Abstract
It is well known that reactive oxygen species (ROS) attack several living tissues and increase the risk of development and progression of serious diseases. In neuronal level, ROS induce cell death in concentration-dependent fashion. However, little is known about the mechanisms of neuronal changes by ROS prior to induction of cell death. Here we found that treatment of cerebellar granule neurons (CGCs) with 0.5 μM hydrogen peroxide induced axonal injury, but not cell death. The number of dendrites remarkably decreased in hydrogen peroxide-treated CGCs, and extensive beading was observed on survival dendrites. In addition, an abnormal band of the original collapsin response mediator protein (CRMP)-2 was detected by Western blotting in hydrogen peroxide-treated CGCs. Treatment with each tocotrienol isoform prevented axonal and dendrite degeneration and induction of the abnormal band of the original band of CRMP-2 in hydrogen peroxide-treated CGCs. These results indicate that treatment with tocotrienols may therefore be neuroprotective in the presence of hydrogen peroxide by preventing changes to the CRMP-2 that occur before neuron death.
Keywords::
Introduction
It is well known that aging is an inevitable and degenerative process. In the process of aging, the cognitive functions of learning and memory gradually attenuate. However, the detailed mechanisms of age-related cognitive dysfunction are not well understood. Recently, several lines of evidence have indicated that age-related cognitive dysfunction is related to oxidative injury in brain. Reactive oxygen species (ROS)including superoxide and hydroxyl radicals, attack living tissues resulting in oxidative injury[Citation1]. The hypothesis that ROS is one actor in the progression of aging, the ‘free-radical theory of aging’, is well known [Citation2]. Using this theory as a start point, we previously investigated and reported the relationship between ROS-derived oxidative injury and cognitive dysfunction during aging using rodent models. We observed gradual, age-dependent degeneration in cognitive functions in several maze tasks [Citation3]. In addition, the cognitive performance scores of vitamin E-deficient young rats and ROS-exposed young rats were similar to those of normal (not vitamin E-deficient or ROS-exposed) older rats. We also found apoptosis and β-amyloid-like proteins in the CA1 region of the hippocampus in these three models [Citation4]. These results strongly indicate that age-related cognitive dysfunction is associated with ROS. However, it is difficult to identify possible treatments for or means of preventing cognitive dysfunction, as the dysfunction manifests only after the neurons have already died. It is necessary to find early events of neuronal change before the induction of cell death in ROS-exposed neurons.
In order to detect early changes in ROS-exposed neurons, we focused on axon and dendrite function, which plays an important role in neurotransmission. A large number of synapses exist on the distal ends of axons, and many substances, including neurotransmitters and other proteins, are carried from the cell body by the axonal transport system. Axonal dysfunction may therefore have serious effects on cognitive function even if cell death does not occur. Using electron microscopy, Urano et al. examined a large number of synaptic vesicles accumulated in synapses in brain slices from aged rats and ROS-exposed young rats [Citation5]. Touma et al. reported that neurite, which is characterized by expansion and contraction, degenerated in nerve-growth factors (NGF)-free medium [Citation6]. These findings indicate that axons are more sensitive than the cell body, because of its flexibility [Citation6,Citation7]. On the other hands, dendrites are receivers of neurotransmitters. Many kinds of receptors exist on the post-synaptic region. Thus, we focused first on axon and dendrite function.
If ROS induce axonal dysfunction, treatment with antioxidant substances may affect neurons. To clarify the possibility of preventing ROS-induced axonal dysfunction, we used tocotrienols. Tocotrienols, which include four different isoforms (α-, β-, γ-, and δ-tocotrienol), comprise a vitamin E family (). Vitamin E is one of the most common natural lipophilic vitamins. One of pivotal functions of vitamin E is as an antioxidant. Vitamin E plays a role in radical scavenger [Citation8–10]. It has been reported that palm and olive oils contain relatively large amount of vitamin E, and widely mixed in food products as an antioxidant substances [Citation9,Citation11]. Recently, several lines of evidence have suggested that vitamin E has neuroprotecitive effect [Citation12,Citation13]. Furthermore, the antioxidant or non-antioxidant function of tocotrienols has proven more effective than that of tocopherols [Citation14,Citation15]. However, the neuroprotective effects of tocotrienols at the cell biology level are not well understood.
Figure 1. Chemical structure of tocotrienols.
The purpose of this study was to determine whether treatment of primary cultures of mice cerebellar granule cells (CGCs) with a low concentration of hydrogen peroxide induces axonal degeneration and to determine the neuroprotective effects, if any, of α- and γ-tocotrienol in low concentration hydrogen peroxide-treated CGCs. We measured collapsin response mediator protein (CRMP)-2, which is part of a family of cytoplasmic proteins in the brain, with important roles in neurite polarity and axon guidance [Citation16–18]. Furthermore, we investigated the autophagyfunction of axonal regions in low concentration hydrogen peroxide-treated CGCs. Our data illustrate that ROS-derived axon and dendrite dysfunction is one of the early signs of ROS-induced cell degeneration. ROS-derived axon and dendrite degeneration, but not cell death, may be one of the processes underlying age-related cognitive dysfunction. Resulting from these investigations may help to prevent neurodegenerative diseases related to axonal dysfunction.
Materials and methods
Animals and reagents
The wild-type C57BL/6 mouse strains were obtained from Japan SLC, Inc. (Hamamatsu, Japan). The α- and γ-tocotrienols were kindly provided by Eisai Food & Chemical Co., Ltd (Tokyo, Japan). All other chemical agents were obtained from either Wako Pure Chemicals Industries, Ltd. (Osaka, Japan) or Sigma–Aldrich Corporation (St. Louis, MO, USA). All animal experiments were performed with the approval of the Animal Protection and Ethics Committee of the Shibaura Institute of Technology. All tissue culture plates and dishes were purchased from Becton Dickinson (Franklin Lakes, NJ, USA). All other reagents were purchased from Sigma–Aldrich.
Cell cultures
Primary cultures of CGCs were prepared from 7-day-old postnatal mice as described previously, with some modifications [Citation19]. Briefly, the cerebellum was dissected after decapitation, cleaned free of meninxes, and then treated with 0.125% (w/v) trypsin in minimum essential medium (MEM) for 10 min at 37°C. The reaction was terminated by adding feed medium consisting of MEM, 25 μg/mL streptomycin, 25 U/mL penicillin, and 5% heat-inactivated fetal calf serum (FCS)(Biological Industries, BeitHaemek, Israel) supplemented with K + to a final concentration of 25 mM. After trituration, the cells were suspended and plated onto 6- or 24-well dishes coated with 0.1% polyethyleneimine. This was designated as Day 0 (DIV0). On Day 1, cytosine-D-arabinofuranoside was added to a final concentration of 1 μM to prevent non-neuronal cell proliferation. CGCs were used in experiments on Day 3.
Analysis of lipid peroxidation
For analysis of lipid oxidation, we measured lipid hydroperoxides (LOOH) and thiobarbituric acid reactive substances (TBARS) after treatment of CGCs with hydrogen peroxide in the presence or absence of each tocotrienol isoform. LOOH were measured using chemiluminescence methods, as described previously with some modifications [Citation20]. One hundredmicroliters of cell lysate was extracted with a mixture of chloroform and methanol (2:1 v/v, 1 mL), and evaporated by N2 gas. The residue was dissolved by 200 μL of methanol, and mixed with 150 μL of a chemiluminescent solution (a mixture of 0.18 mg isoluminol/mL and 1 mg microperoxidase/mL of 70% methanol, 150:1, v/v). Chemiluminescence intensity was analyzed using a Luminometer (Luminescencer-PSN AB-2200, Atto Corp., Tokyo, Japan). Preparation of the standard curve for the analysis was carried out using cumenhydroperoxide as a standard for LOOH. Lipid peroxides were measured by Yagi's method, as previously described with some modifications [Citation21]. One hundredmicroliters of cell lysate was mixed with 200 μL of 5 mM EDTA solution, 2 mL of 1% phosphate acid solution, and 1 mL of 0.7% thiobarbituric acid solution. The mixture was heated in a block heater for 45 min at 100°C. After cooling in an ice box, the sample was then incubated with 2 mL of butanol for 3 min. After centrifugation, we isolated the upper layer and measured the absorbance of 535 nm using spectrophotometry (GeneQuant 100, GE Healthcare UK Ltd, Buckinghamshire, UK). Both oxidized markers were evaluated per mg protein in the samples and were performed at least five times. Data were analyzed using Student's t-test, with findings of p < 0.01 considered significant.
Transfection
CGCs were transfected using Lipofectamin 2000 (Life Technologies Japan Ltd, Tokyo Japan) using the manufacture's protocol. One day before transfection, medium was changed without antibiotics. A pEGFP-C1 mammalian expression vector (Clontech Takara Bio Inc., Tokyo, Japan) was added to 25 μL of opti-MEM I Life Technologies reduced serum medium. Lipofectamine 2000 was diluted with 25 μL of opti-MEM I reduced serum medium. After 5 min incubation, the diluted DNA was combined with diluted Lipofectamin 2000. After 15 min, the solution was added drop-wise to the dishes. After 3 h, the solution was added to the culture medium, and used in the experiment. The transfected cells were observed with a fluorescence microscope (IX81, Olympus, Tokyo, Japan). Transfection experiments were performed at least three times.
Axon and dendrite degeneration
Axon or dendrite degeneration was evaluated by monitoring morphological hallmarks of neurite degeneration such as beading and fragmentation, as described previously with some modifications [Citation19]. To evaluate the extent of axons, pEGFP-C1 vector was transfected to CGCs (see Materials and methods). After 18 h of transfection, CGCs were treated with 0.5 μM hydrogen peroxide in the presence or absence of 5 μM α- or γ-tocotrienol. Twenty-four hours after treatment with hydrogen peroxide, the cells were fixed with 4% paraformaldehyde (PFA)(Merck KGaA, Darmstadt, Germany) in PBS for 15 min at 4°C. Photomicrographs of the cells were taken on an Olympus IX81 phase-contrast microscope (Olympus) equipped with a DP72 digital camera (Olympus), stored and then processed on a personal computer. Axon and dendrite degeneration experiments were performed at least three times.
Western blotting
After 24 h of treatment with hydrogen peroxide, the samples were collected and used in Western blotting as described previously, with some modifications [Citation6]. The lysates were centrifuged, and protein content was determined using a Bio-Rad protein assay (#500-0006JA, Bio-Rad Japan, Tokyo, Japan) according to the manufacturer's procedure. Protein extracts (15 μg) were separated on 10% SDS-polyacrylamide gels and transferred to Immobilon transfer membranes (PVDF; Merck KGaA). The membranes were washed and then incubated in blocking solution [Tris-HCl-buffered saline, pH 7.6 (TBS), containing 0.1% Tween20 and 3% bovine serum albumin (BSA)] for 1 h at room temperature (R/T). The membranes were washed in TBS containing 0.1% Tween20, and then treated with anti-human CRMP-2 (C4G) mouse IgG monoclonal antibody (#11096, Immuno-Biological Laboratories Co., Ltd, Gunma, Japan) at 1:400 dilution overnight at 4°C. Anti-mouse IgG HRP antibody (Promega Corp., Madison, WI, USA) was used as a secondary antibody at 1:4000 dilution for 1 h at R/T.
In LC3 assay, the protein extracts (15 μg) were separated on 15% SDS–polyacrylamide gels. The transferred membranes were treated with anti- human LC3 (APG8B)(N-term) rabbit IgG polyclonal antibody (#AP1802a, Abgent, Inc., CA, USA) at 1:400 dilution overnight at 4°C. Anti-rabbit IgG HRP antibody (Promega) was used as a secondary antibody at 1:2000 dilution for 1 h at R/T. Both Western blotting experiments were performed at least three times. All chemiluminescent signals were generated by incubation with the detection reagents (ECL Prime Western Blotting Analysis Reagent; GE Healthcare) according to the manufacturer's procedure. For normalization of each band of CRMP-2 and LC3, the membranes were reprobed with anti-β actin antibodies (#ab8226, Abcam Plc., Cambridge, UK). The relative intensities of CRMP-2 and LC3 were determined using LAS-3000 (FUJIFILM Corp., Tokyo, Japan).
Immunocytochemic alanlysis
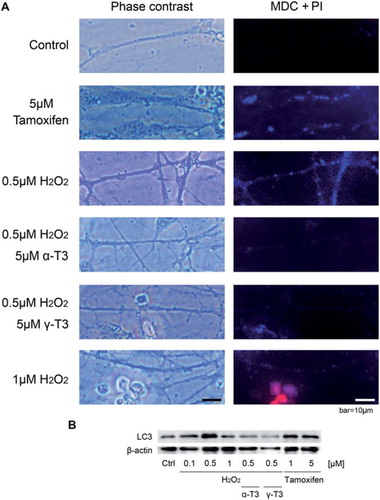
After 24 h of treatment with hydrogen peroxide, we detected autophagy using a Autophgy/Cytotoxicity Dual Staining Kit using the manufacturer's protocol (Cayman Chemical Company, Ann Arbor, MI, USA). For judgment of cell death, the samples were treated with propidium iodide (PI) solution at 1:1000 dilution for 2 min at R/T. After PI staining, the samples were treated with monodansylcadaverine (MDC) solution at 1:1000 dilution for 10 min at 37°C. Tamoxifen, a known inducer of autophagy vacuoles, was used as a positive control. The stained cells were observed with a fluorescence microscope. Immunocytochemical experiments were performed at least three times.
Results
Treatment with hydrogen peroxide induces lipid peroxidation in CGCs
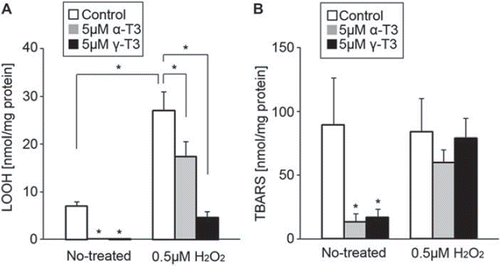
To determine the effect of a low concentration of hydrogen peroxide, we checked the lipid peroxidation levels in CGCs. LOOH and TBARS were used as indices of lipid peroxidation. After treatment with 0.5 μM hydrogen peroxide, the levels of LOOH significantly increased (). Treatment with each tocotrienol isoform significantly inhibited the levels of LOOH in hydrogen peroxide-treated CGCs. After treatment with hydrogen peroxide, TBARS did not significantly differ in the presence or absence of each tocotrienol isoform (). These results indicate that treatment with a low concentration of hydrogen peroxide induced lipid hydroperoxides in CGCs.
Figure 2. Analysis of lipid peroxidation. CGCs were treated with 0.5 μM hydrogen peroxide in the presence or absence of α-tocotrienol (5 μM) or γ-tocotrienol (5 μM). After 24 h, the cells were collected and measured to determine the index of lipid peroxidation (LOOH is shown in (A), TBARS is shown in (B)). Both oxidized markers were evaluated per mg protein in samples. Each column represents the mean of results of five independent experiments. Data were analyzed by Student's t-test, with findings of p < 0.01 considered significant.
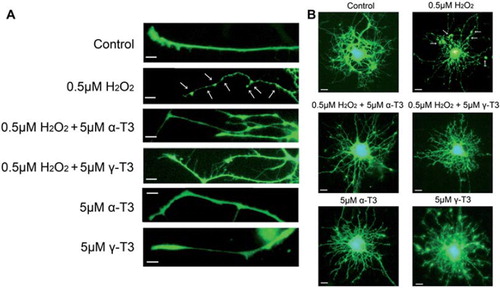
Appearance of axon and dendrite injury after treatment of pEGFP-transfected CGCs with hydrogen peroxide
Treatment with 0.5 μM hydrogen peroxide of CGCs induced abnormal morphology and marked axonal beading (). Fragmented nuclei were not found on Hoechst 33258 staining (data not shown). However, treatment with 5 μM α- or γ-tocotrienol significantly prevented axonal beading on hydrogen peroxide-treated CGCs. This finding indicates that treatment with 0.5 μM hydrogen peroxide induced axonal degeneration of CGCs, but did not inducecell death. After treatment with 0.5 μM hydrogen peroxide, the distal end of the axons appeared to be collapsed. Furthermore, we investigated morphological changes in the dendrites around the cell body. Large numbers of dendrite were observed in a spider-web formation around the cell body (). Whereas these dendrites were densely distributed around the cell bodies of normal CGCs, the number of dendrites was lower in hydrogen peroxide-treated CGCs. In surviving dendrites, extensive beading was observed. Treatment with each tocotrienol isoform prevented dendrite degeneration in the hydrogen peroxide-treated CGCs. These results indicate that treatment of CGCs with a low concentration of hydrogen peroxide induces axonal and dendritic degenerations, but not cell death.
Figure 3. Axon and dendrite degeneration were induced by treatment of EGFP-transfected CGCs with hydrogen peroxide. Each image was taken by fluorescence microscopy. After 18 h of transfection with EGFP, the CGCs were treated with 0.5 μM hydrogen peroxide in the presence or absence of α-tocotrienol (5 μM) or γ-tocotrienol (5 μM). After 24 h, the cells were fixed with 4% PFA in PBS. Photomicrographs of the cells were taken and analyzed in the regions of the axon (A) and the dendrite (B) on a personal computer. The scale bar is 10 μm. Arrows indicate beading on the degeneration of CGCs. At least, three wells were used per experiment. The method of transfection is described in the Materials and Methods.
Hydrogen peroxide induced CRMP-2 alteration on western blot analysis
On Western blotting, two original bands of CRMP-2 (64- and 72-kDa) were detected for a mouse brain homogenate sample on the data sheet of IBL Co., Ltd (http://www.ibl-japan.co.jp/eng/index.htm). We detected the two original bands of CRMP-2 in CGCs. However, the upper side of the original two bands of CRMP-2 was very weak in our experimental model. Treatment of CGCs with hydrogen peroxide enhanced an unusual band located below the two original bands of CRMP-2 in a concentration- dependent fashion (). Treatment with α- or γ-tocotrienol significantly inhibited the appearance of this unknown band of CRMP-2. These results indicate that tocotrienols protect axon and dendrite functions while preventing the induction of the unknown CRMP-2 in hydrogen peroxide-treated CGCs.
Figure 4. Denatured CRMP-2 proteins appeared after treatment of CGCs with hydrogen peroxide. CGCs after treatment with hydrogen peroxide in the presence or absence of α-tocotrienol (5 μM) or γ-tocotrienol (5 μM) were lysed and used for Western blotting analysis. The same membranes were reprobed and used for the detection of β-actin (A). CGCs were treated with various concentrations of colchicine (B). After 24 h, the cells were lysed and used for Western blotting. Each data represents the mean of three independent experiments.
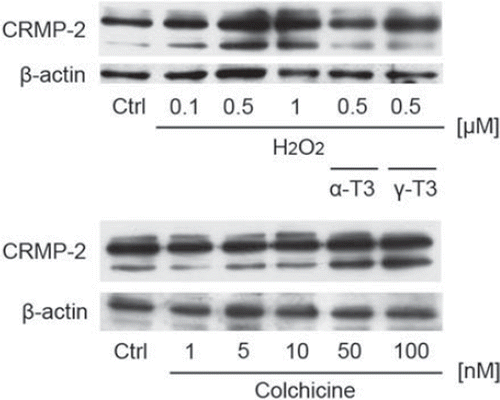
To clarify the relationship between the induction of the unknown band of CRMP-2 and axonal beading after treatment in hydrogen peroxide-treated CGCs, we focused on a cytoskeletal protein. The cells were treated with colchicine, which is used as an inhibitor of microtubule polymerization. Previously, we found that treatment of neuro2a cells with colchicine induced extensive axonal beading [Citation22]. Treatment with colchicine induced an unusual band located below the two original bands of CRMP- 2 in a concentration-dependent fashion (). These results indicate that treatment with hydrogen peroxide induced axonal degeneration via induction of an unknown band of the CRMP-2. Induction of axonal beading may affect dysfunction of microtubule assembly.
Changes in MDC accumulation and LC3 expression in hydrogen peroxide-treated CGCs
To more precisely determine the mechanism by which hydrogen peroxide induces axonal beading in CGCs, we examined autophagy using immunocytochmical and Western blot analysis. MDC, a fluorescent compound, was used as a probe for detection of autophagic vacuoles in cultured cells [Citation23]. In s tested with 0.5 μM of hydrogen peroxide were stained with MDC. Treatment of CGCs with 1 μM hydrogen peroxide did not result in positive MDC staining. However, staining with PI, which is a hall-mark of cell death, was positive. Treatment with each tocotrienol isoform prevented accumulation of MDC in 0.5 μM hydrogen peroxide-treated CGCs.
To more determine about autophagy in hydrogen peroxide-treated CGCs, we checked expression of LC3 protein level on Western blotting (). Treatment of CGCs with 1 or 5 μM Tamoxifen increased LC3 expression. Treatment of CGCs with 0.5 μM hydrogen peroxide, the expression of LC3 was stronger than that of 0.1 μM one. On the other hands, treatment with each tocotrienol isoform prevented enhancement of LC3 expression in 0.5 μM hydrogen peroxide-treated CGCs. Both results indicate that treatment with a low concentration of hydrogen peroxide leads to the accumulation and induction of autophagic vacuoles, but not cell death.
Figure 5. Treatment with hydrogen peroxide induced autophagy in CGCs, and its prevention by tocotrienols. Immunocytochemical analysis of CGCs using the double staining system of MDI and PI. CGCs were treated with 0.5 μM of hydrogen peroxide in the presence or absence of α-tocotrienol (5 μM) or γ-tocotrienol (5 μM). After 24 h, the cells were treated with PI solution (1:1000) for 2 min at R/T. After PI staining, the cells were treated with MDC solution (1:1000) for 10 min at 37°C. Treatment with 5 μM Tamoxifen was used as a control. Photomicrographs of the cells were taken and analyzed on a personal computer. At least, three wells were used per experiment. The scale bar is 10 μm (A). CGCs after treatment with hydrogen peroxide in the presence or absence of α-tocotrienol (5 μM) or γ-tocotrienol (5 μM) were lysed and used for Western blotting analysis. The same membranes were reprobed and used for the detection of β-actin. Two different concentrations of Tamoxifen were used as a control of autophagy. After 24 h, the cells were lysed and used for Western blotting (B). Each data represents the mean of three independent experiments.
Discussion
Treatment with a low concentration of hydrogen peroxide gradually but certainly induces lipid peroxidation in CGCs
It is well known that hydrogen peroxide attacks living tissues. In the present study, we used hydrogen peroxide not only to examine its direct effects on the cell membrane, but also as a trigger for ROS production in the cell. The purpose of this study was to determine the early events of ROS-induced cell degeneration before the induction of cell death. Treatment with 0.5 μM hydrogen peroxide did not induce cell death. However, the levels of LOOH, but not TBARS, significantly increased in hydrogen peroxide-treated CGCs (). ROS, including superoxide, hydroxyl radical, hydro perradical and hydrogen peroxide, attack polyunsaturated fatty acid, and play a crucial role in initiating lipid peroxidation. As a result of this reaction, i.e., a chain reaction of lipid peroxidation, LOOH increases [Citation1,Citation24]. Finally, products of aldehyde substances detected by TBA methods are increased. LOOH may increase more quickly than TBARS. We also analyzed antioxidant enzymes, including superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx). The activities of these enzymes did not change in hydrogen peroxide-treated CGCs (data not shown). These results suggest that treatment with 0.5 μM hydrogen peroxide slowly but certainly induce cell oxidation in CGCs without affecting the functions of antioxidant enzymes.
Hydrogen peroxide induces axon and dendrite degeneration before induction of cell death in pEGFP-transfected CGCs
The density of CGCs in culture is typically very high, and the extended axons of CGCs are very long, more than 20 times longer than the diameter of the cell body. To evaluate cell morphology, the pEGFP-C1 mammalian expression vector was transfected to CGCs using the lipofection method. The transfection efficiency was low, at 10–20%. However, this low transfection efficiency made it convenient for us to check the condition of whole cells. We observed the cell morphology of individual cells from the distal end of the axon to the cell body.
After 18 h of transfection, treatment of CGCs with hydrogen peroxide resulted in extensive axonal beading (). Axonal beading is a major hall mark of neurite degeneration [Citation6,Citation19]. In addition, the number of dendrites was remarkably decreased, with particularly extensive beading around surviving dendrites in hydrogen peroxide-treated CGCs (). It is well known that dendrites are receivers of neurotransmitters. Impairment of axonal and dendritic functions such as damaged neurotransmitter receptors, dysfunction of axonal transport systems and other discrepancies may occur in hydrogen peroxide-treated CGCs. In our present study, both the pre- and post-synaptic sides of synapses were injured in CGCs after treatment with hydrogen peroxide.
Treatment with each tocotrienol isoform prevented axon and dendrite degeneration in hydrogen peroxide-treated CGCs due to their antioxidant effects. Before start of this study, we checked cell survival on hydrogen peroxide–treated neuro2a cells in the presence or absence of each vitamin E isoforms. The preventive effects of tocotrienol were significantly stronger than that of tocopherol. Usually, vitamin E exist in cell membrane because of its hydrophobic formation. Serbinova et al. [Citation25] reported that antioxidant activity of α-tocotrienol is 40–60 times higher than that of α-tocopherol against (Fe2 + + ascorbate)– and (Fe2 + + NADPH)–induced lipid peroxidation in rat liver microsomal membranes. They explained that tocotrienol is more uniformly distributed in membrane bilayer because of its unsaturated isopreniod chain. In this study, treatment with each tocotrienol isoform significantly prevented induction of LOOH in hydrogen peroxide-treated CGCs (). These results strongly suggest that ROS is one of the reasons of the induction of axon and dendrite beading in our experimental model. However, further investigation is needed to determine the relationship between hydrogen peroxide-induced axon and dendrite degeneration and the possibility of impairment of the neurotransmission system. We did not observe fragmented and accumulated nuclei using Hoechst33258 staining. Chronic exposure to ROS, even at low concentrations, may gradually increase the risk of neuronal degeneration.
Treatment with hydrogen peroxide induces unusual CRMP-2 in CGCs
In order to clarify the mechanism of induction of axon and dendrite degeneration in hydrogen peroxide-treated CGCs, we analyzed CRMP-2 using Western blotting. CRMP-2, a cytosolic protein, is highly expressed in the distal ends of axons and plays a crucial role in neurite polarity and axon guidance [Citation26–28]. Inagaki et al. reported that over- expression of the CRMP-2 induces multiple axons in cultured hippocampal neurons [Citation29]. Treatment with hydrogen peroxide induced an unknown band of the CRMP-2 in a concentration-dependent fashion (). Treatment with each tocotrienol isoform prevented induction of the unknown band of the CRMP-2. This result showed that treatment with ROS can directly or indirectly induce changes in the CRMP-2. Jiang et al. examined CRMP-2 where cleavage by ischemia-activated calpain and synaptosomal CRMP-2 were more sensitive to cleavage compared with the cytosolic fraction [Citation27]. Touma et al. reported that the CRMP-2, which is cleaved by calpain 1 in the presence of calcium ions, induces neurite degeneration in mice [Citation6]. Our results may be one of the early events caused by ROS before the induction of cell death in our experimental model. Recently, several lines of evidence highlight the relationship between changes in the CRMP-2 and the progressive neurodegenerative effects characteristic of Alzheimer's disease (AD) [Citation30–32], ischemia [Citation27]and other disorders [Citation28,Citation33]. Furthermore, Castegna et al. reported that the CRMP-2 is significantly oxidized in the AD brain, and that ROS attenuate the function of the CRMP-2 [Citation34]. Consistent with this evidence, our hypothesis suggests that treatment with hydrogen peroxide induces dysfunction in the CRMP-2. However, further investigation is needed to elucidate the relationship between dysfunction of axons and dendrites and ROS-derived CRMP-2 alteration in neurons.
We focused on microtubules in order to obtain more evidence about the induction of the unknown CRMP-2 in hydrogen peroxide-treated CGCs. Microtubules play important roles in axonal extension [Citation26,Citation29,Citation35]. Recently, several lines of evidence have been reported about the importance of the relationship between the CRMP-2 and microtubules [Citation18,Citation36,Citation37]. Uchida et al. reported CRMP-2 which cleaved by calpain bound to α-tubulin [Citation38]. If changes in the CRMP-2 are one of the reasons for axonal beading on hydrogen peroxide-treated CGCs, treatment with hydrogen peroxide also induces dysfunction of cytoskeletal protein. We used colchicine, which acts as an inhibitor of microtubule polymerization by binding to tubulin. Previously, we found extensiveneuronal beading in colchicine-treated neurons [Citation22]. On Western blotting, treatment with colchicine gradually increased induction of the unknown band of the CRMP-2 in a concentration-dependent fashion (). This result was similar to that of hydrogen peroxide-treated CGCs. These results indicate that treatment with low concentrations of hydrogen peroxide induces some changes in microtubule polymerization via induction of the CRMP-2 alteration. We are continuing to investigate the relationship between possible disruption of microtubule assembly and ROS-derived axon and dendrite degeneration.
Treatment with hydrogen peroxide affected autophagy in CGCs
To determine whether transport systems were damaged in the axons of hydrogen peroxide-treated CGCs, we assessed autophagy using immunocytochemical and Western blot analysis. In , treatment with 0.5 μM hydrogen peroxide remarkably increased MDC and LC3 in CGCs. Accumulation of autophagic vacuoles is related to axonal beading, and the regions of axonal beading corresponded to the location of autophagic vacuoles in the NGF deprivation model of PC12 cells [Citation39]. In our experimental model, treatment with 0.5 μM of hydrogen peroxide induced extensive axonal beading.
Several lines of evidence have shown that autophagy is associated with aging [Citation40–43]. Saimonsen et al. discussed the relationship between changes in autophagy levels and accumulation of age-related oxidative damage [Citation41]. Down-regulation of transcriptional levels of autophagy may contribute to normal human brain aging [Citation42]. Following the free radical theory of aging, low concentrations of hydrogen peroxide and involvement of dysfunction of autophagy may induce the accumulation of autophagy vacuoles via specific damage to axon. Treatment with hydrogen peroxide at concentrations higher than 1 μM results in immediate cell death for some cells, and surviving cells may no longer have normal axonal function, including autophagy, as the axons accumulate oxidative damage by ROS. However, further investigation is needed to determine changes in the levels of autophagic vacuoles and ROS-derived axonal damage. Little is known about the relationship between vitamin E and autophagy. We are continuing to study the relationship between the neuroprotecitive effect of tocotrienols and changes in ROS-derived autophagy in neuronal cells.
Conclusion
In summary, treatment with a low concentration of hydrogen peroxide induces axon and dendrite degeneration, but not cell death. It is possible that one of the reasons for the induction of axonal degeneration is changes in the CRMP-2 and disruption of microtubules on hydrogen peroxide-treated CGCs. Treatment with each tocotrienol isoform inhibited axon and dendrite degeneration. These results showed that chronic exposure to ROS, even at low concentrations, may gradually accelerate neuronal degeneration. Several lines of evidence have shown that the CRMP-2 is phosphorylated in Alzheimer's disease [Citation30–32]. In the near future, we plan to examine the relationship between ROS- derived axon and dendrite degeneration and neurodegenerative disorders such as cognitive impairment and Alzheimer's disease.
Acknowledgements
We would like to thank Kouichi Abe and Hiroyuki Yoshimura Eisai Food & Chemical Co., Ltd., for providing us with the tocotrienols. We would also like to thank Satoko Kato, Hokkaido University, for her initial contribution to this project. Finally, the authors thank Dr. Kazuhiko Suzuki for his valuable advice on establishment of the culture samples.
Declaration of interest
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology of Japan and Eisai Food & Chemical Co., Ltd (Tokyo, Japan). This study was also supported by a grant- in-aid for Project Research from the Shibaura Institute of Technology (Tokyo, Japan) and the Sasagawa Scientific Research Grant from The Japan Science Society (#22-401) (Tokyo, Japan).
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
This paper was first published online on Early Online on 23 January 2012.
Related Research Data
References
- Sies H . Oxidative stress: oxidants and antioxidants. Exp Physiol 1997;82:291–295.
- Harman D . Aging; a theory based on free radical and radiation chemistry. J Gerontrol 1956;11 (3):298–300.
- Fukui K , Omoi N , Hayasaka T , Shinkai T , Suzuki S , Abe K , Urano S . Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann NY Acad Sci 2002;959:275–284.
- Fukui K , Takatsu H , Shinkai T , Suzuki S , Abe K , Urano S . Appearance of amyloid beta-like substances and delayed-type apoptosis in rat hippocampus CA1 region through aging and oxidative stress. J Alzheimers Dis 2005;8:299–309.
- Urano S , Sato Y , Otonari T , Makabe S , Suzuki S , Ogata M , Endo T . Aging and oxidative stress in neurodegeneration. Biofactors 1998;7:103–112.
- Touma E , Kato S , Fukui K , Koike T . Calpain-mediated cleavage of collapsing response mediator protein (CRMP)-2 during neurite degeneration in mice. Eur J Neurosci 2007; 26:3368–3381.
- Yang Y , Kawataki T , Fukui K , Koike T . Cellular Zn2 + chelators cause ‘‘dying-back’’ neurite degeneration associated with energy impairment. J Neurosci Res 2007;85:2844–2855.
- Sies H , Stahl W , Sundquist AR . Antioxidant functions of vitamins. Vitamins E and C, beta-carotene, and other carotenoids. Ann NY Acad Sci 1992;669:7–20.
- Niki E , Noguchi N . Dynamics of antioxidant action of vitamin E. Acc Chem Res 2004;37 (1):45–51.
- Samhan-Arias AK , Tyurina YY , Kagan VE . Lipid antioxidants: free radical scavenging versus regulation of enzymatic lipid peroxidation. J Clin Biochem Nutr 2011;48 (1):91–95.
- Sen CK , Rink C , Khanna S . Palm oil-derived natural vitamin E alpha-tocotrienol in brain health and disease. J Am Coll Nutr 2010;29 (3 Suppl):314S–323S.
- Miyazawa T , Shibata A , Sookwong P , Kawakami Y , Eitsuka T , Asai A , Oikawa S , Nakagawa K . Antiangiogenic and anticancer potential of unsaturated vitamin E (tocotrienol). J Nutr Biochem 2009;20 (2):79–86.
- Frank J, Chin XW, Schrader C, Eckert GP, Rimbach G. Do tocotrienols have potential as neuroprotective dietary factors? Ageing Res Rev 2011; doi:10.1016/j.arr.2011.06.006.
- Saito Y , Nishio K , Ogawa-Akazawa Y , Yamanaka K , Miyama A , Yoshida Y , Noguchi N , Niki E . Cytoplrotective effects of vitamin E homologues against glutamate-induced cell death in immature primary cortical neuron cultures: Tocopherols and tocotrienols exert similar effects by antioxidant function. Free Radic Biol Med 2010;49 (10):1542–1549.
- Noguchi N , Hanyu R , Nonaka A , Okimoto Y , Kodama T . Inhibition of THP-1 cell adheshion to endothelial cells by α-tocopherol and α-tocotrienol is dependent on intracellular concentration of the antioxidants. Free Radic Biol Med 2003; 34 (12):1614–1620.
- Wang LH , Strimatter SM . A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci 1996;16:6197–6207.
- Fukata Y , Itoh T , Kimura T , Manager C , Nishimura T , Shiromizu T , . CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 2002;4:583–591.
- Yuasa-Kawada J , Suzuki R , Kano F , Ohkawara T , Murata M , Noda M . Axonal morphologenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci 2003;17:2329–2343.
- Ikegami K , Koike T . Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience 2003;122:81–93.
- Omoi NO , Arai M , Saito M , Takatsu H , Shibata A , Fukuzawa K , . Influence of oxidative stress on fusion of pre-synaptic plasma membranes of the rat brain with phosphatidyl choline liposomes, and protective effect of vitamin E. J Nutr Sci Vitaminol (Tokyo) 2006;52:248–255.
- Ohkawa H , Ohishi N , Yagi K . Assay for lipid peroxides in animal tissue by thiobarbituric acid reaction. Anal Biochem 1979;95:351–358.
- Fukui K , Abe K , Urano S . Vitamin E improve cognitive deficit of rats caused by oxidative stress and aging. J Clin Biochem Nutri 2008;43 (Suppl 1):47–50.
- Niemann A , Takatsuki A , Elässer HP . Thelysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem 2000;48 (2):251–258.
- de Zwart LL , Meerman JH , Commandeur JN , Vermelen NP . Biomarkers of free radical damage: Applications in experimental animal and in humans. Free Radic Biol Med 1999; 26 (1–2):202–228.
- Serbinova E , Kagan VE , Han D , Packer L . Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radic Biol Med 1991;10 (5):263–275
- Arimura N , Menager C , Fukata Y , Kaibuchi K .Role of CRMP-2 in neuronal polarity. J Neurobiol 2004;58(1):34–47.
- Jiang SX , Kappler J , Zurakowski B , Desbois A , Aylsworth A , Hou ST . Calpain cleavage of collapsin response mediator proteins in ischemic mouse brain. Eur J Neurosci 2007;26 (4): 801–809.
- Chrrrier E , Reibel S , Rogemond V , Aguera M , Thomasset N , Honnorat J . Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol 2003;28 (1):51–64.
- Inagaki N , Chihara K , Arimura N , Menager C , Kawano Y , Matsuo N , . CRMP-2 induces axons in cultured hippocampal neurons. Nature Neurosci 2001;4:781–782.
- Cole AR , Noble W , van Aalten L , Plattner F , Meimaridou R , Hogan D , . Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer's disease progression. J Neurochem 2007;103:1132–1144.
- Yoshida H , Watanabe A , Ihara Y . Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer's disease. J Biol Chem 1998;273 (16):9761–9768.
- Petratos S , Li QX , George AJ , Hou X , Kerr ML , Unabia SE , . The β-amyloid protein of Alzheimer's disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2008;131:90–108.
- Vuaillat C , Varrin-Doyer M , Bernard A , Sagardoy I , Cavagna S , Chounlamountri I , Lafon M , Giraudon P . High CRMP2 expression in peripheral T lymphocytes is associated with recruitment to the brain during virus-induced neuroinflammation. J Neuroimmunol 2008;193 (1–2):38–51.
- Castegna A , Aksenov M , Thongboonkerd V , Klein JB , Pierce WM , Booze R , Markesbery WR , Butterfield DA . Proteomic identification of oxidativelymodificated proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heart shock cognate 71. J Neurochem 2002;82 (6):1524–1532.
- Yamada KM , Spooner BS , Wessells NK . Axon growth: roles of microfilaments and microtubules. Proc Natl Acad Sci USA 1970;66 (4):1206–1212.
- Gu Y , Ihara Y . Evidence that collapsing response mediator protein-2 is involved in the dynamics of microtubules. J Biol Chem 2000;275:17917–17920.
- Chae YC , Lee S , Heo K , Ha SH , Jung Y , Kim JH , Ihara Y , Suh PG , Ryu SH . (2009). Collapsin response mediator protein-2 regulates neurite formation by modulating tubulin GTPase activity, Cell Signal, 21(12), 1818–1826.
- Uchida Y , Ohshima T , Sasaki Y , Suzuki H , Yanai S , Yamashita N , . Semaphorin3A signaling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP-2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimr's disease. Gene Cell 2005;10 (2): 165–179.
- Yang Y , Fukui K , Koike T , Zheng X . Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci 2007;26:2979–2988.
- Ma Q , Qiang J , Gu P , Wang Y , Geng Y , Wang M . Age-related autophagy alterations in thebrain of senescence accelerated mouse prone 8 (SAMP8) mice. Exp Gerontol 2011;46 (7): 533–541.
- Simonsen A , Cumming RC , Brech A , Isakson P , Schubert DR , Finley KD . Promoting basa l levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult drosophila. Autophagy 2008;4 (2): 176–184.
- Lipinski MM , Zheng B , Lu T , Yan Z , Py BF , Ng A , . Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and Alzheimer's disease. Proc Natl Acad Sci USA 2010;107(32):14164–14169.
- Cuervo AM . Autophagy and aging: keeping that old broom working. Trends Genet 2008;24 (12):604–612.