Abstract
Objective: To compare genome-wide methylation profiles in maternal leukocyte DNA between normotensive and preeclamptic pregnant women at delivery. Methods: Age, body mass index matched case-control comparison of methylation at 27,578 cytosine— guanine sites in 14,495 genes in maternal leukocyte DNA in women with preeclampsia (PE; n = 14) and normotensive controls (n = 14). Results: PE was associated with widespread differential methylation favoring hypermethylation. Pathway analysis identified the best matched process as a neuropeptide signaling pathway (p < 10−5); best matched disease as eclampsia (p < 9.97 × 10−20). Significantly differentially methylated genes (GRIN2b. GABRA1. PCDHB7, and BEX1) are associated with seizures. Conclusion: Altered maternal leukocyte DNA methylation is associated with PE at delivery, and differential methylation of certain neuronal genes may explain the risk for eclampsia.
Introduction
Preeclampsia, defined as new onset hypertension after 20 weeks gestation, accompanied by proteinuria >300 mg in 24 h, occurs in 5–8% of pregnancies. Eclampsia, its convulsive form, complicates up to 3% of these pregnancies. Preeclampsia is an inflammatory state with significant immune system dysfunction. To date, alterations in multiple proteins which control immune tolerance and inflammatory mechanisms have been noted to be associated with the clinical state of PE; however, it is not clear what mediates these changes and whether DNA methylation may play a role in controlling the maternal expression of these proteins.
Epigenetic phenomena, such as variable methylation, could explain certain differences in gene expression. An epigenetic trait is a stably inherited pheno-type resulting from changes in a chromosome without alterations in its DNA sequence. The most studied epigenetic modification is DNA methylation, which is the addition of a methyl group to the DNA molecule at the sites of cytosine-guanine residue combinations called CpG sites. DNA methylation may result in an altered structure which blocks transcription factors, resulting in gene silencing or downregulation (Citation1).
We hypothesize that altered DNA methylation patterns may contribute to the pathophysiology of PE by driving the differential expressions of maternal genes in preeclamptic compared to normal pregnancies. A review of the literature shows a paucity of studies in the area of DNA methylation changes and their possible roles in the manifestation of PE. What has been done, largely, has been directed at a few specific genes and/or in fetal-derived tissues (Citation2–9). To our knowledge, there have been no previously published reports describing genome-wide methylation patterns from maternal leukocyte DNA in cases of PE.
In this study, we sought to characterize genome-wide differential methylation patterns in maternal blood leukocytes in 14 normotensive and 14 preeclamptic pregnancies at the time of delivery. We analyzed the DNA methylation status of 27,578 CpG dinucleotides in 14,495 genes using the Illumina Human Methylation-27 Assay (Illumina, San Diego, CA, USA). In addition, we performed gene-network analysis using GeneGo Metacore software (Thomson Reuters, Philadelphia, PA, USA) to determine candidate pathways, disease states, and gene sets.
Methods
Ethical approval
This study was approved by the Mayo Clinic Institutional Review Board and informed consent was obtained prior to enrollment.
Sources for case and control DNA
Preeclampsia was defined as the presence of new onset hypertension (>140/90 mmHg), as documented by at least two readings 6 h apart, accompanied by proteinuria, as defined by a 24 h urine protein excretion >300 mg, or the equivalent based on either the protein/creatinine ratio or urine dipstick. Preeclamptic participants (n = 14) and selected normotensive controls (n = 14) were matched for age (age, ±5 years) and body mass index (BMI: ±5 kg/m2). The medical records were abstracted for data, such as gravidity (primigravid being defined as no prior pregnancies), comorbidities (such as chronic hypertension or chronic kidney disease), BMI, ethnicity, intrapartum magnesium exposure, blood pressure readings, proteinuria, and gestational age at the time of delivery. All control and preeclamptic participants were in their first pregnancies, were non-smokers, and were of European descent.
DNA extraction and processing
Blood was drawn within a 24 h window of delivery. Blood was collected in a 10-mL ethylenediaminetetraacetic acid (EDTA) tube, separated into a buffy coat, and stored at −80 °C until processed. Genomic DNA was extracted from the thawed buffy coat using the QIAgen Flexigene Reagent kit (QIAgen, Germantown, MD, USA), purified using the AutoGenFlex DNA purification kit (AutoGen, Holliston, MA, USA), quantified with NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA), normalized with standard PicoGreen methodology (Thermo Fisher Scientific, Wilmington, DE, USA), and plated in 1000 ng aliquots. Bisulfite modification was performed using the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA).
Methylation assay
This article focuses on 28 samples plus technical replicates that were collected at the time of delivery. Plate maps were generated to determine the random location for each sample on the plate, as well as the samples that were run in duplicate. All samples were run in a single batch. We used the Illumina Human Methylation-27 Assay – a platform utilizing bead chip technology to evaluate the methylation status of over 27,578 CpG sites in 14,495 genes.
Quality control
The raw data were processed using the BeadArray Reader from Illumina GenomeStudio (version 2010.2, Illumina, San Diego, CA, USA), with methylation module (version 1.7). Next, a series of quality control metrics were applied. Quality assessment of the array was conducted using the “Control Dashboard” in the software package, which includes a graphical inspection of the eight types of embedded control probes: staining, hybridization, target removal, extension, bisulfite conversion, guanine/thymine (G/T) mismatch, negative control, and non-polymorphic controls.
Overall sample performance was determined by the total number of detected CpGs, the average detection p value across all CpG sites, and the distribution of average fi value for all CpGs. Call rates for each CpG site and sample were determined. Methylation sites and samples were excluded if the unreliable call rate (detection p value >0.05) was greater than 5%. Technical replicate repro-ducibility was estimated by the Pearson correlation coefficient.
All samples were modified, plated, and run concurrently to avoid batch effect. However, BeadChips (Illumina, San Diego, CA, USA), even when processed at the same time, can have variations in assay integrity leading to chip effect. To assess for such effects, data were examined using principal components analysis and unsupervised hierarchical clustering (Citation10–12).
Data analysis
A mean β value between 0 and 1 was generated for each CpG site, representing the average ratio of methylated cytosine residues to the total number of cytosine residues at that site for each sample.
Data were analyzed as quantitative variables. The Student group t-test was used to compare the mean methylation level at each CpG site between the PE versus normotensive pregnant groups. All potential confounders were considered in developing the study design.
All sites with an absolute mean methylation difference of >2% and p < 0.05 were used to characterize global methylation patterns, and the GeneGo Metacore software was used to determine candidate pathways, disease states, and gene sets (Citation13).
Results
Characteristics of cases and controls
By design, the mean values did not differ significantly between preeclamptic and normotensive pregnant control participants in age; BMI, tobacco use, comorbidities, or gravidity. The only exception was a significantly higher gestational age at delivery in the normotensive controls than in the preeclamptic women (mean = 40 versus 36.6 weeks, p < 0.0001). Blood pressures were different reflecting case control status, that is, higher blood pressures, both systolic and diastolic, in women with preeclamptic compared to normotensive pregnancies Magnesium prophylaxis was used in 9 of 14 PE cases who were considered at risk for eclamptic seizures; none of the controls were exposed to magnesium ().
Table 1. Demographics of preeclamptic cases and normotensive controls.
Quality control
Of the 28 samples, three were randomly chosen to be run in duplicate as technical replicates. The technical replicates were highly reproducible, as estimated by a Pearson correlation coefficient (r2 > 0.994). All samples were randomly allocated to one of the eight BeadChips, no two technical replicates randomized to the same BeadChip, and all chips were processed at the same time. Finally, whole genome amplification (WGA) DNA was used as a negative external control, was also run in duplicate, and performed as expected.
Review of the eight types of embedded control probes such as staining, hybridization, target removal, extension, bisulfite conversion, G/T mismatch, negative control, and non-polymorphic controls, demonstrated that the overall assay performance was as expected. Overall sample quality was determined by the total number of detected CpGs at a detection p value <0.05, the average detection p value across all CpG sites, and the distribution of the average β for all CpGs. CpGs that were not reliably detected in 95% samples based on detection p values (n = 475), or samples that did not have >95% reliable calls for all CpG sites (n = 0), were excluded. All 28 patient samples and 98%, or 27,103 CpG sites, met quality control standards and were included for analysis.
All samples were run concurrently, thus eliminating issues of batch effect. However, differences related to chip effect (first chip versus last chip) or placement on the chip, for example, left versus right can exist. All samples were randomly allocated to eight BeadChips. The initial principal components analysis of our entire cohort did not detect any significant chip effect. As normalization/batch correction made a negligible difference in the results, the original data were presented in this article (Citation11).
Differentially methylated CpG sites genome-wide
Our first analytic approach was from a genome-wide perspective. All βs with mean methylation differences >2% and p < 0.05 were included, for a total number of CpG sites of 997. Of note, these differentially methylated genes are located on all 22 autosomes, and the X chromosome as well, demonstrating a true genome-wide distribution. (List as online addendum)
Seven hundred twenty-nine genes were hypermethylated in PE, as compared with normotensive controls. The set of CpG sites that were more methylated in PE had an absolute methylation difference of up to 12%.
Two hundred sixty-eight genes were hypomethylated in PE, as compared with normotensive controls. This set of CpG sites that were less methylated in PE had an absolute methylation percentile difference of up to 20%.
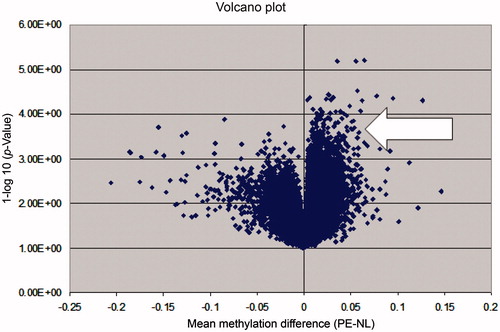
Overall, PE appears to be a hypermethylated state when compared with normotensive pregnancy. This is represented graphically in the Volcano plot in .
Figure 1. A volcano plot illustrating the global methylation differences between preeclamptic and normotensive samples. Each dot represents a comparison of mean methylation at an individual CpG site. The x-axis is the methylation mean difference (preeclampsia-normotensive). The y-axis is the negative log10 of the p value. Note the large number of significant differentially hypermethylated sites in preeclamptic pregnancy (arrow).
Pathway analysis
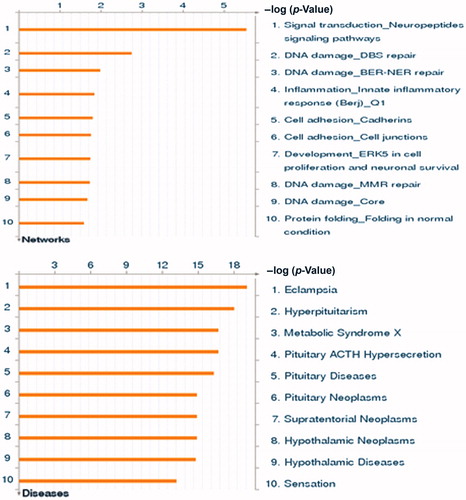
In putting all genes with statistically significant differences in methylation between preeclamptic and normotensive pregnancies into the GeneGo Metacore program identified the cellular and molecular process most likely to be involved as signal transduction involving neuropeptide signaling (p < 10−5). Other pathways identified were as follows: macrophage differentiation and vascular tone (p < 10−3). Using the same gene set analysis, the best-matched disease state was identified as eclampsia (p < 9.97 × 10−20) ().
Figure 2. Pathway analysis of most highly differentially methylated genes. The GeneGo program compared the genes that were differentially methylated (defined as >2% different and p < 0.05) in our data with >500 diseases and >100 cellular and molecular processes to generate a list of best matched pathways without a prior knowledge of the phenotype. The best matched process was the neuropeptide signaling pathway (p < 10−5). The best matched disease was eclampsia (p < 9.97 × 10−20). See http://www.genego.com
Differential methylation identifies potential candidate genes
There are 19 CpG sites in 18 genes that have statistically significant (p < 0.001) differences in methylation in maternal leukocyte DNA, when measured at the time of delivery in normotensive and preeclamptic pregnancies. Of these 18 genes, four genes are highly expressed in the brain and associated with certain neurologic diseases, including seizure disorders. GRIN2b is 6% more methylated in PE (p = 0.00006). PCDHB7 is 6% more methylated in PE (p = 0.00007). BEX1 is 3% more methylated in PE (p = 0.0003). GABRA1 is 5% more methylated in PE (p = 0.00066; see and ). Differential methylation measured at the time of delivery did not appear to be associated with peripartum exposure to magnesium (data not shown).
Table 2. Differentially methylated genes in maternal leukocyte DNA from preeclampsia cases compared with normotensive controls at delivery.
Table 3. Four genes differentially methylated in preeclampsia cases compared with normotensive controls at the time of delivery.
Discussion
Our study indicates that genome-wide methylation profiles in maternal leukocyte DNA at the time of delivery are more methylated in PE cases than in matched normotensive controls. The presence of a methyl group at key CpG sites can lead to structural changes in the DNA molecule, which block gene transcription. Increased methylation at promoter sites in maternal DNA in PE is consistent with the idea that many genes may be either turned off or down-regulated compared to normal pregnancy. Our previous work has identified normal pregnancy as a hypomethylated state compared with non-pregnancy. It is possible that the relative hypermethylation, we see in this study, is actually a state of failed hypomethylation that accompanies normal maternal adaptation to pregnancy (Citation14).
Many factors can influence global DNA methylation – both in the establishment of methylation, and also in the maintenance of methylation over time. Putative factors investigated to date have included age, ethnicity, nutrition, chemical exposures, and tobacco (Citation1). As our sample size was too small to stratify by these factors, we controlled for potential confounding factors by matching the two groups of women for age (within 5 years), tobacco status (all non-smokers), BMI (within 5 kg/m2), and ethnicity. All women were free of major medical comorbidities.
It is unclear what level of differential methylation is clinically meaningful when defining “significantly different,” as this area of study in epigenetics is in its infancy. We chose an absolute change of 2% as the lower limit for this initial exploratory analysis, as our goal was not to exclude any statistically significant genes, accepting that some of these changes might not be clinically significant. This absolute change may represent a relative change in methylation at any given CpG, ranging from 2% to >100%. For example, a specific CpG site in the PCDHB7 gene is methylated 43% of the time in preeclamptic subjects, but only 38% of the time in normotensive pregnancy, representing a 5% absolute difference, but a 15% relative change in methylation.
The GeneGo Metacore program examined our differential methylation gene data and compared these biomarkers with over 500 diseases and 110 cellular and molecular processes to come up with likely matches. The program has no information about the patients from whom our data was derived. Even without a priori knowledge of our patient population, the best matched disease state identified was eclampsia (p < 9.97 × 10−20). The best matched cellular and molecular process was signal transduction in neuropeptide signaling pathways (p < 10−5).
Given the above results, we closely examined our top differentially methylated sites. When ranked by p value, an unexpected finding was that 4 of the top 18 differentially methylated genes are highly expressed in brain, and are associated with cortical excitability and neuronal connections. These genes, the related proteins, and a description of their basic functions are summarized in . The relationships between these genes and seizure disorders are particularly intriguing given the development of eclamptic seizures in a subset of preeclamptic patients, suggesting the possibility that altered methylation may be involved. Three of these genes, GABRA1. BEX1, and GRIN2B, have also been shown to be expressed in blood at low levels. Though their role in normal leukocyte physiology is not established, BEX1 and GRIN2B have putative roles in hematopoietic cancers (Citation15,Citation16).
There has been limited work by others addressing the methylation changes found in PE. Most have focused on specific genes (Citation2,Citation5–8) rather than a genome-wide approach (Citation3,Citation4). The changes in methylation were described in fetal-derived tissues, such as placenta, (Citation3–5,Citation8,Citation17–19) or free fetal DNA in maternal plasma, (Citation5,Citation7) but not in maternal leukocyte DNA.
Maternal leukocyte DNA from blood can be easily isolated from the buffy coat containing white blood cells. Neutrophils and lymphocytes produce circulating cytokines which promote immunotolerance of the antigenic challenge associated with the developing fetus, thus helping to establish and maintain pregnancy. Alterations in maternal immune system function have been associated with PE. In fact, the inflammatory actions mediated by leukocytes have been associated with the endothelial dysfunction seen in PE. If differential methylation proves predictive of or diagnostic for pathologic states such as PE, knowledge of these patterns in a readily accessible tissue, such as blood, will be essential.
We recognize that although some of these genes have been shown to be expressed in leukocytes, most are primarily expressed in neurons (Citation20). Blood cells interact with every tissue in the body, including leukocyte transmigration across the blood–brain barrier. Peripheral changes in the leukocyte epigenome could be part of the pathophysiology of disease or, alternatively, may mirror epigenetic changes of distant tissues, including the brain (Citation21,Citation22). A growing body of literature suggests that the presence of alterations in the peripheral blood epigenome or transcriptome may serve as a biomarker for non-hematologic diseases (Citation22). Given the inaccessibility of brain tissue for analysis, this concept of developing a marker from peripheral blood for central neurologic diseases or treatments has been widely explored. For example, microarray analysis has demonstrated unique patterns of differentially expressed genes in peripheral blood mononuclear cells associated with amyotrophic lateral sclerosis, migraines, and schizophrenia (Citation23,Citation24). Altered methylation of neuropsychiatric genes in DNA obtained from blood in patients with borderline personality disorder has been demonstrated (Citation25). Altered gamma-aminobutyric acid receptor expression in peripheral blood mononuclear cells in patients with schizophrenia after treatment with selective serotonin re-uptake inhibitors has also been demonstrated (Citation26).
Differential methylation analysis can be confounded by many factors including age, BMI, and race; we matched on the factors we could control (age and BMI), or excluded them as variables (homogeneous ethnicity in our study). The possibility still exists though that there is confounding by factors for which we were unable to completely control. For example, we attempted to match for gestational age but still there was almost a 4-week difference (cases 36.6 gestational weeks versus 40.0 in controls), which reflects widely accepted notion that delivery, which is the gold standard for treatment of PE, frequently results in prematurity. However, our longitudinal data for normal pregnancy suggest that methylation changes occur over months, not weeks, making this an unlikely explanation for the differential methylation (unpublished data).
Also the presence of cell-free DNA from the fetus could vary between cases and controls, however, this DNA is typically derived from the plasma fraction, not the buffy coat, and comprises only up to 6% of the total DNA measured (Citation27). As we utilized the cellular buffy coat, although some fetal DNA contamination was possible, it likely represented only a small fraction of the total DNA measured and, therefore, is unlikely to significantly alter our results.
The major limitation of our study is the relatively small number of women in each group. There are hundreds of genes differentially methylated at p < 0.05. The genes discussed above, with p values ranging from 0.0003 to 0.00006, did not remain significant after correction for multiple comparisons using the false discovery rate method. The false discovery rate, commonly used in genomics, produces a q value, which is the expected proportion of false discoveries among all results deemed significant. Type II error is inherent in pilot experiments involving high throughput platforms whose expense on a per sample basis limit the number of samples that can be assessed.
Given the Type 2 error associated with high throughput platforms, candidate genes identified by genome-wide association studies involving single nucleotide polymorphisms (SNPs) often require replication utilizing either an independent sample set or a different methodology. Although replication in epigenome-wide studies is not a requirement for data to be published, and (Citation28) to establish an association between these candidate genes and the preeclamptic phenotype, additional confirmatory studies are needed. Although the best mechanism for confirming an association is not clear, one possible approach is to repeat genome-wide analysis on a larger independent sample, utilization of pyrosequencing to confirm altered methylation in individual genes, or exploration of downstream transcriptional and translational implications via gene expression arrays such as quantitative real time reverse transcriptase polymerase chain reaction and protein product quantification (Citation29). However, the fact that the most pronounced methylation changes were observed in genes which were, in a blind fashion, matched subsequently to eclampsia, may represent indirect evidence of the validity of our findings. In addition, these genes are involved in biologically plausible neurologic pathways that are likely affected in both PE and eclampsia.
Another limitation includes the possible confounding effect by SNPs in the CpG sites, which may falsely implicate or obscure differential methylation patterns in individual genes. In future studies, we plan to use the latest version of the Illumina chip, the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA), which takes SNPs into account when analyzing output data.
Despite the small sample size and lack of replication, there is a high likelihood that some of our identified genes are in fact differentially methylated in PE. We hope to replicate the present findings in a larger, independent sample, and identify differentially methylated genes that are crucial to human reproduction –both normal and pathologic.
Conclusion
In summary, we have demonstrated a widespread trend toward hypermethyla-tion in maternal leukocyte DNA in preeclamptic compared to normotensive pregnancies at the time of delivery. Pathway analysis of these differentially methylated genes implicates the involvement of neuropeptide signaling and the eclampsia disease state. Several of the genes with the most pronounced methylation changes between preeclamptic and normotensive pregnancies produce proteins that have previously been implicated in seizure disorders, suggesting possible novel pathways in the pathogenesis of eclampsia. The study of genome-wide differential methylation patterns, in both normal pregnancy and in pregnancy disease states, may be a new and powerful technique to identify novel genes involved in these processes.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgments
We thank the Mayo Clinic Center for Individualized Medicine for access to the Biobank samples.
Funding
This work was funded by the Mayo Clinic Foundation: Mary Kathryn and Michael B. Panitch Career Development Award and K08HD051714 (Vesna D. Garovic) from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health.
Methylation data can be found at http://www.ncbi.nlm.nih.gov/geo/ using
GEO submission ID: GSE37722.
References
- Tost J. DNA methylation: an introduction to the biology and the disease-associated changes of a promising biomarker. Mol Biotechnol 2010;44:71–81
- Bourque DK, Avila L, Penaherrera M, et al. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 2010;31:197–202
- Yuen RK, Avila L, Penaherrera MS, et al. Human placental-specific epipolymorphism and its association with adverse pregnancy outcomes. PLoS One 2009;4:e7389
- Yuen RK, Penaherrera MS, von Dadelszen P, et al. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet 2010;18:1006–12
- Zhao A, Cheng Y, Li X, et al. Promoter hypomethylation of COMT in human placenta is not associated with the development of pre-eclampsia. Mol Hum Reprod 2011;17:199–206
- Chiu RW, Chim SS, Wong IH, et al. Hypermethylation of RASSF1A in human and rhesus placentas. Am J Pathol 2007;170:941–50
- Bellido ML, Radpour R, Lapaire O, et al. MALDI-TOF mass array analysis of RASSF1A and SERPINB5 methylation patterns in human placenta and plasma. Biol Reprod 2010;82:745–50
- Avila L, Yuen RK, Diego-Alvarez D, et al. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta 2010;31:1070–7
- Kulkarni A, Chavan-Gautam P, Mehendale S, et al. Global DNA methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA Cell Biol 2011;30:79–84
- Bell CG, Teschendorff AE, Rakyan VK, et al. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics 2010;3:33
- Sun Z, Chai HS, Wu Y, et al. Batch effect correction for genome-wide methylation data with Illumina Infinium platform. BMC Med Genomics 2011;4:84
- Teschendorff AE, Menon U, Gentry-Maharaj A, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 2010;20:440–6
- Available online at: www.genego.com [last accessed Jul 2010]
- White WM, Brost BC, Sun Z, et al. Normal early pregnancy: A transient state of epigenetic change favoring hypomethylation. Epigenetics 2012;7:729–34
- Quentmeier H, Tonelli R, Geffers R, et al. Expression of BEX1 in acute myeloid leukemia with MLL rearrangements. Leukemia 2005;19:1488–9
- Pike BL, Greiner TC, Wang X, et al. DNA methylation profiles in diffuse large B-cell lymphoma and their relationship to gene expression status. Leukemia 2008;22:1035–43
- Bouchard L, Thibault S, Guay SP, et al. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010;33:2436–41
- Katari S, Turan N, Bibikova M, et al. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet 2009;18:3769–78
- Muller HM, Ivarsson L, Schrocksnadel H, et al. DNA methylation changes in sera of women in early pregnancy are similar to those in advanced breast cancer patients. Clin Chem 2004;50:1065–8
- Dionisio L, Jose De Rosa M, Bouzat C, Esandi Mdel C. An intrinsic GABAergic system in human lymphocytes. Neuropharmacology 2011;60:513–19
- Nicholson AC, Unger ER, Mangalathu R, et al. Exploration of neuroendo-crine and immune gene expression in peripheral blood mononuclear cells. Brain Res Mol Brain Res 2004;129:193–7
- Mohr S, Liew CC. The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends Mol Med 2007;13:422–32
- Mougeot JL, Li Z, Price AE, et al. Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med Genomics 2011;4:74
- Plummer PN, Colson NJ, Lewohl JM, et al. Significant differences in gene expression of GABA receptors in peripheral blood leukocytes of migraineurs. Gene 2011;490:32–6
- Dammann G, Teschler S, Haag T, et al. Increased DNA methylation of neuropsychiatric genes occurs in borderline personality disorder. Epigenetics 2011;6:1454–62
- Silver H, Susser E, Danovich L, et al. SSRI augmentation of antipsychotic alters expression of GABA(A) receptor and related genes in PMC of schizophrenia patients. Int J Neuropsychopharmacol 2011;14:573–84
- Bischoff FZ, Sinacori MK, Dang DD, et al. Cell-free fetal DNA and intact fetal cells in maternal blood circulation: Implications for first and second trimester non-invasive prenatal diagnosis. Hum Reprod 2002;8:493–500
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011;12:529–41
- Liu YJ, Papasian CJ, Liu JF, et al. Is replication the gold standard for validating genome-wide association findings? PLoS One 2008;3:e4037