the penetration of the delivered drug into the systemic circulation limiting adverse side effects of the treatment on other organs and tissues. This review is focused on different approaches to the treatment of respiratory manifestations of CF as well as on methods of pulmonary delivery of therapeutics.
Introduction
Cystic fibrosis (CF) is an autosomal recessive monogenetic disease that afflicts nearly 70 000 patients worldwide [Citation1]. The mutation results in the accumulation of vicious mucus in multiple organs especially in the lungs, liver and pancreas. The disease is mainly attributed to recessive mutations in the CF transmembrane conductance regulator (CFTR), which is a chloride transport channel. The channel is also involved in the transport of sodium and bicarbonate. The involvement of bicarbonate transport may play an important role in gastrointestinal (GI) manifestations of CF; however, animal models tend not to develop GI disease [Citation2]. Over 1800 mutations have been identified, but most are very rare. The most common mutations are identified in . The mutations cause qualitative or quantitative reduction in CFTR activity. CFTR is a transmembrane glycoprotein that is a member of the ATP binding cassette (ABC) family [Citation3,Citation4]. It is composed of 1480 amino acids and has a molecular weight of 170 kDa. The structure contains five domains: two transmembrane domains consisting of six alpha helices each; two nucleotide-binding domains (NBDs) on the cytoplasmic side; and one regulatory domain (). ATP hydrolysis by the first NBD results in channel opening and hydrolysis by NBD2 closes the channel. Qualitative mutations result in CFTR proteins that are mature and reached the target location, apical plasma membrane of epithelial cells [Citation5]. The most common mutation (>70% of alleles) involves the deletion of phenylalanine at position 508 of the CFTR protein, termed ΔF508 CFTR. This mutated protein is misfolded and is recycled before reaching the cell membrane. The ΔF508 mutated form is believed to retain only 30% activity of wild type CFTR.
Figure 1. A schematic representation of the structure of the CFTR protein. The protein consists of five domains: two transmembrane domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2), and a regulatory domain (RD).
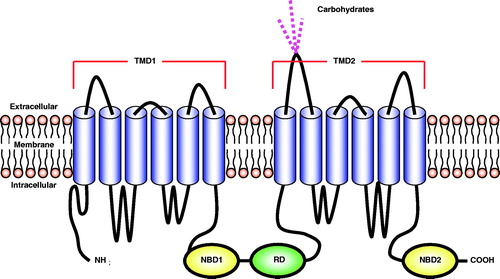
Table 1. Classification of CFTR mutations*.
High associated morbidity and mortality is caused by CF due to the lack of effective therapies, and the average lifespan for patients is approximately 40 years. Most available therapies only treat the symptoms of CF and do not correct the underlying cause of the disease. Because CFTR is found on the apical surface of many different types of epithelial cells, patients suffer numerous symptoms, and have a high medication burden. CF patients are also unable to recover salt from their sweat; this forms the basis of the sweat chloride diagnosis test for CF. CFTR dysfunction in the pancreas leads to numerous effects such as autodigestion, malabsorption of fat soluble vitamins, poor growth, gallstones and hepatobiliary disease.
It is widely accepted that morbidity and mortality caused by CF is primarily due to the respiratory manifestations of the disease. The deficiency in the CFTR protein causes decreased chloride secretion from lung epithelium cells and alters ionic and water transport via the apical membrane of these cells (). These disturbances led to increased water absorption and abnormal mucus composition. Mucus in CF patients becomes thick and sticky, builds up in the lungs causing bronchial obstruction and provoking bacterial infections and inflammation. Finally, patients with a lung form of CF suffer from several lung dysfunctions including bronchiectasis and lung insufficiency. CF patients need to take many therapies for respiratory manifestations. These patients are unable to clear phlegm from their airways. Unlike normal patients, the phlegm in CF patients consists primarily of bacteria, inflammatory cells, polymeric DNA and contains nearly no mucin. Patients must perform chest physiotherapies, take hypertonic saline and dornase alfa to clear the mucus. Patients also suffer from inflammation, which is treated using prednisone, ibuprofen and N-acetyl cysteine. Because they are unable to clear mucus, patients tend to have higher incidence of respiratory infections particularly due to pseudomonas infections and must take antibiotics [Citation1,Citation6]. The present review is focused on different approaches to the treatment of respiratory manifestations of CF as well as on methods of pulmonary delivery of therapeutics.
Figure 2. Development and progression of the pulmonary form of cystic fibrosis. Lung manifestations are the primary cause of morbidity and mortality in patients. Modified from Ref. [Citation3].
![Figure 2. Development and progression of the pulmonary form of cystic fibrosis. Lung manifestations are the primary cause of morbidity and mortality in patients. Modified from Ref. [Citation3].](/cms/asset/4dd55c24-db13-473d-8566-35a61cc3f25d/idrt_a_829078_f0002_b.jpg)
Advantages and challenges of lung delivery
Recently, the administration via the respiratory route attracts considerable attention due to the several distinct advantages. For the systemic delivery, the respiratory route might be beneficial for the delivery of peptides and proteins, which cannot be delivered by the oral route. Peptides and proteins must be administered by parenteral injection and may lead to patient compliance issues. The rich vasculature at the level of the alveoli provides a mean for the systemic exposure. As a local delivery, the pulmonary delivery is ideal for the treatment of respiratory diseases such as asthma, chronic obstructive pulmonary disease (COPD), respiratory infections and lung manifestations of CF. To reduce systemic adverse side effects and reach efficacious concentrations at the target airway, many inhalable drug formulations have been developed for respiratory diseases.
Pulmonary delivery may provide an additional advantage due to altered pharmacokinetic parameters in CF patients [Citation7]. The majority of patients have increases in total body clearance and volume of distribution. Increased clearance may be the result of increased activity of hepatic metabolizing enzymes or increased renal and pulmonary clearance. Pulmonary delivery can ensure proper dosage of therapies for treatment of lung manifestations. An example is TOBI®, an inhalable formulation of tobramycin, an antibiotic for treatment of Pseudomonas aeruginosa infection.
Lung deposition depends on inertial impaction, sedimentation and diffusion [Citation8]. The location of deposition can be determined by calculating the aerodynamic diameter (da) of the inhaled substance. Particles with a da exceeding 5 µm are either filtered in the nose or impacted in the nasal and oral pharynx and then cleared by coughing or sneezing. The particles with da between 1 and 5 µm are trapped in mucus blanket in the conducting airways and moved cephalad by ciliary action. At the level of the larynx they are either swallowed or expectorated. Smaller particles are deposited in the deep lung and in most cases are phagocytosed by alveolar macrophages. In addition, a low epithelia thickness and high surface area of the respiratory zone of the lungs allow the access of non-phagocytosed substances to vasculature for systemic absorption. However, these mechanisms are altered in CF patients [Citation9]. Because the diameter of airways is decreased, the influence of impaction is increased. Deposition of particles greater than 1 µm in the tracheobronchial airway is nearly tripled when compared with healthy individuals.
In addition to ciliated epithelium as a barrier to pathogens and chemicals, the lumen of the respiratory system is covered in a layer of airway surface liquid (ASL) [Citation5,Citation10,Citation11]. The ASL consists of two layers: periciliary layer (PCL) and the upper mucus layer. The PCL is approximately 7 µm thick, is watery, and in contact with airway epithelia. The mucus layer in normal patients consists of mucin proteins, which are actually decreased in CF patients. Hydration is a vital part of mucociliary clearance. The PCL must maintain a certain thickness and low viscosity to act as a lubricant and allow ciliary beat. The dysfunction of CFTR leads to loss of inhibitory function of epithelial sodium channels and increased sodium absorption. The result is a decrease in PCL, mucociliary clearance, bacterial colonization and ultimately respiratory failure. To prevent low sodium concentration in the luminal surface of the airways, researchers have attempted to inhibit sodium channels using blockers such as amiloride or use hyperosmotic agents such as mannitol and hypertonic saline [Citation1]. These strategies aim to correct ion transport through alternative mechanisms not involving CFTR.
Sputum of CF patients is laden with bacteria (mainly Pseudomonas aeruginosa), cellular debris, DNA and filamentous actin (F-actin) with a decrease in the amount of mucin [Citation12–14]. These polymers tend to be negatively charged and connect to each other forming entanglements via electrostatic, hydrogen and hydrophobic interactions. Because of the lack of ion transport, deficits in water and mucus, airways in patients with CF are not cleared as readily as in normal conditions. Delivered particles become trapped within the sputum network and are not able to penetrate in order to reach target cells.
As defense from the environment, the airways contain immune cells such as macrophages and T helper cells [Citation15]. Delivery of particles to the lung must overcome non-specific immune response as well as humoral response resulting in neutralizing antibodies. The immune response may occur after the transgene has been inserted. If a viral vector is used, it will likely be a cytotoxic T lymphocyte response due to expression of viral proteins. Non-viral vectors with ligands have a higher likelihood of eliciting an immune response than those without an attached ligand.
Choosing an appropriate animal model is critical for pre-clinical studies. Interspecies difference in anatomy and function may not give the true representation to design human clinical trials. Compared to other species, human lungs are more spherical, there is relatively symmetrical branching at all levels of the airways, and the trachea is shorter [Citation16]. In addition, small rodent species are not ideal for deposition experiments because they are nose-breathers. Transgenic animal models have been developed to study many diseases including CF mice models. Unfortunately, application of these models has not been optimal because they do not always spontaneously develop the disease [Citation17]. Aerosol delivery for CF is typically performed in healthy animals. Sheep lungs have been shown to be the best model to replicate the shape, size and cellular morphology of human lungs. Another challenge in animal models is the delivery of the aerosol. Animal studies require some level of invasive procedure such as use of a bronchoscope.
Small molecules targeted to CFTR
Recently, a novel drug Ivacaftor (Kalydeco™) was approved by the FDA for administration in CF patients [Citation18,Citation19]. This is the first medication that targets the underlying cause of CF. Although a step in the positive direction, Ivacaftor works in patients with at least one copy of the G551D mutation, which is present in a small minority of patients. The G551D mutation is a qualitative reduction in CFTR activity; therefore, ivacaftor is classified as a CFTR potentiator. Ivacaftor does not present a viable option for patients homozygous for the common ΔF508 mutation [Citation20]. Clinical trials demonstrated that Ivacaftor treatment resulted in a statistically significant improvement in forced expiratory volume in 1 s (FEV1) and sweat chloride [Citation19].
CFTR corrector is a class of drug candidates that rescues mutated CFTR from intracellular degradation and promote their incorporation into the apical plasma membrane [Citation3]. There are three classes of CFTR correctors: chemical chaperones (mimic molecular chaperones); compounds that interact with molecular chaperones to affect their levels or binding to defective proteins; and target-specific pharmacochaperones. Chemical chaperones include dimethyl sulfoxide, glycerol and taurine. High amounts of chemical chaperones are needed because they are very non-specific. Curcumin is an example of a compound that affects the binding of chaperones to defective CFTR protein.
There are current clinical trials evaluating a CFTR corrector drug candidate, VX-809, that targets the ΔF508 mutation, which is found in a majority of patients [Citation20]. The ΔF508 mutation results in a misfolded, yet functional protein. Due to the misfolding, the protein is destroyed prior to reaching the cell membrane leading to a quantitative reduction in CFTR activity. VX-809 is being studied in phase 3 clinical trials in combination with Ivacaftor for patients homozygous for the ΔF508 mutation and in patients who have one copy of ΔF508 mutation and one copy of a mutation that is not expected to respond to either VX-809 or Ivacaftor [Citation21].
Approximately 10% of CF patients suffer from disease as the result of a nonsense mutation due to a premature stop codon [Citation22]. This unexpected stop codon leads to the production of a truncated CFTR protein, which typically causes the loss of its function. The common aminoglycoside antibiotic, gentamicin, has been demonstrated to promote readthrough of the stop codon. However, gentamicin must be administered parenterally and has ototoxic and nephrotoxic adverse side effects. Ataluren (PTC124) is an investigational oral small drug candidate that has been designed to promote readthrough of premature stop codons, but not of terminal stop codons. Ataluren can promote the production of full length CFTR and improvement of chloride transport in the airways. This drug is also being investigated as a treatment option for patients suffering from Duchenne muscular dystrophy. A single arm, dose-ranging study of Ataluren was performed in 30 patients aged 6–18 [Citation22]. Administration of ataluren resulted in increased apical side CFTR expression in respiratory epithelia and increase in nasal trans-epithelial potential difference. However, the treatment did not lead to a statistically significant improvement in pulmonary function.
Targeting receptors in the respiratory tract
Drugs which are used for treating respiratory diseases such as asthma and COPD interact with receptors found in the airways [Citation23]. Examples of the receptors in the airway include β2 and β1 adrenergic, muscarinic, and leukotriene receptors. Because β2 and β1 adrenergic and muscarinic receptors are found throughout the body, inhalable delivery formulations of drugs targeting these receptors have been developed. Leukotriene receptors are primarily located in the respiratory tract and systemic administration shows little side effects.
β2 adrenergic receptors are found in airway smooth muscle (throughout the airways), epithelium, vascular smooth muscle and submucosal glands [Citation24]. Alveolar walls contain both β1 and β2 receptors. β2 agonists have been shown to decrease plasma exudation, possess anti-inflammatory and antiedema effects, and may increase mucociliary clearance.
Inhaled anticholinergic medications for the treatment of COPD target muscarinic receptors [Citation23]. Muscarinic receptors are found on smooth muscle throughout the airways, but increased amounts of these receptors are found in larger airways. Muscarinic receptors are also localized on epithelial cells and submucosal gland, which secrete mucus in response to acetylcholine stimulation. There are four types of muscarinic receptors in the airways. Types M1 and M3 mediate mucus secretion; however, M3-subtype also mediates bronchoconstriction and releases nitric oxide which causes vasodilatation.
Glucocorticoid receptors are cytoplasmic receptors that are found in all cell types of the airway and may also be found in immune cells (eosinophils, dendritic cells, macrophages and T cells) [Citation23]. Glucocorticoid receptors are widely distributed throughout the body, so there is a high likely of systemic effects of drugs targeting these receptors after their systemic injection.
Gene therapy
Despite the CFTR gene being cloned in 1989, most patients do not have an option for successful treatment of the disease. Because of the monogenetic nature of CF, gene therapy held promise for treatment. Besides lung transplant, gene therapy is the only option as a cure for CF. However, there were numerous shortcomings of this type of treatment such as low gene expression levels and failure of repeated administration [Citation24].
CFTR protein is not expressed in significantly high amounts in normal patients (20–100 CFTR proteins/cell) [Citation25]. Therefore, even a small increase in CFTR expression may be beneficial to patients with CF. Since CFTR is involved in the regulation of other ion channels, gene therapy that dramatically increases cellular expression of CFTR may alter these other ion channels and overall cellular physiology.
Studies have shown that full restoration of CFTR expression in the lung is not needed to restore its disease-free chloride transport activity. In vitro cell mixing experiments demonstrated that if the cell population consisted of 6–10% of non-CF cells restored chloride secretion to non-CF levels [Citation26]. A 5% correction of CFTR gene expression restores nearly 50% of normal chloride transportation, thus demonstrating the non-linear relationship between phenotype and genotype [Citation27]. For restoration of sodium transport, nearly 100% of cells affected would need to be corrected. It is less clear, what percentage of cells with unaffected CFTR function is needed to restore its other functions to non-CF levels. Zhang et al. [Citation28] transfected a human CF ciliated surface airway epithelium using an engineered human parainfluenza virus expressing CFTR. Normal mucus transport was restored when CFTR was delivered to 25% of the epithelial cells.
Another important characteristic of gene therapy is duration of transgene expression. Optimal gene therapy would induce gene expression for the life of the target cell in order to avoid repetitive dosing. This would be even more beneficial when viral vectors are used because there is a likelihood of these vectors eliciting immunogenic responses. In addition, integrating viral vectors would be most suited to induce lifelong transgene expression.
The human airway is composed of a heterogeneous cell population. There is no consensus as to which cell types should be targeted to correct CFTR in CF. CFTR is expressed in ciliated cells and cells in the submucosal gland ducts and acini. Ciliated airway epithelium has a reported lifespan of 3 months [Citation29], epithelium in the trachea has a lifespan of 6 months, and for that in the lung it is 17 months [Citation30]. Certain progenitor cells have been reported to express CFTR; this would confer long-term CFTR gene expression when using integrating viruses. Many groups believe that the ciliated epithelium should be the primary targets for CF gene therapy [Citation31].
Another approach is to perform gene therapy ex vivo in stem cells that can differentiate into airway epithelial cells [Citation15]. Patient derived mesenchymal stem cells or induced pluripotent stem cells can be isolated, transfected in vitro, and administered back to the patient. There is a severe lack of information on this process. Early studies showed that the efficiency of engraftment was less than 1% [Citation32].
Viral gene delivery
A recent study has shown that the CF condition presents an advantageous environment for viral infection [Citation33]. As a result, this provides a possible avenue to exploit for viral gene therapy. As mentioned before, the ΔF508 mutation results in a misfolded CFTR protein, which undergoes proteasomal degradation. The degradation process compounds with subcellular stress, inflammation and endoplasmic reticulum expansion. The sum of these events depresses cellular defense against viral replication, particularly adeno-associated virus 2 (AAV2) infection.
DNA viruses such as adenovirus and adeno-associated virus (AAV) have been tested as gene delivery vectors for CF (). These vectors are non-integrating and therefore, transduction may be transient [Citation24]. A challenge to pulmonary delivery of adenoviral and adeno-associated viral vectors is that receptors for these viruses such as coxsackie-adenovirus receptor are located on the basolateral side of airway epithelial cells [Citation25,Citation31]. In addition, adenoviral vectors have low transduction efficiency in airway epithelia and induce robust immune responses [Citation34]. A phase I clinical trial was performed in 11 CF patients, who had at least one copy of ΔF508 mutation [Citation35]. Patients received a single of dose of 2.1 × 109 and 2.1 × 1011 adenoviral particles via a bronchoscope. Four days after the dose, gene transfer was detected in <1% of harvested bronchial epithelial cells; however, gene expression was undetectable 43 d post-dose. The adenoviral vector induced mild humoral immune response and T-cell response as well.
Table 2. Viral vectors studied for gene therapy of cystic fibrosis [Citation36].
Although AAVs have been shown to induce long-term gene expression in non-human primates and are not as immunogenic as adenoviruses, the cloning capacity of AAVs is very low. A solution has been to develop a truncated form of CFTR, which is missing the first 264 amino acids (Δ264CFTR). This modified form maintained biological activity and correction of chloride transport in vitro. However, it is unknown whether this form will perform all functions of CFTR in vitro and in vivo [Citation36,Citation37]. Another proposed solution used two plasmids, each encoding partial CFTR cDNA [Citation38]. Each partial CFTR plasmid was nonfunctional alone, but together they produce functional CFTR protein. One plasmid encodes the 5′ untranslated, upstream coding sequence through exon 14a and the other encodes the downstream coding sequence from exon 14b through 3′ untranslated region. Both plasmids were delivered with AAV6.2, a derivative that has high transduction of respiratory epithelium due to a one amino acid change in the capsid (F129L).
Recent studies have investigated different serotypes of AAVs. AAV2 serotype was the first tested. However, a lack of targeted receptors on the apical airway epithelia restricted the use of this gene delivery vehicle. Using serotypes AAV1, AAV5 and AAV6 could be advantageous because they can target the apical epithelium, have more enhanced uptake, and evade the immune system more efficiently [Citation37,Citation39,Citation40]. However, Hida et al. [Citation41] showed () that adenoviruses and AAVs are unable to sufficiently diffuse through mucus collected from CF patients. The group calculated that the diffusivities of adenovirus and AAV5 were 3000- and 12 000-fold slower in mucus than in water, respectively.
Figure 3. Transport mode distributions of adenovirus (AdV) adeno-associated virus (AAV5, serotype 5), muco-adhesive PS control nanoparticles (PS) and muco-inert control PS-PEG nanoparticles. Particles were classified into either (i) immobile or hindered and (ii) diffusive. Mean ± SD are shown. *Statistically significant difference when compared with AAV5, AdV or PS within the same transport mode classification (p < 0.05). Redrawn using data from Ref. [Citation41].
![Figure 3. Transport mode distributions of adenovirus (AdV) adeno-associated virus (AAV5, serotype 5), muco-adhesive PS control nanoparticles (PS) and muco-inert control PS-PEG nanoparticles. Particles were classified into either (i) immobile or hindered and (ii) diffusive. Mean ± SD are shown. *Statistically significant difference when compared with AAV5, AdV or PS within the same transport mode classification (p < 0.05). Redrawn using data from Ref. [Citation41].](/cms/asset/83bc823e-e74b-4e20-a40e-0139bd16fca4/idrt_a_829078_f0003_b.jpg)
The Sendai virus, a single-stranded RNA virus of the Paramyxoviridae family, has attracted great interest as a vector for treatment of CF. The Sendai virus binds to sialic acid and cholesterol receptors on the apical surface of airway epithelia, has highly efficient gene transfer, and using the cytoplasmic expression system. Although CF mucus still presents a barrier, the impact is minimized due to the high gene expression levels. Therapy with the Sendai virus is hindered by immunogenicity and short gene transduction [Citation31]. The virus can be modified to not express the fusion protein; this results in transmission incompetent second generation viruses. To capitalize on the Sendai virus' ability to enter respiratory cells and overcome immunogenicity issues, Mitomo et al. [Citation42] used a derived simian immunodeficiency virus (SIV) lentiviral vector. One of biggest advantage of SIV is the ample packaging capacity of 8–10 kb and as a lentivirus, it is integrating and transfects both dividing and non-dividing cells [Citation24]. This is advantageous because of the lifespan of airway epithelial cells. The vector was pseudotyped with Sendai virus envelope proteins, hemagglutinin-neuraminidase, which attach to sialic acids and fusion proteins. The group was able to transduce mice nasal epithelia using the vector. The evaluation of the in vivo gene transduction efficiency of the SIV vector in the nasal airway epithelium of mice [Citation42] showed that this vector was capable to transduce cells in both the respiratory and olfactory epithelium of the nose (). Moreover, the expression of the targeted protein persisted for more than one year after transduction ().
Figure 4. Determination of transduced cell types. The transduced green fluorescent protein (GFP)-positive cells were identified using fluorescent microscopy (original magnification × 63) 30 d after administration of SIV-GFP (4 × 108 TU/mouse) vector to the mouse nose. (A) Ciliated respiratory epithelial cell, (B) neuronal cell in olfactory epithelium, (C) squamous epithelial cell and (D) non-neuronal cell in olfactory epithelium. The central image shows a cross-section through the mouse nose and red boxes indicate regions in mouse nasal epithelium where respective transduced cell types were found. Panels A, B and D were rotated ∼45°, 130° and 180° counter clockwise, respectively, to improve clarity of the figure. Modified from Ref. [Citation42].
![Figure 4. Determination of transduced cell types. The transduced green fluorescent protein (GFP)-positive cells were identified using fluorescent microscopy (original magnification × 63) 30 d after administration of SIV-GFP (4 × 108 TU/mouse) vector to the mouse nose. (A) Ciliated respiratory epithelial cell, (B) neuronal cell in olfactory epithelium, (C) squamous epithelial cell and (D) non-neuronal cell in olfactory epithelium. The central image shows a cross-section through the mouse nose and red boxes indicate regions in mouse nasal epithelium where respective transduced cell types were found. Panels A, B and D were rotated ∼45°, 130° and 180° counter clockwise, respectively, to improve clarity of the figure. Modified from Ref. [Citation42].](/cms/asset/bf741798-ca71-4992-a03d-e25d19478255/idrt_a_829078_f0004_b.jpg)
Figure 5. Duration of green fluorescent protein (GFP) expression after transduction with F/HN-SIV-GFP. Mouse nasal tissue was perfused with SIV-GFP (4 × 108 TU/mouse or phosphate-buffered saline (PBS) and gene expression was analyzed at indicated time points after transduction. GFP-positive cells were quantified on histological sections taken 2 mm into the nasal tissue of the nose. Data from 30 to 360 d after transduction are represented both by mean ± SEM and individual values (ratio to GFP cells positive on day 30). The number (n) per group are 13 (day 30), 3 (day 50), 12 (day 90), 14 (day 160–180), 10 (day 220–270) and 17 (day 360) [Citation42].
![Figure 5. Duration of green fluorescent protein (GFP) expression after transduction with F/HN-SIV-GFP. Mouse nasal tissue was perfused with SIV-GFP (4 × 108 TU/mouse or phosphate-buffered saline (PBS) and gene expression was analyzed at indicated time points after transduction. GFP-positive cells were quantified on histological sections taken 2 mm into the nasal tissue of the nose. Data from 30 to 360 d after transduction are represented both by mean ± SEM and individual values (ratio to GFP cells positive on day 30). The number (n) per group are 13 (day 30), 3 (day 50), 12 (day 90), 14 (day 160–180), 10 (day 220–270) and 17 (day 360) [Citation42].](/cms/asset/274a8ec4-5f54-4ec9-a7b9-22f1e04bc9cc/idrt_a_829078_f0005_b.jpg)
Viruses including the Norwalk virus, polio virus and human papilloma virus have dense coating of both positive and negative charges on the surface and have an overall neutral charge. This feature makes the surface hydrophilic and avoids hydrophobic interactions and the neutral charge does not have any electrostatic interaction adhesion [Citation41,Citation43]. These viruses can diffuse through mucus as easily as they diffuse in water and may demonstrate improved transduction compared to other viruses.
Respiratory syncytial virus has a natural tropism for airways. Although immunogenic, subsequent infections are not thwarted. Kwilas et al. designed a respiratory syncytial virus containing a 4.5 kb CFTR gene [Citation44]. The researchers examined inserting the gene in four locations in the genome. Two of the four vectors were successful in inserting the gene in differentiated human airway epithelial cells derived from CF patients and improved chloride transport in vitro.
Non-viral gene delivery
Most non-viral vectors are designed to possess a positive charge. Such cationic carriers can form electrostatic bonds with negatively charged nucleic acids. However, most clinical and pre-clinical animal studies focus on delivering the particle to the nasal epithelium as proof-of-concept trials because it has a similar cell composition to the lower respiratory tract, easier access and safer than lung administration [Citation31]. These studies do not take into consideration the fact that effective clinical implementation of therapy requires delivery to the lungs, which imposes additional barriers to effective transfection. The first barrier is the complex airway architecture and the differences in architecture between a pre-clinical animal model and human. Although higher order animals such as chimpanzees better represent the human airway, these experiments would be more costly than those performed in mice and rats. The second major barrier is the presence of CF mucus, which is more viscous and purulent than normal mucus. Studies on mucus penetration have shown that charged and hydrophobic molecules and nanoparticles do not diffuse CF mucus and that therapy with such particles is ineffective for majority of cases [Citation14,Citation45].
Matsui et al. [Citation46] performed a study to detect internalization mechanism of liposome–DNA complexes in differentiated and non-differentiated airway epithelia cells. Although the liposome–DNA complexes entered non-differentiated cells via phagocytosis, this mechanism was absent in differentiated cells. One possible explanation of this phenomenon is that non-differentiated cells tend to be more negatively charged than the differentiated cells. The authors hypothesized that entry of liposome-DNA complexes into differentiated cells requires receptor mediated endocytosis.
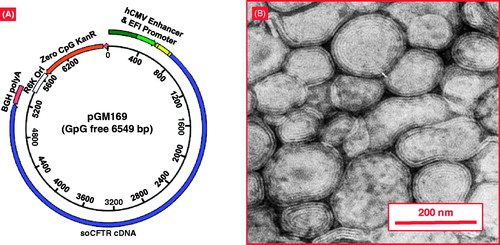
The biggest trial for non-viral gene therapy for CF is being sponsored by the UK CF Gene Therapy Consortium. A plasmid encoding the CFTR protein (pGM169) was delivered by cationic liposomes [Citation47,Citation48] (). The pGM169 plasmid is under the control of the hybrid elongation factor 1α promoter and contains no CpG islands, which can cause inflammation [Citation49]. Pringle et al. [Citation50] showed that incorporation of a murine cytomegalovirus (CMV) enhancer in the plasmid resulted in higher transfection efficacy when compared with human CMV enhancer. In a mouse model, CFTR gene expression was registered 4 weeks after a single dose treatment. Previous pre-clinical evaluation of aerosol delivery to sheep lung demonstrated that the GL67A liposomes induced the highest gene expression when compared with 25 kDa-branched polyethyleneimine (PEI) and compacted DNA nanoparticle with polyethylene glycol (PEG)-substituted lysine 30-mer [Citation51].
Figure 6. The combination of cationic liposome (GL67A) and plasmid DNA expressing CFTR (pGM169). (A) pGM169 plasmid encoding the gene for CFTR protein contains no CpG dinucleotides, which are unmethylated in bacterial derived plasmids and induce inflammatory response. The plasmid also includes a CMV promoter, which aids in increased gene expression. (B) Electron microscope image of pGM169-GL67A liposomal complex. Modified from http://www.genemedresearch.ox.ac.uk/ukcfgtc/product.html.
The pGM169/GL67A delivery system has been previously tested in human clinical trials [Citation52]. However in these studies, the system was delivered intra-nasally to patients. Although this method is easier to administer and evaluate when compared with inhalation, it does not result in effective treatment of CF. CFTR protein is not being expressed in tissues that need to be targeted to improve CF respiratory manifestations and the delivery system does not have to traverse the complicated respiratory architecture and penetrate the purulent mucus to reach epithelial cells.
Manunta et al. [Citation53] designed and delivered a nanoparticle delivery system consisting of cationic liposomes (DHDTMA/DOPE), plasmid DNA and a targeting peptide. The peptide sequence was K16GACSERSMNFCG and showed similar receptor binding as rhinovirus and Listeria monocytogenes. The K16 aids DNA packaging and the SERSMNF motif is similar to the intercellular adhesion molecule 1 sequence of rhinovirus. The nanocomplexes were 150 nm in size and had a zeta-potential of +50 mV. The nanocomplexes were delivered using different nebulizers, which had no effect on physico-chemical properties of the complexes. β-Galactosidase was used as the reporter gene and there was no statistically significant difference between treatment and control groups.
Compacted plasmid DNA nanoparticles using a PEG-substituted lysine 30-mer peptide as a vector has also been studied [Citation54]. Twelve patients were assigned to receive one of three doses (0.8 mg, 2.67 mg or 8.0 mg of DNA) administered to the nasal epithelium. A difference in nasal potential was used to assess CFTR restoration. Eight of twelve patients had partial or complete restoration; however, the effect lasted for only six days.
Nanoparticles for treatment of fibrosis
Although nanoparticles for inhalation have been studied by many research groups, nanoparticle based delivery of therapeutics is still not being used in clinical practice for the treatment of fibrosis. This is in part due to the absence of an effective drug for treatment of fibrosis. While gene therapy strategies have entered clinical trials for treatment of CF, they have primarily been focused on viral vectors as gene delivery vehicles. Most of studied non-viral vectors demonstrated lower transfection efficacy compared to viral counterparts. Also, conventional nanoparticles have a tendency to be trapped in mucus and rapidly cleared by mucociliary action [Citation43].
Mucoadhesion is desirable in many drug delivery applications and there are numerous methods to achieve this. The adhesion between mucus and particles can be due to various non-specific forces such as hydrogen bonding, van der Waals forces, hydrophobic or electrostatic interactions. Studies have shown that cationic surface charges due to polymers can increase the retention time of nanoparticles in mucus [Citation55,Citation56]. Targeted mucoadhesion can be achieved using tomato lectin, which specifically binds to N-acetyl glucosamine on cell surfaces [Citation57]. However, it is still unknown whether mucoadhesion can improve CF therapy. Mucoadhesive particles have not been shown to penetrate the mucus layer and enter the underlying epithelia (). Therefore, they are likely to be unsuitable systems for drug or gene delivery for the treatment of lung form of CF [Citation43]. Consequently, mucoadhesive properties may be unnecessary for nanoparticles in CF therapy. CF lung disease is characterized by increased viscosity of mucus and decreased amount of ASL. In addition, there is buildup of purulent sputum and obstruction due to decreased clearance.
Figure 7. Behavior of mucoadhesive and muco-inert particles in mucus layers. Muco-inert particles do not possess any attractive forces with CF mucus like mucoadhesive conventional particles and therefore have greater penetration. Muco-inert particles present as an optimal option for inhalation delivery for patients with cystic fibrosis. Modified from Ref. [Citation43].
![Figure 7. Behavior of mucoadhesive and muco-inert particles in mucus layers. Muco-inert particles do not possess any attractive forces with CF mucus like mucoadhesive conventional particles and therefore have greater penetration. Muco-inert particles present as an optimal option for inhalation delivery for patients with cystic fibrosis. Modified from Ref. [Citation43].](/cms/asset/5badf58c-1f02-45ca-95c6-04731df03803/idrt_a_829078_f0007_b.jpg)
A correlation between physical characteristics of particles and ability to penetrate CF sputum was found. Dawson et al. demonstrated that neutrally charged polystyrene particles less than 200 nm in diameter have faster transport through CF sputum than charged particles [Citation58]. Treatment with recombinant human DNase (Pulmozyme®) reduced macroscopic elasticity and viscosity of CF sputum by 50% and 40%, respectively. Pulmozyme treatment resulted in more homogenous diffusion of particles, but there was no change in the mean particle diffusion rate. A possible reason is that resulting DNA fragments may have increased microviscosity of the sputum.
It appears to be advantageous to develop muco-inert particles to penetrate CF sputum and reach target epithelial cells. Suk et al. [Citation59] modeled that the average mesh spacing in CF sputum was 140 ± 50 nm with a range of 60–300 nm. They further demonstrated that particles up to 200 nm in diameter that were densely coated with low molecular weight PEG traversed CF sputum up to 90-fold faster than uncoated particles with a similar size (). In another study, transportation of these nanoparticles was examined after CF sputum was treated with N-acetylcysteine (NAC) [Citation60]. NAC increased the average spacing between sputum mesh elements from 145 ± 50 nm to 230 ± 50 nm () and the transport of the densely coated 200 nm particles in CF sputum approached their theoretical speed in water. However, when NAC was used in combination with mucoadhesive cationic liposomes and polymers for gene therapy, there was no observable correction of nasal transepithelial potential difference in CF-null mice [Citation43].
Figure 8. Penetration of polystyrene 200 nm nanoparticles through CF sputum. Polystyrene nanoparticles densely coated with low MW PEG (PS-PEG) have greater penetration through CF sputum compared to non-PEGylated particles (PS). Treatment with N-acetyl cysteine (NAC) increases penetration of both coated and non-coated particles. Mean ± SD are shown. Redrawn using data from Ref. [Citation60].
![Figure 8. Penetration of polystyrene 200 nm nanoparticles through CF sputum. Polystyrene nanoparticles densely coated with low MW PEG (PS-PEG) have greater penetration through CF sputum compared to non-PEGylated particles (PS). Treatment with N-acetyl cysteine (NAC) increases penetration of both coated and non-coated particles. Mean ± SD are shown. Redrawn using data from Ref. [Citation60].](/cms/asset/c640aa34-64a9-472b-b812-993731fc1ecf/idrt_a_829078_f0008_b.jpg)
Figure 9. An influence of mucolytic treatment on the mucus mesh. Treatment with hypertonic saline increases the spacing within the mucus mesh. Treatment with a mucolytic such as N-acetyl cysteine breaks disulfide bonds within the mucosal mesh network. The increase in openings can increase particle penetration [Citation12].
![Figure 9. An influence of mucolytic treatment on the mucus mesh. Treatment with hypertonic saline increases the spacing within the mucus mesh. Treatment with a mucolytic such as N-acetyl cysteine breaks disulfide bonds within the mucosal mesh network. The increase in openings can increase particle penetration [Citation12].](/cms/asset/2c752f06-3461-468b-8f9f-87d604c7ed80/idrt_a_829078_f0009_b.jpg)
To increase biocompatibility and hydrophilicity, nanoparticles are typically coated with PEG. The PEG coating can have a big influence on the transport of particles through mucus. Lai et al. [Citation61] investigated coating of polystryene nanoparticles with either 2 kDa PEG or 10 kDa PEG. High-density coating with 2 kDa PEG resulted in neutrally charged particles with relatively unhindered transport through mucus. High-density coating with 10 kDa PEG increased the adhesiveness of nanoparticles to mucus. To examine the role of coating density, the researchers also assess low-density coating with 2 kDa PEG. Low-density 2 kDa PEG coating had a cationic zeta potential (−10 ± 3 mV) and the particles were mucoadhesive.
Small interfering RNA (siRNA) may present an option to rescue mutated CFTR such as ΔF508 protein from proteasomal degradation [Citation62]. However, delivery of siRNA to the target cells will face many of the same delivery barriers as gene therapy. Also, the proteasomal degradation is an integral part of normal cellular function. Therefore, off target effects of siRNA therapy must be minimized either through local delivery or use of a targeting moiety.
Although currently there are no siRNA based proteasomal inhibition therapies of CF, investigations were carried out to use small molecules to inhibit CFTR from the proteasomal degradation. Researchers have proposed to rescue the ΔF508 CFTR protein from the degradation using treatment with curcumin [Citation63]. Curcumin has low affinity as an inhibitor of sarco(endo)plasmic reticulum calcium ATPase (SERCA) pump. Inhibition of the SERCA pump would allow the mutated CFTR protein to escape from the endoplasmic reticulum and possibly reach the membrane. Curcumin was encapsulated within PLGA nanoparticles to increase absorption and decrease metabolism of curcumin. Nanoparticles were synthesized with a diameter of 77 ± 16 nm and drug loading of 7.6% w/w. The curcumin nanoparticles were administered orally to homozygous and heterozygous ΔF508 mice and the nasal potential difference was measured. The curcumin nanoparticle conjugates showed improved efficacy compared to free curcumin.
Bortezomib (PS-341; Velcade®) is a FDA approved proteasome inhibitor for the treatment of refractory multiple myeloma. Whereas normal cells overcome proteasome inhibition, cancer cells undergo apoptosis after treatment with Bortezomib. The drug was encapsulated in PLGA/PEG matrix and administered intranasally to mice with a model of CF [Citation64]. A two-fold decrease in proteasome activity and subsequently control of inflammation caused by Pseudomonas lipopolysaccharide were registered after the treatment.
Nanotechnology-based approach for inhalation treatment of different lung diseases including the pulmonary form of fibrosis has been developed in our laboratory [Citation65–70]. This nanomedicine platform is currently based on the use of three types of nanocarriers: mesoporous silica nanoparticles, nanostructured lipid nanocarriers (NLC), and liposomes (). NLC-based system () was successfully used for the delivery of different drugs and nucleic acids. Liposomal drug delivery system () was used for inhalation treatment of lung diseases using both hydrophilic and lipophilic drugs. In particular, alpha-tocopherol and prostaglandin E2 (PGE2) were used for treatment of lung edema and idiopathic pulmonary fibrosis (IPF), respectively. A special instillation unit consisting of a collison nebulizer connected to four-port nose-only exposure chambers was used for inhalation delivery of NLC and liposomal systems in mice (). It was found that the selected regimen of nebulization and delivery via the unit did not change significantly the characteristics of the liposomes and NLC. In particular, the size of NLC before and after the nebulization was practically the same (). It should also be stressed that nebulization did not influence functionality and silencing efficiency of siRNA. Organ content of fluorescently labeled NLC and liposomes after intravenous or intratracheal delivery was examined. It was found that 24 h after intravenous instillation, NLC and liposomes accumulated predominately in the liver, while substantially smaller amount of nanoparticles was found in the lungs (). In contrast, after pulmonary delivery, NLC and liposomes were retained in the lungs with much smaller amounts found in other organs, including the liver. It should be stressed that NLC demonstrated more favorable lung accumulation after inhalation when compared with liposomes. It was confirmed by in vivo experiments that the NLC and liposomes could effectively deliver drugs and siRNA by inhalation locally to the lungs and limit possible adverse systemic effects of the treatment. Liposomal PGE2 delivered by inhalation was very effective in treatment of IPF. The data obtained showed that inhalation of animals with liposomal PGE2 substantially decreased accumulation of hydroxyproline (one of the major indicator of lung fibrosis) in the lungs () substantially limited lung damage caused by IPF () and finally prevented the mortality of experimental animals with IPF.
Figure 10. Nanomedicine approach for inhalation treatment of pulmonary fibrosis. (A) Nanostructured lipid carrier (NLC)-based drug delivery system (DDS). (B) Structure of liposomal DDS. (C) Installation for inhalation delivery of DDS. (D) NLC particle size distribution by dynamic light scattering. (E) Organ distribution of liposomes after intravenous and inhalation administration. (F) Organ distribution of NLC after intravenous and inhalation administration. (G) Hydroxyproline content in the lungs. Means ± SD are shown. (H) Lung tissue histology. The tissues were stained with eosin-hematoxylin. (I) Survival of experimental animals. Mice were treated with liposomal PGE2 by inhalation twice per week for three weeks starting one day later after the bleomycin administration. *p < 0.05 when compared with control (healthy animals). †p < 0.05 when compared with untreated animals with fibrosis. Modified from Refs. [Citation66,Citation67,Citation70].
![Figure 10. Nanomedicine approach for inhalation treatment of pulmonary fibrosis. (A) Nanostructured lipid carrier (NLC)-based drug delivery system (DDS). (B) Structure of liposomal DDS. (C) Installation for inhalation delivery of DDS. (D) NLC particle size distribution by dynamic light scattering. (E) Organ distribution of liposomes after intravenous and inhalation administration. (F) Organ distribution of NLC after intravenous and inhalation administration. (G) Hydroxyproline content in the lungs. Means ± SD are shown. (H) Lung tissue histology. The tissues were stained with eosin-hematoxylin. (I) Survival of experimental animals. Mice were treated with liposomal PGE2 by inhalation twice per week for three weeks starting one day later after the bleomycin administration. *p < 0.05 when compared with control (healthy animals). †p < 0.05 when compared with untreated animals with fibrosis. Modified from Refs. [Citation66,Citation67,Citation70].](/cms/asset/67cffbaf-4f10-4200-8464-89141aa11b8a/idrt_a_829078_f0010_b.jpg)
Declaration of interest
The research was supported in part by grants from the National Institutes of Health (R01 CA111766 and R01 CA138533).
References
- Flume PA, Van Devanter DR. State of progress in treating cystic fibrosis respiratory disease. BMC Med 2012;10:88
- Pier GB. The challenges and promises of new therapies for cystic fibrosis. J Exp Med 2012;209:1235–9
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol Sci 2007;28:334–41
- Kreindler JL. Cystic fibrosis: exploiting its genetic basis in the hunt for new therapies. Pharmacol Ther 2010;125:219–29
- Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev 2002;54:1359–71
- Hoffman LR, Ramsey BW. Cystic fibrosis therapeutics: the road ahead. Chest 2013;143:207–13
- Garcia-Contreras L, Hickey AJ. Pharmaceutical and biotechnological aerosols for cystic fibrosis therapy. Adv Drug Deliv Rev 2002;54:1491–504
- Sung JC, Pulliam BL, Edwards DA. Nanoparticles for drug delivery to the lungs. Trends Biotechnol 2007;25:563–70
- Martonen T, Katz I, Cress W. Aerosol deposition as a function of airway disease: cystic fibrosis. Pharm Res 1995;12:96–102
- Hirsh AJ. Altering airway surface liquid volume: inhalation therapy with amiloride and hyperosmotic agents. Adv Drug Deliv Rev 2002;54:1445–62
- Thelin WR, Boucher RC. The epithelium as a target for therapy in cystic fibrosis. Curr Opin Pharmacol 2007;7:290–5
- King M, Rubin BK. Pharmacological approaches to discovery and development of new mucolytic agents. Adv Drug Deliv Rev 2002;54:1475–90
- Rubin BK. Mucus structure and properties in cystic fibrosis. Paediatr Respir Rev 2007;8:4–7
- Sanders NN, De Smedt SC, Van Rompaey E, et al. Cystic fibrosis sputum: a barrier to the transport of nanospheres. Am J Respir Crit Care Med 2000;162:1905–11
- Kotzamanis G, Kotsinas A, Papalois A, et al. Gene therapy – tools and potential applications. 22. targeting the lung: challenges in gene therapy for cystic fibrosis. InTech 2013. Available from: http://www.intechopen.com/books/gene-therapy-tools-and-potential-applications/targeting-the-lung-challenges-in-gene-therapy-for-cystic-fibrosis [last accessed 9 Jun 2013]
- Cryan SA, Sivadas N, Garcia-Contreras L. In vivo animal models for drug delivery across the lung mucosal barrier. Adv Drug Deliv Rev 2007;59:1133–51
- Pringle IA, Hyde SC, Gill DR. Non-viral vectors in cystic fibrosis gene therapy: recent developments and future prospects. Expert Opin Biol Ther 2009;9:991–1003
- Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 2010;363:1991–2003
- Condren ME, Bradshaw MD. Ivacaftor: a novel gene-based therapeutic approach for cystic fibrosis. J Pediatr Pharmacol Ther 2013;18:8–13
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009;106:18825–30
- Vertex Pharmaceutical Incorporated. Study of VX-809 alone and in combination with VX-770 in cystic fibrosis (CF) patients homozygous or heterozygous for F508del-CFTR mutation. ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). 2010. Available from: http://clinicaltrials.gov/ct2/show/NCT01225211 [last accessed 30 Jun 2013]
- Sermet-Gaudelus I, Boeck KD, Casimir GJ, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 2010;182:1262–72
- Barnes PJ. Distribution of receptor targets in the lung. Proc Am Thorac Soc 2004;1:345–51
- Oakland M, Sinn PL, McCray PB Jr. Advances in cell and gene-based therapies for cystic fibrosis lung disease. Mol Ther 2012;20:1108–15
- Ferrari S, Geddes DM, Alton EW. Barriers to and new approaches for gene therapy and gene delivery in cystic fibrosis. Adv Drug Deliv Rev 2002;54:1373–93
- Johnson LG, Olsen JC, Sarkadi B, et al. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet 1992;2:21–5
- Dorin JR, Farley R, Webb S, et al. A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther 1996;3:797–801
- Zhang L, Button B, Gabriel SE, et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol 2009;7:e1000155
- Borthwick DW, Shahbazian M, Krantz QT, et al. Evidence for stem-cell niches in the tracheal epithelium. Am J Respir Cell Mol Biol 2001;24:662–70
- Rawlins EL, Hogan BL. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol 2008;295:L231–4
- Griesenbach U, Inoue M, Hasegawa M, et al. Viral vectors for cystic fibrosis gene therapy: what does the future hold? Virus Adaptation Treatment 2010;2:159–171
- Loi R, Beckett T, Goncz KK, et al. Limited restoration of cystic fibrosis lung epithelium in vivo with adult bone marrow-derived cells. Am J Respir Crit Care Med 2006;173:171–9
- Johnson JS, Gentzsch M, Zhang L, et al. AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis. PLoS Pathog 2011;7:e1002053
- Yei S, Mittereder N, Wert S, et al. In vivo evaluation of the safety of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator cDNA to the lung. Hum Gene Ther 1994;5:731–44
- Zuckerman JB, Robinson CB, McCoy KS, et al. A phase I study of adenovirus-mediated transfer of the human cystic fibrosis transmembrane conductance regulator gene to a lung segment of individuals with cystic fibrosis. Hum Gene Ther 1999;10:2973–85
- Conese M, Ascenzioni F, Boyd AC, et al. Gene and cell therapy for cystic fibrosis: from bench to bedside. J Cyst Fibros 2011;10:S114–28
- Fischer AC, Smith CI, Cebotaru L, et al. Expression of a truncated cystic fibrosis transmembrane conductance regulator with an AAV5-pseudotyped vector in primates. Mol Ther 2007;15:756–63
- Song Y, Lou HH, Boyer JL, et al. Functional cystic fibrosis transmembrane conductance regulator expression in cystic fibrosis airway epithelial cells by AAV6.2-mediated segmental trans-splicing. Hum Gene Ther 2009;20:267–81
- Halbert CL, Allen JM, Miller AD. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J Virol 2001;75:6615–24
- Zabner J, Seiler M, Walters R, et al. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J Virol 2000;74:3852–8
- Hida K, Lai SK, Suk JS, et al. Common gene therapy viral vectors do not efficiently penetrate sputum from cystic fibrosis patients. PLoS One 2011;6:e19919
- Mitomo K, Griesenbach U, Inoue M, et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol Ther 2010;18:1173–82
- Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev 2009;61:158–71
- Kwilas AR, Yednak MA, Zhang L, et al. Respiratory syncytial virus engineered to express the cystic fibrosis transmembrane conductance regulator corrects the bioelectric phenotype of human cystic fibrosis airway epithelium in vitro. J Virol 2010;84:7770–81
- Khanvilkar K, Donovan MD, Flanagan DR. Drug transfer through mucus. Adv Drug Deliv Rev 2001;48:173–93
- Matsui H, Johnson LG, Randell SH, et al. Loss of binding and entry of liposome-DNA complexes decreases transfection efficiency in differentiated airway epithelial cells. J Biol Chem 1997;272:1117–26
- Alton EW, Boyd AC, Cheng SH, et al. A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax 2013. [Epub ahead of print]
- Oxford University Gene Medicine. GL67A/pGM169 – The Consortium's first clinical trial product. 2013. Available from: http://www.genemedresearch.ox.ac.uk/ukcfgtc/product.html [last accessed 30 Jun 2013]
- Hyde SC, Pringle IA, Abdullah S, et al. CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat Biotechnol 2008;26:549–51
- Pringle IA, Hyde SC, Connolly MM, et al. CpG-free plasmid expression cassettes for cystic fibrosis gene therapy. Biomaterials 2012;33:6833–42
- McLachlan G, Davidson H, Holder E, et al. Pre-clinical evaluation of three non-viral gene transfer agents for cystic fibrosis after aerosol delivery to the ovine lung. Gene Ther 2011;18:996–1005
- Davies J, Davies G, Voase N, et al. Evaluation of safety and gene expression with a single dose of PGM169/GL67A administered to the nose and lung of individuals with cystic fibrosis: the UK Gene Therapy Consortium “Pilot Study”. Thorax 2009;64:A70
- Manunta MD, McAnulty RJ, Tagalakis AD, et al. Nebulisation of receptor-targeted nanocomplexes for gene delivery to the airway epithelium. PLoS One 2011;6:e26768
- Konstan MW, Davis PB, Wagener JS, et al. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther 2004;15:1255–69
- Jubeh TT, Barenholz Y, Rubinstein A. Differential adhesion of normal and inflamed rat colonic mucosa by charged liposomes. Pharm Res 2004;21:447–53
- Prego C, Torres D, Alonso MJ. The potential of chitosan for the oral administration of peptides. Expert Opin Drug Deliv 2005;2:843–54
- Woodley J. Bioadhesion: new possibilities for drug administration? Clin Pharmacokinet 2001;40:77–84
- Dawson M, Wirtz D, Hanes J. Enhanced viscoelasticity of human cystic fibrotic sputum correlates with increasing microheterogeneity in particle transport. J Biol Chem 2003;278:50393–401
- Suk JS, Lai SK, Wang YY, et al. The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles. Biomaterials 2009;30:2591–7
- Suk JS, Lai SK, Boylan NJ, et al. Rapid transport of muco-inert nanoparticles in cystic fibrosis sputum treated with N-acetyl cysteine. Nanomedicine (Lond) 2011;6:365–75
- Lai SK, O'Hanlon DE, Harrold S, et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A 2007;104:1482–7
- Sinn PL, Anthony RM, McCray PB, Jr. Genetic therapies for cystic fibrosis lung disease. Hum Mol Genet 2011;20:R79-86
- Cartiera MS, Ferreira EC, Caputo C, et al. Partial correction of cystic fibrosis defects with PLGA nanoparticles encapsulating curcumin. Mol Pharm 2010;7:86–93
- Vij N, Min T, Marasigan R, et al. Development of PEGylated PLGA nanoparticle for controlled and sustained drug delivery in cystic fibrosis. J Nanobiotechnology 2010;8:22
- Garbuzenko OB, Saad M, Betigeri S, et al. Intratracheal versus intravenous liposomal delivery of siRNA, antisense oligonucleotides and anticancer drug. Pharm Res 2009;26:382–94
- Garbuzenko OB, Saad M, Pozharov VP, et al. Inhibition of lung tumor growth by complex pulmonary delivery of drugs with oligonucleotides as suppressors of cellular resistance. Proc Natl Acad Sci U S A 2010;107:10737–42
- Ivanova V, Garbuzenko OB, Reuhl KR, et al. Inhalation treatment of pulmonary fibrosis by liposomal prostaglandin E2. Eur J Pharm Biopharm 2013;84:335–44
- Minko T, Stefanov A, Pozharov V. Selected contribution: lung hypoxia: antioxidant and antiapoptotic effects of liposomal alpha-tocopherol. J Appl Physiol 2002;93:1550–60; discussion 1549
- Taratula O, Garbuzenko OB, Chen AM, et al. Innovative strategy for treatment of lung cancer: targeted nanotechnology-based inhalation co-delivery of anticancer drugs and siRNA. J Drug Target 2011;19:900–14
- Taratula O, Kuzmov A, Shah M, et al. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J Control Release 2013. [Epub ahead of print]