
Abstract
Objective: To assess how routine clinical use of the Roche fully automated Elecsys® sFlt-1/PlGF test changes decision-making of physicians to hospitalize pregnant women with suspected preeclampsia. Methods: The Preeclampsia Open Study (PreOS) study is a multicenter, prospective, open-label, non-interventional study in 150 women showing signs and symptoms of preeclampsia (suspected preeclampsia). Physicians record their intended procedures before and after knowledge of participants’ sFlt-1/PlGF ratio. The study is conducted at five investigational sites in Germany and Austria. Conclusion: The PreOS study will provide evidence on how sFlt-1/PlGF ratio testing influences clinical decision-making in women with suspected preeclampsia in real-world clinical practice.
INTRODUCTION
The Preeclampsia Open Study (PreOS) is the first prospective, multicenter study to evaluate the clinical utility of the fully automated Elecsys® (Roche Diagnostics GmbH, Mannheim, Germany) sFlt-1/PlGF test in the diagnosis of preeclampsia in pregnant women with signs and symptoms of preeclampsia and their subsequent clinical management. Early detection of preeclampsia, enabling monitoring and supportive care, is beneficial to both mother and fetus. However, the accuracy and prognostic performance of the current “gold standard” diagnosis, relying on blood pressure and urinary protein measurement, are relatively poor (Citation1,Citation2). Therefore, there is a high-unmet medical need for reliable confirmation or exclusion of diagnosis and short-term prediction of preeclampsia in pregnant women with suspected preeclampsia/eclampsia/HELLP syndrome (hemolysis, elevated liver enzymes and low-platelet count).
Preeclampsia is a serious complication of pregnancy characterized by the new onset of hypertension and proteinuria after 20 weeks of gestation (Citation3,Citation4) and is a major cause of maternal, fetal and neonatal morbidity and mortality, affecting 2–8% of pregnancies (Citation5), and accounting for 50 000 maternal deaths per year, worldwide (Citation6). No therapeutic or preventive strategy other than induction of delivery is yet available. Accurate diagnosis and prediction of preeclampsia would have a significant clinical impact, allowing closer prenatal monitoring and more timely intervention to improve maternal and perinatal outcomes. In addition, improved diagnosis and prediction of preeclampsia could also reduce costs by minimizing the number of unnecessary hospitalizations and procedures due to uncertain diagnosis (Citation7,Citation8).
In recent years, increased understanding of the pathogenesis of preeclampsia has led to the identification of several biomarkers that could be used in the diagnosis and/or prediction of the disease. In particular, it has been shown that increased levels of the anti-angiogenic factor, soluble fms-like tyrosine kinase 1 (sFlt-1) and reduced levels of pro-angiogenic placental growth factor (PlGF) in maternal serum are associated with preeclampsia (Citation9,Citation10). The ratio between serum levels of sFlt-1 and PlGF has also been shown to be elevated in pregnant women prior to the clinical onset of preeclampsia and in women diagnosed with preeclampsia (Citation11–14). The sFlt-1/PlGF ratio is considered to be a better predictor of preeclampsia than either sFlt-1 or PlGF alone since it reflects changes in the pathophysiological dynamics of both biomarkers (Citation11). Moreover, the sFlt-1/PlGF ratio has been shown to be superior to the established clinical features such as blood pressure and uric acid in the prediction of adverse pregnancy outcome (Citation15). The Elecsys® sFlt-1 and Elecsys® PlGF assays (Roche Diagnostics GmbH, Mannheim, Germany) are the first fully automated immunoassays in maternal serum used as an aid in the diagnosis of preeclampsia. The assays provide convenient, rapid and reliable quantification of sFlt-1 and PlGF and are currently CE-IVD (Conformité Européenne-in vitro diagnostics) approved for use as an aid in the diagnosis of preeclampsia in conjunction with other clinical findings (Citation12,Citation16–18). An investigation into the routine clinical management of pregnant women with suspected preeclampsia demonstrated that an sFlt-1/PlGF ratio of ≥85 may be useful to confirm a diagnosis of hypertensive pregnancy disorder, whereas a ratio of ≤33 was useful to rule out a diagnosis of preeclampsia (Citation13).
Although the sFlt-1/PlGF ratio is a useful aid in the diagnosis of hypertensive disorders of pregnancy, the clinical utility of the Roche fully automated Elecsys® sFlt-1/PlGF test is not yet generally established. The aim of the PreOS study is to determine the influence of the sFlt-1/PlGF ratio on clinical decision-making in the management of pregnant women with suspected preeclampsia.
METHODS
Study Objectives
The primary study objective is to assess the influence of the sFlt-1/PlGF ratio on the decision-making of physicians to hospitalize pregnant women with suspected preeclampsia. Hospitalization was chosen as the primary objective as this is the key consideration of physicians assessing women who present with signs and symptoms of preeclampsia. Although most women presenting with suspected preeclampsia do not develop preeclampsia, hospitalization is commonly employed as a safety measure until preeclampsia can be ruled out. The decision whether or not to admit a woman with suspected preeclampsia to hospital has important implications not only for the woman herself but also in terms of costs for the healthcare system (Citation19).
The secondary study objectives are exploratory and aim to assess the influence of the sFlt-1/PlGF ratio on the decision-making of physicians to induce delivery, to request further diagnostic and therapeutic procedures, to predict adverse outcomes of preeclampsia, eclampsia and HELLP syndrome, and to predict other preeclampsia-related adverse outcomes of mother, fetus and neonate. In addition, analyses will include assessments of blood pressure, proteinuria and laboratory parameters, ultrasonography/Doppler sonography data, and other incidents and indirect harms.
Study Design
PreOS is a multicenter, prospective, open, non-interventional study. It is planned to recruit 150 participants with suspicion of preeclampsia from gestational week 24 + 0 days until delivery. Investigators use a predefined set of diagnostic criteria to identify women with suspected preeclampsia (outlined in ). The study protocol does not stipulate any procedures to be followed, allowing treating investigators complete freedom in their clinical decisions. The study design has been reviewed and approved by the competent local Institutional Review Boards (IRBs) of all five investigational sites. One of the key selection criteria for the study sites was the routine clinical use of the sFlt-1/PlGF ratio as an aid in the diagnosis of preeclampsia.
Table 1. Criteria contributing to suspicion of clinical diagnosis of preeclampsia; the presence of at least one of these clinical criteria is required for inclusion in the study.
Study Participants
Inclusion Criteria
Pregnant women presenting with suspected preeclampsia are eligible for the study if they are 18 years or over at gestational week 24 + 0 days until delivery and cannot be diagnosed with preeclampsia or pregnancy-related hypertensive disorder using the diagnostic criteria for preeclampsia as defined in the study protocol (). To preserve the non-interventional nature of the study, only participants for whom determination of the sFlt-1/PlGF ratio is planned, but not yet carried out, are included. Written, signed, informed consent is required to participate in the study.
Table 2. Definitions of preeclampsia-associated conditions and maternal and fetal outcomes.
Exclusion Criteria
Women with manifest preeclampsia, eclampsia or HELLP syndrome, and women with proteinuria ≥2+ by dipstick urinalysis who are receiving anti-hypertensive treatment are excluded from the study. Women in whom sFlt-1 and/or PlGF levels have already been assessed in their current pregnancy, or who have received any investigational medicinal product (i.e. have participated in an interventional trial of a drug product) within the last 3 months (90 days), are not eligible. Concomitant participation in another clinical study, employment at one of the investigational sites, or relationship (relative or spouse) with a study investigator also excludes women from participation in the PreOS study.
Definition of Diagnostic Criteria for Preeclampsia
In the PreOS study, preeclampsia is defined as the concomitant occurrence of proteinuria ≥2+ by dipstick urinalysis and elevated blood pressure (≥140 mmHg systolic and/or ≥90 mmHg diastolic, reproducible on two occasions). defines the criteria for suspicion of preeclampsia as applied for inclusion of women in the study. Suspicion of preeclampsia includes one of the following: new onset of elevated blood pressure, new onset of hypertension, aggravation of pre-existing hypertension, new onset of protein in urine, new onset of proteinuria, aggravation of pre-existing proteinuria, or one or more other reason for clinical suspicion of preeclampsia.
Definitions of Diagnostic Criteria for Preeclampsia-Associated Conditions and Maternal and Fetal Outcomes
As there is still a lack of consensus on classification and diagnosis of hypertensive disorders in pregnancy, investigators will use a pre-defined set of established criteria to identify preeclampsia-associated conditions and maternal and fetal outcomes (; 20–25).
Study Procedures
Assessment Schedule
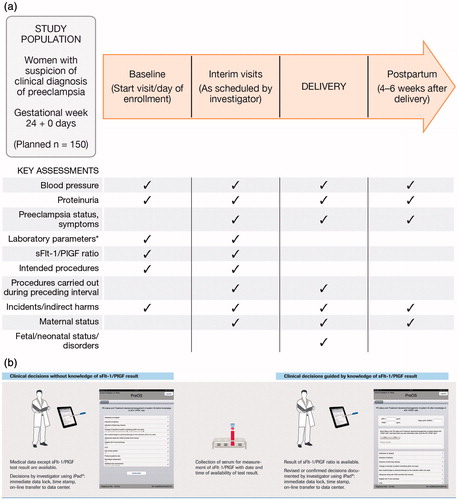
The schedule of assessments defines a baseline visit, interim visits (defined as any occasion after baseline where sFlt-1/PlGF is determined), a delivery visit and a postpartum visit 4–6 weeks after delivery (). Maternal sFlt-1/PlGF determination, intended procedures, body weight measurement, laboratory parameters and sonographic/Doppler data are performed at baseline and at interim visits. Intended procedures carried out in the interval preceding a visit are documented including all clinical assessments, the maternal and neonatal status, and incidents and indirect harms. Details of pre-specified drug classes (anti-hypertensive drugs, acetylsalicylic acid, low-molecular-weight heparin, anti-convulsive drugs, corticosteroids for induction of fetal lung maturation, corticosteroids for therapy of HELLP syndrome) are documented. In addition, demographic data, medical history and history of preeclampsia-related conditions and outcomes in previous pregnancies, and routine pregnancy assessments, are documented at the baseline visit. Blood pressure and protein in urine are measured at each visit. At all visits except at baseline, the preeclampsia status, the date of diagnosis and the diagnosis [e.g. no preeclampsia, suspected preeclampsia (except at postpartum assessment), preeclampsia, severe preeclampsia, superimposed preeclampsia, eclampsia, HELLP syndrome] are recorded. In addition, data on neurologic symptoms (e.g. headache, visual disturbances), epigastric pain, severe edema and oliguria are documented at each visit.
Figure 1. (a) Study design and key assessments. (b) Study flow chart: influence of sFlt-1/PlGF test result on clinical decision-making. Study design (a) and data collection overview (b). *Laboratory parameters tested include hematocrit, thrombocyte counts and serum levels of aspartate aminotransferase, lactate dehydrogenase, bilirubin (indirect), uric acid, haptoglobin and creatinine.
Sample Collection, Processing and Measurement
Sample collection and processing are carried out according to the instructions in the package insert of the fully automated Roche Elecsys® sFlt-1 and Roche Elecsys® PlGF immunoassays. Measurements of sFlt-1 and PlGF are performed at a designated laboratory as previously described (Citation12,Citation16,Citation17) and as outlined in the package insert (Citation26,Citation27). To avoid any potential bias, Elecsys sFlt-1 and Elecsys PlGF tests were purchased by the respective study sites and were not provided free of charge.
Documentation of Physicians’ Decision-Making via iPad™ App
An iPad™ Application (App) containing a questionnaire on treatment decisions is used to document the investigator’s decision-making on intended procedures for patient management at baseline (start visit/day of enrollment) and at any interim visit(s) between baseline and delivery (). This documentation method was chosen to allow results to be recorded in real time, in a consistent manner between investigators and study sites. Information entered into the App is immediately “locked”, time-stamped and transferred online to the data center (the electronic case report form, eCRF). Clinical decisions made without knowledge of the sFlt-1/PlGF test result, but based on all other available medical data, are first entered into the App by the investigator. Maternal serum is then collected for measurement of the sFlt-1/PlGF result, and the time and date of availability of the test result recorded. Following availability of the sFlt-1/PlGF test result, clinical decisions guided by knowledge of the sFlt-1/PlGF ratio and revised or confirmed decisions are also documented by the investigator via the App.
Intended procedures outlined in the App questionnaire include: admission to hospital (the investigator inputs an answer of yes or no into the questionnaire); induction of delivery; induction of fetal lung maturity; time interval to next visit; change in intensity of patient monitoring (intervals of blood pressure measurement; blood pressure home monitoring); assessment of protein in urine; new onset or changes in (pharmaco-)therapy for the indication; ultrasound study [for intrauterine growth restriction (IUGR), amniotic fluid volume] and/or uterine Doppler [for pulsatility index (PI), notching]; cardiotocography (CTG); 24-h urinary protein; protein/creatinine ratio; additional laboratory-parameter assessments [for example, hematocrit, thrombocytes, aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), bilirubin (indirect), uric acid, serum creatinine and haptoglobin]; neurologic assessment.
Electronic Data Capture
Clinical trial data are entered into a web-based electronic data capturing (EDC) system using eCRF version 2.2 and proprietary software provided by Amedon GmbH (Lübeck, Germany) in cooperation with IST GmbH (Mannheim, Germany). The eCRF is linked to a validated database, which is compliant with all applicable privacy and legal requirements and regulations. eCRF data (but not those collected via the iPad™ App) are reviewed by the data management team at IST GmbH and queried if considered inconsistent. The eCRF also provides an interface for capturing of the data collected by use of the dedicated iPad™ App documenting the investigator’s decision-making on intended procedures. The decisions entered by the investigator are transferred to the eCRF as soon as the iPad™ is connected to the internet.
Setting/Locations
The study started in July 2012 and is being conducted at five hospitals (four in Germany and one in Austria) that have implemented sFlt-1/PlGF testing in routine clinical practice as an aid in the diagnosis of preeclampsia.
Primary Endpoint
The primary endpoint is defined as the difference between the proportions of appropriate decisions of hospitalization/no hospitalization before and after the investigator learns the sFlt-1/PlGF ratio result during the baseline visit, which is equivalent to the proportion of appropriate changes of decisions of hospitalization/no hospitalization minus the proportion of inappropriate changes. An adjudication committee will determine whether any of the changed decisions were or were not appropriate.
Secondary Endpoints
The secondary endpoints of the study are as follows:
As in the case of the primary endpoint, but limited to the decision “no hospitalization” after learning the sFlt-1/PlGF ratio.
As in the case of the primary endpoint, but limited to the decision “hospitalization” after learning the sFlt-1/PlGF ratio.
As in the case of the primary endpoint, but with hospitalization/no hospitalization replaced with induction/no induction of delivery.
As in the case of the primary endpoint, but extended to decisions on intended procedures of any kind.
As in the case of the primary endpoint, but extended to all visits with sFlt-1/PlGF ratio determination.
As in the case of the secondary endpoint no. 1, but extended to all visits with sFlt-1/PlGF ratio determination.
As in the case of the secondary endpoint no. 2, but extended to all visits with sFlt-1/PlGF ratio determination.
As in the case of the secondary endpoint no. 3, but extended to all visits with sFlt-1/PlGF ratio determination.
As in the case of the secondary endpoint no. 4, but extended to all visits with sFlt-1/PlGF ratio determination.
Value of the sFlt-1/PlGF ratio in predicting preeclampsia/eclampsia/HELLP syndrome within the further course of observation.
Value of the sFlt-1/PlGF ratio in predicting preeclampsia-related adverse outcomes other than preeclampsia/eclampsia/HELLP syndrome of mother and fetus within the further course of observation.
Occurrence of maternal death, pulmonary edema, acute renal failure, cerebral hemorrhage, cerebral thrombosis, disseminated intravascular coagulation (DIC) and hepatic rupture.
Frequency of neurologic symptoms (headache, visual disturbances, vomiting), epigastric pain, severe edema, oliguria.
Occurrence of perinatal/fetal death, IUGR, small for gestational age (SGA), placental abruption.
Occurrence of neonatal respiratory distress syndrome, necrotizing enterocolitis, intraventricular hemorrhage.
Course of systolic, diastolic blood pressure.
Occurrence and course of proteinuria (protein dipstick, spot urine protein, spot urine protein/creatinine ratio, protein in 24-h urine).
Preeclampsia status.
Course of laboratory parameters: hematocrit, thrombocytes, AST, ALT, LDH, bilirubin (indirect), uric acid, serum creatinine, haptoglobin.
Date and mode of delivery, delivery <37 weeks, data of the newborn: weight, length, Apgar score.
Ultrasonography/Doppler sonography data.
Adjudication Committee
An adjudication committee will be used to judge whether changes to investigators’ intended decisions were appropriate or inappropriate. For example, a changed decision to not hospitalize a participant who goes on to develop preeclampsia shortly thereafter would be classified as an inappropriate change; whereas a changed decision to not hospitalize a study participant with suspicion of preeclampsia who does not then develop preeclampsia or HELLP syndrome would be classified as an appropriate change.
The adjudication committee comprises three independent experts who are not otherwise involved in the study, but who have experience in using the sFlt-1/PlGF ratio in clinical practice. To make their adjudication on whether changes in investigators’ decisions were appropriate, the adjudicators will review patient listings of all data available in the eCRF and collected by the iPad™ App, except for the sFlt-1/PlGF ratio results. They will take into consideration the outcomes in the individual participant and the temporal relationship between outcomes and the decision to change the intended procedures based on the sFlt-1/PlGF ratio. Each case in which intended procedures were changed will be adjudicated by two randomly-selected committee members. If their adjudication decisions are not identical, the case will also be adjudicated by the third member. The majority judgment (appropriate or inappropriate) will be used for the statistical analysis for each change in an intended decision.
Safety Assessment
As the study is strictly non-interventional, adverse events will not be documented. Incidents and indirect harms will be determined by the investigator during participants’ visits and will be documented as per MEDDEV 2.12.–1 rev. 6 guidance (Citation28) and local regulations and guidelines. Incidents are defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or have led to the death of a participant, or of a user or of other persons, or to a serious deterioration in their state of health. Indirect harm may occur as a consequence of the medical decision, action taken or not taken on the basis of information or result(s) provided by the device. Examples of indirect harm include misdiagnosis, delayed diagnosis, delayed treatment or inappropriate treatment.
All incidents and indirect harms are to be recorded by each investigation site via the VKN-Form (incidents and harms) within the eCRF. A description of the event, including the start date, resolution date, action taken, and the outcome are to be provided. Investigators are encouraged to report any occurrence where an incident or indirect harm may have occurred, even if information is incomplete.
Sample Size and Statistical Analysis
Sample Size Calculation
The sample size is based on the primary endpoint. In total, 143 evaluable participants were considered necessary to detect with 90% power and a two-sided McNemar test (alpha = 0.05) with a difference of 15% between the proportion of appropriate changes (P10) and the proportion of inappropriate changes (P01), assuming that the percentage of discordant decisions (P10 + P01) is 30% in the analysis population. Assuming that not all participants will be evaluable, at least 150 participants were to be included into the study.
Analysis Populations
The primary analysis regarding the primary endpoint and secondary endpoints 1–9 will be performed in the per-protocol population, i.e. in all participants fulfilling the following requirements: (i) eligible according to all inclusion criteria and most relevant exclusion criteria, (ii) the sFlt-1/PlGF ratio result is available, (iii) data on decision-making by the investigator before and after the sFlt-1/PlGF test result is recorded with the iPad™, and (iv) adjudication by an independent expert committee is completed. The remaining secondary endpoints will be analyzed for the full analysis population, i.e. in all participants fulfilling the criteria (i) and (ii), mentioned above.
The safety analysis will include a record of incidents and indirect harms to participants, rather than a collection of predefined “adverse events”. The safety population is defined as all participants with signed informed consent and in whom at least one blood sample was taken to determine the sFlt-1/PlGF ratio.
General Methods
The main focus of the study is to analyze endpoints recorded before and after the measurement of the sFlt-1/PlGF ratio at the baseline visit. Similar data recorded at the interim visits (see endpoints 5–9) will only be analyzed if paired investigator decisions are available for at least 10% of the participants. All study data will be summarized using appropriate descriptive statistical methods (e.g. appropriate frequency tables for nominal and ordinal data) and standard statistical parameters (mean, standard deviation, standard error of the mean, median, interquartile ranges, minimum, maximum) for continuous variables.
Analysis of the Primary Study Objective
The statistical null hypothesis to be tested is:
with the following proportions: P10 = percentage of appropriate changes in investigator decision regarding hospitalization from prior to measurement of the sFlt-1/PlGF ratio to the decision regarding hospitalization after the measurement. P01 = percentage of inappropriate changes in investigator decision regarding hospitalization from prior to measurement of the sFlt-1/PlGF ratio to the decision regarding hospitalization after the measurement. The difference P10 − P01 is identical to the difference Ppost − Pprior, where Ppost is the proportion of appropriate decisions after the measurements of the sFlt-1/PlGF ratio and Pprior is the proportion of appropriate decisions prior to the measurement.
The proportion of appropriate and inappropriate changes will be compared using a two-sided McNemar test at a significance level of 0.05. In addition, 95% confidence intervals will be determined for odds ratio P10/P01.
Multiple logistic regression analysis will be performed to investigate the impact of various explanatory factors on the endpoint “investigator decision regarding hospitalization after measurement of the sFlt-1/PlGF ratio at the baseline visit”. In addition to various disease characteristics, the decision of the investigator prior to the measurement and the sFlt-1/PlGF ratio will be considered as covariates in the models. A stepwise selection procedure will be applied to identify the most relevant and independent predictors for the investigator decision after measurement of the sFlt-1/PlGF ratio.
Analysis of Secondary Study Objectives
The secondary study objectives are exploratory. The secondary endpoints based on paired investigator decisions before and after measurement of the sFlt-1/PlGF ratio at the baseline visit will be analyzed with the same statistical method as specified for the primary endpoint.
Ethics Statement
Each participating study site provided Ethics Committee/Institutional Review Board approval of the study protocol and associated documents (participant informed consent, participant information) before the start of the clinical part of the study.
Conclusion
There is a high-unmet need for the reliable prediction of the development of preeclampsia and the risk of related maternal and fetal adverse outcomes in women with a suspected but not yet fully established diagnosis. Improved diagnostic accuracy of the syndrome offers the potential to vastly improve pregnancy care. A test that can reliably exclude preeclampsia, for example, based on a low sFlt-1/PlGF ratio, would result in fewer hospitalizations for pregnant women with suspected preeclampsia, as well as a reduction in costs incurred, leaving more resources available for those women most in need.
The results of PreOS aim to provide evidence on the clinical value of sFlt-1/PlGF testing in indeterminate cases of suspected preeclampsia and may help to guide management of pregnant women with suspected preeclampsia with regard to hospitalization, delivery and other clinical procedures in real world clinical practice for improved maternal, fetal and neonatal outcomes.
Declaration of interest
MH, WV and MR are employed by Roche Diagnostics. DM and RvdD are employed by IST Studien Therapeutica GmbH. HS has received consultancy payments from Roche Diagnostics regarding his role as a Medical Advisor for the PROGNOSIS study. MH and RvdD designed the study with input from DM. MR is the study manager of PreOS. MH, WV, MR, DM, RvdD and HS contributed to the drafting of the initial manuscript. All authors contributed to critical revision of the manuscript and approved the final manuscript. Support for third-party writing assistance for this manuscript was provided by Roche Diagnostics GmbH. ELECSYS is a trademark of Roche. The study is funded by Roche Diagnostics GmbH, Sandhofer Straβe 116, 68305 Mannheim, Germany.
References
- Hagmann H, Thadhani R, Benzing T, et al. The promise of angiogenic markers for the early diagnosis and prediction of preeclampsia. Clin Chem 2012;58:837–45
- Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet 2010;376:631–44
- Lim K-H. Preeclampsia. Medscape Reference: Diseases & Conditions. Available from: http://emedicine.medscape.com/article/1476919-overview#aw2aab6b3 [last accessed 29 May 2014]
- Milne F, Redman C, Walker J, et al. The pre-eclampsia community guideline (PRECOG): how to screen for and detect onset of pre-eclampsia in the community. BMJ 2005;330:576–80
- Duley L. Pre-eclampsia and the hypertensive disorders of pregnancy. Br Med Bull 2003;67:161–76
- Duley L. Maternal mortality associated with hypertensive disorders of pregnancy in Africa, Asia, Latin America and the Caribbean. Br J Obstet Gynaecol 1992;99:547–53
- Hadker N, Garg S, Costanzo C, et al. Financial impact of a novel pre-eclampsia diagnostic test versus standard practice: a decision-analytic modeling analysis from a UK healthcare payer perspective. J Med Econ 2010;13:728–37
- Scazzocchio E, Figueras F. Contemporary prediction of preeclampsia. Curr Opin Obstet Gynecol 2011;23:65–71
- Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 2004;350:672–83
- Vatten LJ, Eskild A, Nilsen TI, et al. Changes in circulating level of angiogenic factors from the first to second trimester as predictors of preeclampsia. Am J Obstet Gynecol 2007;196:239.e1–6
- Levine RJ, Lam C, Qian C, et al. (for the CPEP Study Group). Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006;355:992–1005
- Verlohren S, Galindo A, Schlembach D, et al. An automated method for the determination of the sFlt-1/PlGF ratio in the assessment of preeclampsia. Am J Obstet Gynecol 2010;202:e1–11
- Verlohren S, Herraiz I, Lapaire O, et al. New gestational phase-specific cutoff values for the use of the soluble fms-like tyrosine kinase-1/placental growth factor ratio as a diagnostic test for preeclampsia. Hypertension 2014;63:346–52
- Villa PM, Hämäläinen E, Mäki A, et al. Vasoactive agents for the prediction of early- and late-onset preeclampsia in a high-risk cohort. BMC Pregnancy Childbirth 2013;13:110
- Rana S, Powe CE, Salahuddin S, et al. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation 2012;125:911–19
- Schiettecatte J, Russcher H, Anckaert E, et al. Multicenter evaluation of the first automated Elecsys sFlt-1 and PlGF assays in normal pregnancies and preeclampsia. Clin Biochem 2010;43:768–70
- Schneider E, Gleixner A, Hänel R, et al. Technical performance of the first fully automated assays for human soluble fms-like tyrosine kinase 1 and human placental growth factor. Z Geburtshilfe Neonatol 2009;213:A8. Available from: https://www.thieme-connect.com/ejournals/abstract/10.1055/s-0029-1216308 [last accessed November 2014]
- Verlohren S, Herraiz I, Lapaire O, et al. The sFlt-1/PlGF ratio in different types of hypertensive pregnancy disorders and its prognostic potential in preeclamptic patients. Am J Obstet Gynecol 2012;206:e1–8
- Hund M, Verhagen-Kamerbeek WDJ, Reim M. PreOS (Preeclampsia Open Study) – prospective, open, non-interventional study evaluating the influence of the angiogenic biomarkers sFlt-1 and PlGF on decision-making of physicians in pregnant women with suspicion of preeclampsia. Presented at the 18th World Congress on Controversies in Obstetrics, Gynecology and Infertility (COGI); 2013 Oct 24–27; Vienna, Austria
- ACOG Committee on Practice Bulletins – Obstetrics. ACOG Practice Bulletin: diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet Gynecol 2002;99:159–67
- Brown MA, Lindheimer MD, de Swiet M, et al. The classification and diagnosis of the hypertensive disorders of pregnancy: statement from the International Society for the Study of Hypertension in Pregnancy (ISSHP). Hypertens Pregnancy 2001;20:IX–XIV
- Magann EF, Martin NJ. Twelve steps to optimal management of HELLP syndrome. Clin Obstet Gynecol 1999;42:532–50
- Simhan HN, Caritis SN. Prevention of preterm delivery. N Engl J Med 2007;357:477–87
- Stepan H. Intrauterine Wachstumsretardierung. In: Wacker J, Bastert G, Sillem M, Beckmann MW, eds. Therapiehandbuch Gynäkologie und Geburtshilfe. Heidelberg: Springer Medizin; 2007:45–50
- Von Dadelszen P, Magee LA, Roberts JM. Subclassification of preeclampsia. Hypertens Pregnancy 2003;22:143–8
- Roche Diagnostics GmbH. Elecsys PlGF immunoassay. Package insert. 2013–08, V 5. Available from: http://www.rochecanada.com/fmfiles/re7234008/package_inserts/05144671190_PLGF_EN_PI_V5.pdf [last accessed October 2014]
- Roche Diagnostics GmbH. Elecsys sFlt-1 immunoassay. Package insert. 2013–08, V 5. Available from: http://www.rochecanada.com/fmfiles/re7234008/package_inserts/sFlt-1-05109523190-English-CAN-V5.pdf [last accessed October 2014]
- European Commission DG Enterprise and Industry Guidelines on a Medical Devices Vigilance System. MEDDEV 2.12.–1 rev. 6. 2009. Available from: http://ec.europa.eu/health/medical-devices/files/meddev/2_12_1-rev_6-12-2009_en.pdf [last accessed October 2014]