Abstract
Precipitation of etoposide and adverse events associated with the co-solvents in intravenous solutions can be avoided by using liposomal etoposide (LE). The pharmacokinetics and distribution of the commercial formulation (ETPI) and LE were compared in rats. The pharmacokinetic profiles were biphasic and similar in the initial phase (Cmax, Vd, and t1/2α). However, LE showed a 60% increase in AUC with a 35% decrease in clearance (p < 0.05). This decreased clearance resulted in a 70% increase in the MRT of etoposide. The uptake of etoposide from LE was higher in macrophage-phagocytic endowed tissues indicating that LE is superior to ETPI for targeted delivery of etoposide.
Introduction
Etoposide (VP16), an epipodophyllotoxin alkaloid, has been established in the treatment of several malignancies, and is the first line drug in the treatment of small cell lung cancer, germ cell tumors, and lymphomas (CitationHande, 1998; CitationHatta et al., 2004; CitationHerzog, 2004; CitationKelland, 2005). This drug has also found use in the treatment of autoimmune syndromes such as idiopathic thrombocytopenia (CitationWood & Jacobs, 1988; CitationFigueroa et al., 1993) and LCH (CitationMayou et al., 1991; CitationViana et al., 1991; CitationYu et al., 1994; CitationGadner et al., 2001). Etoposide’s mechanism of action is based on cell phase specific toxicity (late S and early G2 phase of the cell cycle) and is attributed to deoxyribonucleic acid topoisomerase II inhibition (CitationWozniak & Ross, 1983; CitationHande, 1998).
Effective therapy with etoposide has been limited by inadequate drug delivery to the target lesion, because of the poor solubility of the drug in water. Oral dosage forms gave rise to low bioavailability (mean 57%) and non-linear absorption with increasing doses in the clinically used ranges. Moreover, poor physiochemical properties of etoposide stemming from the molecule’s hydrophobicity led to the use of complex intravenous formulations of etoposide (CitationHande, 1998). The present intravenous formulation is supplied as a concentrate, containing benzyl alcohol, Polysorbate 80, and ethanol as co-solvents for etoposide. The formulation has to be diluted carefully (to prevent precipitation of the drug) in a large volume and hypersensitivity reactions and hypotension have been reported (CitationHande, 1998).
The bio-distribution and, consequently, tumor levels of drugs can be manipulated by the use of controlled release or targeted drug delivery systems. Liposomes provide an attractive drug delivery option because they are biodegradable, hypoallergenic, and do not cause toxicity at the dosage levels used. This drug delivery system is expected to increase the duration of action of drug at the tumor, either by shielding the drug from metabolic degradation or increased uptake by tumor cells (CitationMassing & Fuxius, 2000; CitationBrandl, 2001). Increased understanding of the nature, behavior, and safety of liposomes has led to an increase in the number of approved liposomal formulations like Doxil™, AmbiSome™, and DaunoXome™ (CitationGregoriadis, 1995).
Previous studies with etoposide-loaded liposomes (CitationSengupta et al., 2000) were aimed towards avoiding the macrophagic phagocytic system (MPS) and increasing circulation times. In addition the studies were carried out in mice. The aim of this study was to prepare, characterize, and evaluate the pharmacokinetic profile of etoposide-loaded liposomes in rats, a different rodent species used in pre-clinical evaluations. Furthermore, the in vivo behavior of the liposomal product such as tissue distribution was investigated to confirm the ability of the product to effectively target the MPS organs for use in the treatment of these organ related cancers.
Materials and methods
Materials
Etoposide and phosphate buffered saline tablets (pH 7.4) were purchased from Sigma Chemical Company (St. Louis, MO). Lipids; egg phosphatidyl choline was purchased from Avanti Polar Lipids (Birmingham, AL), hydrogenated soy phosphatidylcholine (Phospholipon 90H), and phosphatidyl glycerate were gifts from the American Lecithin Company (Oxford, CT). Teniposide was a gift from Bristol Myers Squibb (Princeton, NJ). Etoposide Injection (Gensia Laboratories Ltd., Irvine, CA) was supplied by Roger Maris Cancer Center, MeritCare Hospital (Fargo, ND). Egg phosphatidyl choline was > 95% pure, whereas all the other lipids were of > 99% purity. All lipids were stored at −70°C under nitrogen and/or chloroform until further use. All solvents and other material were analytical or HPLC grade and were obtained from Fisher Scientific (Fairlawn, NJ) or Spectrum Fine Chemicals (Gardena, CA). The lipids and chemicals were used without further purification. Male Sprague-Dawley rats were obtained from Harlan Sprague-Dawley (Indianapolis, IN) and maintained at North Dakota State University according to the Institutional Animal Care and Use Committee (IACUC) guidelines.
Methods
Preparation of liposomes
Liposomes were prepared in a clean room environment using the conventional thin lipid film hydration method. Neutral liposomes were prepared with a mixture of hydrogenated phosphatidyl choline (H) and egg phosphatidyl choline (E, 4:6, molar ratio). Negatively-charged liposomes were prepared by incorporation of phosphatidyl glycerate (PG, 0.1 molar ratio) into the neutral liposomes. The lipids (50 mM) were dissolved in chloroform whereas methanol (20%) was used to dissolve the PG present in the formulation. Etoposide (1.67 mM) was mixed as a methanolic solution with the organic phase. The mixture was then dried at temperatures above the phase transition temperature (Tc) of the lipids using a rotary evaporator. The residual solvent was removed under a stream of oxygen-free nitrogen (Medipore™, USP). Phosphate buffered saline (PBS, pH 7.4) was then added, and hydration of the lipid film was carried out by shaking at 1600 rpm for 30–60 min at temperatures above the Tc to obtain multilamellar liposomes. The preparations were mixed using a vortex generator, representative samples withdrawn, and scanned under a phase contrast microscope for the presence of drug crystals or lipid drug aggregates. Drug crystals or aggregates, when present, were removed by passing (single pass) the preparation through double stacked polycarbonate membranes (1 μm pore size) using a Liposofast Basic® extruder (Avestin, Canada). The passages were carried out at temperatures above the Tc of the lipids. All liposomal preparations were stored at 4°C and used within 24–48 h.
Visualization and size determination
The preparations were observed under a phase contrast microscope for any distinct qualitative features including presence of crystals, lipid aggregates, lamellarity, and level of aggregation. The time taken for the samples to settle was also observed. The liposomal size distribution was determined using a Coulter Counter (Coulter Electronics, Haileah, FL) by introduction of 100–150 μl samples of the liposomal suspensions.
Determination of the encapsulation percentage
The unencapsulated drug was separated by centrifuging the liposomal suspension at ~ 49,500 −57,000 × g for 60 min at 4°C. The sediment was then resuspended with fresh PBS. The drug content in the initial preparation and liposomal sediment was determined using a HPLC method, which was based on a modified procedure of CitationFleming and Stewart (1991). A phenyl (C-5 matrix) column (250 × 4.6 mm, 5 μm, Varian Inc., Palo Alto, CA) with an RP-18 (10 μm, Varian Inc., Palo Alto, CA) guard column was used. The mobile phase consisted of acetonitrile:water (acetic acid) [35:65(1)] at a flow rate of 1.4 ml/min The chromatographic eluents were monitored at ultraviolet absorbance at 233 nm. Samples (20 μl) were diluted to 2 ml with methanol:water (60:40), and 100 μl of this mixture injected. Concentrations were calculated on the basis of a standard curve. The encapsulation percentage was calculated as the percentage ratio of drug in sediment to drug in original suspension.
In vitro stability in rat plasma
Samples of the liposomal suspension were diluted with rat plasma to obtain a 1:30 dilution. These diluted samples were then incubated in a shaking water bath (100 rpm) at 37 ± 2°C and aliquots of 0.5 ml withdrawn at 0, 2, 4, 6, and 8 h. The aliquots were centrifuged at 40,000–50,000 × g for 60 min at 4°C to sediment the liposomes. The plasma supernatant was carefully separated with a Pasteur pipette and mixed vigorously with 1.5 ml of acetonitrile. This mixture was then centrifuged at 3000 × g for 10 min and 100 μl of the plasma-acetonitrile supernatant injected onto the HPLC column for analysis of drug content.
Pharmacokinetics and tissue distribution in male Sprague-Dawley rats
Approval for the animal studies was obtained from the IACUC, North Dakota State University. The male Sprague-Dawley rats (weighing 250–300 g) were obtained from Harlan Sprague-Dawley, housed in animal rooms as specified by the IACUC, and given food and water ad libitum. Before administration of the formulations, the jugular vein of the rats was cannulated under 1% halothane anesthesia. The cannula patency was maintained by flushing with 100–250 μl of heparin solution (100 U/ml). The animals were allowed to recover from anesthesia for at least 4 h before the drug was administered. The rats were housed in Bollman cages and received either the free drug or the liposomal formulation (~ 4.2 mg/kg) via the cannula. The current formulation (Etoposide Injection, ETPI) concentrate was diluted with 0.5 ml PBS, and injected within 5 min of dilution. This injection was administered over a period of 30 s to 1 min (fast injections were toxic, especially in the case of the ETPI formulation, due to the solvents involved and the potential for precipitation at site of injection). Following administration of the drug formulation, blood (200 μl) was collected at ~ 5 and 10 min, 0.25, 0.5, 0.75, 1, 2, 3, 4, and 6 h via the cannula. All blood samples were stored at −70°C, and assayed within 72 h. The samples were analyzed for drug content using HPLC following extraction. The etoposide in the blood samples (50 μl) was extracted with 2 ml of 1,2-dichloroethane after the addition of 50 μl of teniposide (internal standard, 10 μg/ml). The mixture was shaken for 15 min and centrifuged at 277 × g for 5–10 min. The organic phase was separated and evaporated to dryness in a centrifugal evaporator at 37°C. The residue was reconstituted with 100 μl of methanol and 50 μl was injected onto the column. The conditions for the detection of etoposide in the blood samples were as follows: An ODS (C18, 250 × 4.6 mm, 5 μm, Brownlee, Applied Biosystems, Inc., San Jose, CA) with a RP-18 (10 μm, Varian Inc., Palo Alto, CA) guard column was used. The mobile phase consisted of acetonitrile:water (acetic acid) [40:60(1)] at a flow rate of 1.2 ml/min. Fluorescence detection was based on excitation at 283 nm and emission at 330 nm. Concentrations were calculated on the basis of a standard curve.
For the tissue distribution studies, the animals were sacrificed at 0.5, 3 and 6 h. The tissue samples (~ 50–200 mg) were homogenized with 3 ml PBS and etoposide extracted with 3 ml of 1,2-dichloroethane after addition of 50 μl of teniposide (internal standard, 10 μg/ml) as indicated for the blood samples. The residue was reconstituted with methanol (till clear) and analyzed using the HPLC method described in the previous paragraph.
Data analysis
Statistical Analysis Software® (Version 6.07.01, SAS Institute Inc., Cary, NC) and Microsoft Excel® (Version 6.0/7.0, Microsoft, Seattle, WA) were used for the statistical analysis of the results. Student’s t-test and analysis of variance were used for comparisons. WinNonlin Standard Edition (Pharsight, Mountain View, CA) was used for fitting the pharmacokinetic data and calculating the parameters using a non-compartmental or a two compartmental model.
Results and discussion
Characterization of the formulations
The results of the characterization of the liposomal (LE) formulations are given in . Microscopic observations of the vesicles following preparation indicated a multilamellar nature for the liposomes. All preparations showed the presence of drug particulates following equilibration and were passed through double-stacked polycarbonate membranes (pore size 1 μm) to remove these particulates. PG was introduced into one of the LE formulations to provide a negative charge to the particles, thereby reducing agglomeration via like-charge repulsion. Size analysis was carried out using the Coulter Counter following the removal of drug particulates. The mean diameter of the liposomes ranged from 3–3.5 μm with a standard deviation of 0.2 (n = 6) indicating reproducibility of the preparation. A slight decrease in mean size, though insignificant (p = 0.06), was observed with introduction of PG and is attributed to the negative charge introduced on the liposomes. The charge results in a higher curvature and smaller sizes due to repulsion between the like-charges at the surface. The lack of significant difference is attributed to the low proportion of PG used. Encapsulation studies indicated that > 70% of the etoposide was encapsulated in the liposomes and was not affected by introduction of PG.
Table 1. Characterization of the liposomal formulations.
In vitro stability in rat plasma
The kinetics of etoposide release from liposomes was studied in rat plasma in order to understand the release characteristics following dilution in the circulatory system. The liposomal preparations were diluted 30 times with rat plasma and maintained at 37°C. The study was terminated after 8 h as the drug was observed to degrade. The percentage etoposide released from H:E and H:E:PG was 49.1 ± 0.6 and 50.3 ± 0.6% after 4 h and 74.8 ± 0.2 and 82.4 ± 1.5% after 8 h, respectively. Release of drug from the liposomes in the presence of plasma has been reported (CitationAllen & Cleland, 1980; CitationKim et al., 1994) and is due to the interaction of the liposomal bilayers with lipoproteins and other plasma constituents. However, based on these results the LE formulations are not likely to cause etoposide ‘dumping’ following administration, but effect a gradual release.
Pharmacokinetic studies in male Sprague-Dawley rats
Both the free drug (ETPI) and the LE formulations were administered to rats at a dose of 4.2 mg/kg. The data were analyzed using appropriate compartmental and non-compartmental models in an attempt to obtain as much information as possible. The weighting method of the reciprocal of the predicted concentration was used, since the reciprocal of the predicted value is the right weighting to use if the true variance is proportional to the value itself (CitationGabrielsson & Weiner, 1994).
The blood concentration-time curves of etoposide in the Sprague-Dawley rats after intravenous administration of ETPI and LE formulations are shown in . The decay profile of ETPI in blood was biphasic in nature, showing a rapid initial phase of distribution within 25–35 min followed by a slower elimination phase. The pharmacokinetic parameter estimates of all the formulations are given in and . The distribution half-life (t1/2α) and elimination half-life (t1/2β) for ETPI were calculated as 6.6 min and 92 min, respectively, and are consistent with previous studies (CitationSinkule, 1984; CitationSaita et al., 1990). CitationSinkule (1984) indicated a t1/2α of 28–43 min and a t1/2β of 102–128 minutes, whereas CitationSaita et al. (1990) obtained a t1/2α of 7.8 min and a t1/2β of 73.1 min for the free drug. The clearance of etoposide from the ETPI formulation in our study was observed to be 47 ml/min/kg, which is similar to the reported value of 44 ml/min/kg (CitationBurgio et al., 1996).
Figure 1. Blood concentration-time profiles of etoposide after intravenous injection of 4.2 mg/kg etoposide to male Sprague-Dawley rats. Procedure detailed in Materials and Methods. Formulations administered were Etoposide Injection (ETPI, solid line), hydrogenated phosphatidyl choline: egg phosphatidyl choline (H:E; 4:6) without (broken line) and with phosphatidyl glycerate (H:E:PG; 4:6:1, dotted line). Data points represent mean ± SE (n ≥ 3). Line represents a two-compartmental fit to the data using WinNonlin, SCI Software.
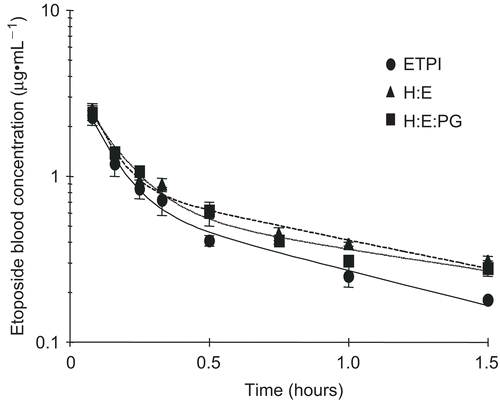
Table 2. Pharmacokinetic parameters after IV administration of etoposide to male Sprague-Dawley rats (dose 4.2 mg/kg).
Table 3. Micro-constants obtained from fitting data to a 2-compartment model following IV administration of etoposide to male Sprague-Dawley rats (dose 4.2 mg/kg).
The blood pharmacokinetic profile of the LE formulations showed similar biphasic elimination profiles (). This behavior is attributed to a combination of the biphasic distribution typically observed following administration of any liposomal formulation and the biphasic nature of free etoposide. Multilamellar vesicles, similar to those used in the present study, have been reported (CitationPalatini, 1992; CitationPardue & White, 1997) to show a biphasic elimination profile when administered intravenously to rats. This profile is attributed to the two different size populations within the multilamellar vesicles; the large vesicles (> 0.5 μm) and the small vesicles (0.2–0.5 μm). While the large vesicles are cleared rapidly from circulation, typically by the macrophagic-phagocytic system (MPS), the smaller vesicles persist for longer periods of time resulting in a biphasic profile.
LE formulations showed significantly longer elimination half-lives (p < 0.03) and a significant increase in the AUC (p < 0.05), which can be explained by the significantly lower clearance rates (p < 0.02) in comparison to the ETPI formulation. Similar results have been reported in mice following administration of etoposide containing liposomes (CitationSengupta et al., 2000). Clearance of etoposide has been reported to be largely via renal excretion (35% unchanged, CitationHande et al., 1988) and through glucoronide formation (35%, CitationHande et al., 1988), both of which are dependent on the free drug concentration in the blood (CitationGibaldi & Perrier, 1982; CitationBenet et al., 1996). In our study and most cases, liposomes, due to encapsulation of the drug, lower the free drug concentration in the blood and decrease the amount of drug available for metabolic or renal clearance from blood (CitationMartin, 1998).
The lack of higher concentrations in blood for the LE formulations in spite of lower clearance in the initial phase may be due to the enhanced uptake of the liposomes into tissues. The increased uptake in tissues (secondary compartments) with LE formulations is supported by the k12/k21 ratios, which were calculated as 2.6 ± 1.5, 4.7 ± 1.3, and 7.4 ± 1.5 for ETPI, H:E, and H:E:PG, respectively. Since k12/k21 are calculated values based on best fits to the models, tissue distribution studies were carried out to confirm the hypothesis.
Tissue distribution studies in male Sprague-Dawley rats
The tissue distribution of etoposide from ETPI and LE formulations was determined after 0.5, 3, and 6 h following administration of these formulations at a dose of 4.2 mg/kg. The results are given in and the tissue distribution of etoposide 3 h after intravenous administration is shown in . As expected, a consistently higher amount of drug was observed in the MPS-associated tissues (lungs, liver, and spleen), when administered as a LE formulation in comparison to ETPI (37% vs 11% and 20% vs 2% of administered dose after 0.5 and 3 h, respectively). The negatively charged LE formulation showed an uptake of 42% of the administered dose into the analyzed tissues after 0.5 h. This observation confirms the uptake of liposomal etoposide into tissues of interest in the initial distribution phase and is consistent with a previously reported value, where 50–70% of the lipid dose was removed from circulation into tissues within 1 h of administration (CitationSenior, 1987). In addition, the concentration of etoposide was lower in kidneys for the LE formulation in comparison to ETPI (), hinting at decreased renal clearance with the LE formulation (35% of etoposide is excreted unchanged when administered as free etoposide, CitationHande et al., 1988).
Figure 2. Tissue distribution of etoposide, 3 h after intravenous administration of the formulations, in male Sprague-Dawley rats. Each bar with error bar depicts mean + SE.
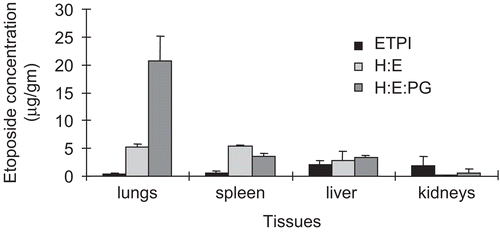
Table 4. Tissue distribution of etoposide following IV administration of the formulations to male Sprague-Dawley rats (dose 4.2 mg/kg).
The results also showed that the lungs accounted for the major uptake with 17 and 3% (94 and 21 μg/g, respectively) of the total dose after 0.5 and 3 h, respectively, when administered as a negatively-charged LE formulation. This uptake is more than observed for the neutral LE formulation (1%, 5 μg/gm in lungs after 3 h). Previous studies (CitationAbra and Hunt, 1981; CitationAbra et al., 1984; CitationSenior, 1987; CitationKim et al., 1994) have shown that liposomes bearing a negative charge are taken up effectively by the lung, liver, spleen, and lymph nodes and accumulate in the lung to a larger extent compared to neutral or positively-charged liposomes of the same size. Also, the average diameter of the liposomes in our study was 3–3.5 μm and the formulations were administered through the jugular vein. Therefore lungs are the first of the fenestrated organs encountered leading to higher entrapment and recovery of etoposide from this organ. Consequently, liposomes are effective for targeted delivery of anti-cancer agents in MPS or MPS-organ oriented therapy.
Conclusion
This study indicates that the liposomal product has a similar PK profile in comparison to the current commercial intravenous formulation of etoposide. The formulations show similar Cmax values and a rapid initial clearance from the blood, thus minimizing the potential for Cmax-related toxicity. The liposomal preparation differs from ETPI with respect to the elimination phase kinetics with longer residence times in the system. The liposomal formulation may be a better alternative to the current commercial intravenous formulation. Moreover, the liposomal formulations show higher accumulation in the organs associated with the macrophagic-phagocytic system (MPS), making this an attractive delivery system for targeting these organs. Further studies are required to investigate if the targeting of the MPS results in improved therapy.
Acknowledgements
Funding for the study provided by Children’s Hospital Research Foundation, MeritCare Hospitals, Fargo, ND 58122.
Declaration of interest: The authors declare that there are no conflicts of interest.
References
- Abra, R.M., Hunt, C.A. (1981). Liposome disposition in vivo. III. Dose and vesicle-size effects. Biochim Biophys Acta. 666, 493–503.
- Abra, R.M., Hunt, C.A., Lau, D.T. (1984). Liposome disposition in vivo. VI: delivery to the lung. J Pharml Sci. 73, 203–2066.
- Allen, T.M., Cleland, L.G. (1980). Serum-induced leakage of liposome contents. Biochim Biophys Acta. 597, 418–26.
- Benet, L.Z., Kroetz, D.L., Sheiner, L.B. (1996). Pharmacokinetics: the dynamics of drug absorption, distribution, and elimination. In: Goodman and Gilman’s The pharmacological basis of therapeutics. eds. Hardman, J.G., Limbird, L.E., Gilman, A.G. 9th Ed. New York: McGraw Hill, 3–27.
- Brandl, M. (2001). Liposomes as drug carriers: a technological approach. Biotechnol Annu Rev. 7, 59–85.
- Burgio D.E., Gosland, M.P., McNamara, P.J. (1996). Modulation effects of cyclosporine on etoposide pharmacokinetics and CNS distribution in the rat utilizing microdialysis. Biochem Pharmacol. 51, 987–92.
- Figueroa, M., Gehlsen, J., Hammond, D., Ondreyco, S., Piro, L., Palmary, T., Williams, F., McMillan, R. (1993). Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med. 328, 1226–9.
- Fleming, R.A., Stewart, F.C. (1991). High performance liquid chromatographic determination of etoposide in plasma. J Liq Chromatog. 14, 1275–83.
- Gabrielsson, J., Weiner, D. (1994). Pharmacokinetic and pharmacodynamic data analysis. Concepts and applications. Stockholm, Sweden: Swedish Pharmaceutical Press, 13.
- Gadner, H., Grois, N., Arico, M., Broadbent, V., Ceci, A., Jakobson, A., Komp, D., Michaelis, J.N., Nicholson, S., Potschger, U., Pritchard, J., Ladisch, S. (2001). A randomized trial for treatement of multisystem Langerhans’ cell histiocytosis. J Pediat. 138, 728–34.
- Gibaldi, M., Perrier, D. (1982). Pharmacokinetics. Vol I. New York: Marcel Dekker Inc.
- Gregoriadis G. (1995). Engineering liposomes for drug delivery: progress and problems. Trends Biotechnol 13, 527–37.
- Hande K.R. (1998). Etoposide: four decades of development of a topoisomerase II Inhibitor. Eur J Cancer. 34, 1514–21.
- Hande, K., Bennett, R., Hamilton, R., Grote, T., Branch, R. (1988). Metabolism and excretion of etoposide in isolated, perfused rat liver models. Cancer Res. 48, 5692–5.
- Hatta, Y., Takahashi, M., Enomoto, Y., Takahashi, N., Sawada, U., Horie, T. (2004). Adenosine phosphate (ATP) enhances the antitumor effect of etoposide (VP-16) in lung cancer. Oncol Rep. 12, 1139–42.
- Herzog, T.J. (2004). Recurrent ovarian cancer:how important is it to treat to disease progression? Clin Cancer Res. 10, 7439–49.
- Kelland, L.R. (2005). Emerging drugs for ovarian cancer. Expert Opin Emerging Drugs. 10, 413–24.
- Kim, C.K., Lee, M.K., Han, J.H., Lee, B.J. (1994). Pharmacokinetics and tissue distribution of methotrexate after intravenous injection of differently charged liposomes-encapsulated methotrexate to rats. Int J Pharm. 108, 21–29.
- Martin F.J. (1998). Clinical pharmacology and antituor efficacy of DOXIL (pegylated liposomal doxorubicin). Medical applications of liposomes. New York: Elsevier Science BV, 635–88.
- Massing, U., Fuxius, S. (2000). Liposomal formulation of anticancer drugs: selectivity and effectiveness. Drug Resist Updates. 3, 171–7.
- Mayou, S.C., Chu, A.C., Munro, D.D., Plowman, N. (1991). Langerhans’ cell histiocytosis—an excellent response to etoposide. Clin Exp Dermatol. 16: 292–4.
- Palatini P. (1992). Disposition kinetics of phospholipid liposomes. Neurobiology of essential fatty acids. New York: Plenum Press, 375–91.
- Pardue R.L., White C.A. (1997). Pharmacokinetic evaluation of liposomal encapsulated ampicillin in male and female rats. Biopharm Drug Dispos. 18, 279–92.
- Saita, T., Fujiwara, K., Kitagawa, T., Mori, M., Takata, K. (1990). A highly sensitive enzyme-linked immunosorbent assay for etoposide using β-D-galactosidase as a label. Cancer Chemo Pharmacol. 27, 115–20.
- Sengupta S., Tyagi, P., Velapandian, T., Gupta, Y.K., Gupta, S.K. (2000). Etoposide encapsulated in positively charged liposomes: pharmacokinetic studies in mice and formulation stability studies. Pharmacol Res. 42, 459–64.
- Senior, J.H. (1987). Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 3, 128–93.
- Sinkule, A. (1984). Etoposide: a semisynthetic epipodophyllotoxin. Chemistry, pharmacology, pharmacokinetics, adverse effects and use as an antineoplastic agent. Pharmacotherapy. 4, 61–73.
- Viana, M.B., Oliveira, B.M., Silva, C.M., Rios-Leite, V.H. (1991). Etoposide in the treatment for six children with Langerhans’ cell histiocytosis (histiocytosis X). Med Pediat Oncol. 19, 289–94.
- Wood, L., Jacobs, P. (1988). Durable response to etoposide-loaded platelets in refractory immune thrombocytopenic purpura: a case report. Am J Hematol. 27, 63–4.
- Wozniak, A.J., Ross, W.E. (1983). DNA damage as a basis for 4’-demethyl epipodophyllotoxin-9-(4, 6-O-ethylidene-β-D-glucopyranoside) (etoposide) cytotoxicity. Cancer Res. 43, 120–4.
- Yu, L.C., Shenoy, S., Ward, K., Warrier, R.P. (1994). Successful treatment of multisytem Langerhans’ cell histiocytosis (histiocytosis X) with etoposide. Am J Pediat Hematol Oncol. 16, 275–7.