Abstract
Nanoparticles loaded with two different commercial insulins (Actrapid®, Novorapid®) and based on different blends of a biodegradable polyester (poly-ε-caprolactone) and a polycationic non-biodegradable acrylic polymer (Eudragit® RS) were characterized in vitro. The zeta potential was positive whenever Eudragit® RS was part of the nanoparticles matrix. The encapsulation efficiency was ~ 96% except for Novorapid®-loaded particles of poly-ε-caprolactone (only 35%). In vitro release studies revealed a burst release from nanoparticles, which may be of interest for oral delivery. Novorapid-loaded nanoparticles were orally administered to diabetic rats and allowed the glycemia to be decreased when compared with free nanoparticles.
Introduction
Diabetes mellitus is a general health problem around the world. For the treatment of insulin-dependent diabetes mellitus, sub-cutaneous insulin administration is necessary. This mode of administration requires frequent daily subcutaneous injections in order to maintain adequate control of serum glucose levels: this is far from being the ideal method of treatment since insulin does not immediately reach the liver following injection. In addition, this route induces some disadvantages as pain, risk of hypoglycemia (due to a narrow therapeutic window), peripherical hyperinsulinemia (CitationKennedy, 1991), lipoatrophy, lipohypertrophy (CitationMonaco et al., 1996), obesity with intensive therapy (CitationCarlson & Campbell, 1993), insulin neuropathy, and insulin presbyopia. Therefore, there is a continuous search for improved insulin formulations in order to reproduce the physiological pattern of insulin secretion as close as possible and, thereby, to minimize the complications of diabetes mellitus. Numerous attempts to deliver insulin by routes avoiding injections have been reported in the literature: buccal, nasal, oral, rectal, pulmonary, ocular, transdermal, vaginal, and intrauterine (CitationBrange & Langkjaer, 1997; CitationOwens et al., 2003; CitationPeppas & Kavimandan, 2006). Of all these routes, oral administration would be the most desirable on the long-term basis. However, enzymatic degradation by proteolytic enzymes and low absorption rates limit the oral bioavailability of insulin in humans to less than 1% (CitationPauletti et al., 1996). Particles in the nanosized range, based on biodegradable and muco-adhesive polymers, are of particular interest as they could provide protection of sensitive proteins against enzymatic degradation and prolong intestinal residence time (CitationChen & Langer, 1998). Such a formulation would contribute not only to an improvement in the patient’s condition, but also to a reduction of the risk of catching additional diseases associated with diabetes mellitus. Furthermore, the delivery of insulin to the liver via the portal circulation would be especially desirable as the normal physiology of insulin will produce a similar effect to pancreas-secreted insulin (CitationLewis et al., 1996).
We have prepared insulin-loaded nanoparticles by the double emulsion method. Two types of insulin were investigated: regular human insulin (Actrapid®) and aspart insulin (Novorapid®), which acts more rapidly and presents duration of action shorter than regular human insulin after subcutaneous injection. We have investigated the influence of polymers ratio on the in vitro characterization (mean diameter, zeta potential, encapsulation efficiency, release kinetic) of nanoparticles composed of a biodegradable polyester (poly-ε-caprolactone) and a polycationic non-biodegradable acrylic polymer (Eudragit® RS). Due to its polycationic groups, Eudragit® RS could favor mucoadhesion and has already demonstrated its properties to enhance oral delivery of insulin from nanoparticles (CitationDamgé et al., 2007). As a preliminary test, one formulation was tested in vivo to evaluate the capacity of such preparations to release insulin in the gastro-intestinal tract and to decrease glycemia on diabetic rats.
Materials and methods
Materials
Recombinant human insulins (Actrapid® and Novorapid®), provided by Novo Nordisk (Denmark) were used as model drugs. Poly-ε-caprolactone (PCL; Mw 42,000 Da) was supplied by Aldrich Chemical Company (Milwaukee, USA) and a copolymer of acrylic and methacrylic acids containing 0.5–0.8% of a quaternary ammonium group (Eudragit® RS, MW 150,000 Da) was kindly supplied by Evonik (Darmstadt, Germany). Polyvinylalcohol (PVA, Mw 30,000 Da, 88% hydrolyzed) was purchased from Sigma (Saint-Louis, MO) and chosen as a surface active agent for the second emulsion. All other chemical reagents were of analytical grade.
Methods
Preparation of nanoparticles
The preparation of nanoparticles was based on a water-in-oil-in-water (w/o/w) emulsification solvent evaporation method. The technique was optimized as follows: 1 mL of insulin aqueous solution (100 IU/mL) was first emulsified by sonication, using an ultrasound probe (Vibra cell 72 434, BioBlock Scientific, Strasbourg, France) at amplitude 60 and 50 W output (15 s) into methylene chloride (10 mL) containing 250 mg of dissolved polymers (Eudragit® RS and PCL in different proportions—RS 100%, RS/PCL 75/25, RS/PCL 50/50, RS/PCL 25/75, PCL 100%; w/w). This first emulsion (w/o) was emulsified by sonication (60 s) in the same conditions into 40 mL of an aqueous PVA solution (0.1% w/v) to obtain the second emulsion (w/o/w). After solvent and water evaporation under reduced pressure for 20 min, nanoparticles were further isolated by centrifugation (Biofuge Stratos, Heraeus Instruments, Germany) at 42,000 g for 20 min. The sedimented nanoparticles were resuspended in 3 mL of purified water and immediately frozen before freeze-drying. Free nanoparticles were prepared according to the same preparation method.
For all formulations, the supernatant obtained after centrifugation was kept for the assay of insulin. Each formulation was made in triplicate.
Particle size and potential
Nanoparticles were analyzed for their size distribution using laser diffusion in a Zetasizer (Malvern Instruments, UK) and for their surface charge using photon correlation spectroscopy with the same instrument. Each sample was measured in triplicate.
Encapsulation efficiency
The amount of entrapped insulin into nanoparticles was determined by high performance liquid chromatography (HPLC) followed by UV detection by measuring the amount of free substance recovered in the supernatant after centrifugation of the nanoparticle suspension at 42,000 g for 20 min.
The HPLC system consisted of a SCL 10A system controller, a FCV 10AL pump, SIL 10AD autoinjector, a CTO 10A column oven, and a SPD 10A UV detector (Shimadzu). Separation was performed at 40°C on a C-18 reversed phase column (250 × 4.6 mm, 5 μm, 300 å - Vydac). An elution gradient was carried out with 0.1% (v/v) trifluoracetic acid in water (eluent A) and 0.1% (v/v) trifluoracetic acid in acetonitrile (eluent B). The gradient started at 25% of B and was increased to 70% in 10 min and then decreased to 25% in 5 min. The flow rate was 1.2 mL/min, the injection volume was 50 μL, and the detection wavelength was 214 nm. The data analysis was performed by using the Class-VP software from Shimadzu. The standard curve (0–1 IU/mL, r2 = 0.999) was prepared by diluting the stock standard solutions in 0.1% PVA solution (w/v).
In vitro release experiments
Fifty milligrams of freeze-dried nanoparticles were suspended in 20 mL of phosphate buffer (PBS pH 7.4) containing 0.1% (v/v) of Tween® 80 and incubated into a bath at 37°C under gentle magnetic stirring (150 rpm). At determined intervals, 1 mL samples were removed, filtered through a 0.22 μm Millipore® filter and assayed for released drug. One milliliter of fresh phosphate buffer was added to the suspension of nanoparticles after each sampling. The amount of insulin released was determined as described previously by HPLC at 214 nm. The standard curve (0–1 UI/mL, r2 = 0.998) was prepared by diluting the stock standard solutions in PBS. All release experiments were performed in triplicate.
In vivo study
The in vivo studies were carried out on adult male Wistar rats (250 mg, Charles River laboratories, L’Arbresle, France) which were housed in air-conditioned quarters under a photoperiod schedule of 12 h light/12 h dark. They received standard laboratory chow diet and tap water ad libitum. All experiments were carried out in accordance with the European Community Council Directive of November 24, 1986 (86/609/EEC).
Diabetes was induced in animals by intravenous injection of streptozotocin (65 mg/kg diluted in phosphate buffer pH 4.5) as previously described (CitationDamgé et al., 1988). Animals were considered diabetic when glycemia was over 300 mg/dL 2 weeks after streptozotocin treatment.
Before experiments, rats were overnight fasted (12 h). Novorapid-loaded nanoparticles (RS/PCL 50/50, 50 IU/kg) aqueous suspension were orally administered to diabetic rats. In order to evaluate the effect of administration on glycemia, unloaded nanoparticles in aqueous suspension were orally administered as control. Blood samples were withdrawn from the tail vein at determined intervals: 0, 30 min, 1, 2, 3, 4, 5, 6 and 8 h, and glycemia was measured with a glucometer.
Statistical analysis
Statistical comparisons were performed by one-way analysis of variance (ANOVA) followed by a Dunnett or a Bonferroni multicomparison test using the Instat 2.00 Macintosh software (Graph Pad Software, San Diego, CA). The difference was considered as significant when p < 0.05.
Results
Characterization of nanoparticles
As shown in , the mean diameter of loaded and unloaded nanoparticles prepared with Eudragit® RS alone or with different amounts of PCL ranged from 315–695 nm. Actrapid®-loaded nanoparticles were not significantly different from unloaded ones (340 nm), whereas the mean diameter of the Novorapid®-loaded nanoparticles was larger (from 480–695 nm). Nanoparticles prepared with PCL alone displayed no major difference in their mean diameter (510 nm) whatever the type of encapsulated insulin.
Table 1. Main physicochemical properties of the unloaded nanoparticles or insulin-loaded nanoparticles prepared with different blends of Eudragit® RS and PCL. Data are shown as mean ± SD (n = 3).
Loaded or unloaded insulin nanoparticles prepared with Eudragit® RS alone or in combination with PCL were positively charged (from + 36 to + 44 mV) due to the quaternary ammonium groups of Eudragit® RS which were directed toward the continuous aqueous phase. However, nanoparticles prepared with PCL alone were quite negatively charged (−14 mV).
As reported in , at the exception of nanoparticles of Novorapid® prepared with PCL alone (36%) the encapsulation efficiency of insulin in each formulation was very high (96%) for Actrapid®- or Novorapid®-loaded nanoparticles.
Table 2. Encapsulation efficiency of insulin loaded nanoparticles prepared with different blends of Eudragit® RS and PCL. Data are shown as mean ± SD (n = 3).
In vitro drug release
and illustrate the in vitro release profiles of insulin in PBS at 37°C and pH 7.4 from Actrapid®- and Novorapid®-loaded nanoparticles, respectively. A biphasic release was observed for each formulation and for each type of insulin. Indeed, after an initial burst during which insulin was rapidly released over 1 h, the drug release profiles displayed a plateau characterized by a slow and incomplete release for an extended period of time. Nanoparticles prepared with Eudragit® RS alone or in combination with PCL exhibited higher drug release (from 55–70%) than those prepared with PCL alone (from 17–35%).
Figure 1. Release kinetics of Actrapid®-loaded nanoparticles prepared with different blends of Eudragit® RS and PCL. Data are shown as mean ± SD (n = 3).
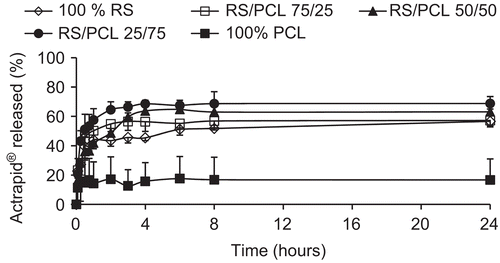
Figure 2. Release kinetics of Novorapid®-loaded nanoparticles prepared with different blends of Eudragit® RS and PCL. Data are shown as mean ± SD (n = 3).
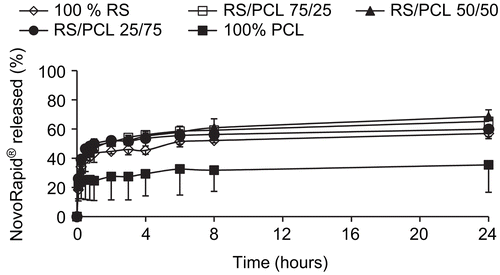
In vivo experiment
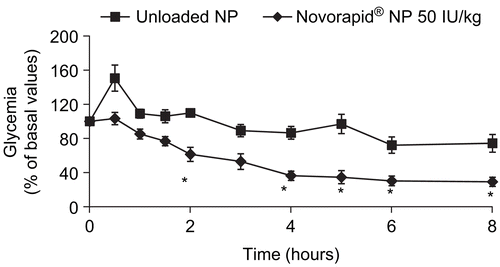
As reported in , Novorapid®-loaded nanoparticles orally administered in diabetic rats allowed a significant decrease of glycemia compared to rats treated with insulin-free nanoparticles from 2–8 h after force-feeding. Glycemia was reduced by 39% at 2 h and 70% at 8 h.
Figure 3. Glycemia after a single oral administration of unloaded nanoparticles (NP) and Novorapid®-loaded nanoparticles (50 IU/kg) based on Eudragit® RS and PCL (50/50). Data are shown as mean ± SEM (n = 6). Statistically different from free nanoparticles. * p < 0.05.
Discussion
For the preparation of nanoparticles, the choice of the method is generally determined by the solubility characteristics of the drug. Since insulin is a water-soluble drug, the double emulsion solvent evaporation technique was selected. This process involves two steps: the first step consists of the formation of the water-in-oil emulsion and leads to the formation of droplets of insulin dispersed into the organic phase of polymers (Eudragit® RS and PCL alone or their blends) followed by the water-in-oil-in-water emulsion obtained by mixing the first emulsion into an aqueous solution of PVA (0.1% w/v). The second step is the solvent evaporation from the droplets of the second emulsion leading to the precipitation of polymers which are insoluble in water and consequently the solidification of the core of the particles and the entrapment of the drug.
The mean diameters of free and Actrapid®-loaded nanoparticles prepared with Eudragit® RS alone or in combination with PCL were approximately the same (340 nm), whereas the Novorapid®-loaded ones were larger (from 480–690 nm) (). Due to their quaternary ammonium groups, Eudragit® RS has surface active properties able to stabilize the first emulsion, and consequently hampers the coalescence of the droplets, leading to minimize the diameter of the particles (CitationChernysheva et al., 2003). Since the particle size is related to a great extent to the stability of the first emulsion, it can be assumed that some excipients included in the commercial presentation of Novorapid® could interfere with Eudragit® RS. Indeed, Actrapid® contains zinc chloride, glycerol, metacresol, and either sodium hydroxide or hydrochloric acid. Novorapid® contains the same excipients as Actrapid® but also phenol, sodium chloride, and disodium phosphate dihydrate which could act on the ionic strength of the medium, destabilize the first emulsion, and consequently induce some coalescence of the droplets, and finally increase the diameter of the nanoparticles comparatively with Actrapid®-loaded nanoparticles. On the other hand, nanoparticles prepared with PCL alone were larger than nanoparticles containing Eudragit® RS (510 nm). It is emphasized that the lack of the surface active properties associated when Eudragit® RS is present induced the coalescence rate of the droplets of the first emulsion and increased the size of the nanoparticles. Insulin itself seems to have no effect on the mean diameter of nanoparticles prepared with PCL alone. Moreover, all nanoparticles formulations prepared with PCL alone showed a quite negative zeta potential (−14 mV). The magnitude of the measured zeta potential is an indication of the repulsive forces that are present and can be used to predict the long-term stability of the nanoparticles suspension (CitationBenita & Levy, 1993). When particles have a high negative or positive zeta potential, they tend to repel each other and have no tendency to aggregate. On the contrary, when particles have low absolute zeta potential values, there is no counteracting force to prevent their aggregation and flocculation. Since PCL nanoparticles displayed a lower zeta potential, their stability was lower. Indeed, they have the tendency to aggregate and their mean diameter was higher.
On the other hand, shows that nanoparticles prepared with Eudragit® RS alone or in combination with PCL were positively charged (from +36 to +44 mV), due to the quaternary ammonium groups of Eudragit® RS. Although insulin (pI 5.4) is mainly negatively charged in the aqueous solution of PVA 0.1% (w/v) (pH 6.5), we can observe that the addition of insulin in the formulation did not affect the zeta potential of the nanoparticles, thus proving that insulin was effectively encapsulated into the nanoparticles. Indeed, some authors have demonstrated that encapsulation of heparin (which is negatively charged) decreased the zeta potential of microparticles (Jiao et al., Citation2002a; Citationb; CitationHoffart et al., 2003); heparin was mainly adsorbed on the surface of the particles, involving a decrease of the zeta potential of unloaded particles (from +40 mV to −30 mV). In the case of nanoparticles prepared with Eudragit® RS alone or in combination of PCL, the lack of zeta potential difference between unloaded and loaded nanoparticles is in favor of the insulin encapsulation in the nanoparticles matrix.
As shown in , the encapsulation efficiency of insulin was very high (96%) except for nanoparticles of Novorapid® prepared with PCL alone (36%). Since Eudragit® RS is a positively charged polymer and insulin is mainly negatively charged in our experimental conditions, electrostatic interactions occurred during the preparation which led to a low release of the drug and high entrapment efficiency. Indeed, all formulations containing Eudragit® RS (from 25–100%) showed high encapsulation efficiency of insulin. The PCL polymer seemed to have no effect on the encapsulation efficiency of insulin in nanoparticles prepared with the blends of polymers. The formulation prepared with PCL alone and Novorapid® encapsulated only 36% of drug. As discussed previously, excipients contained in Novorapid® and not present in Actrapid® could interfere during the preparation and could explain the lower encapsulation efficiency of Novorapid® in nanoparticles prepared with PCL. The lower efficiency could be explained by (i) the absence of electropositive charges of PCL that could retain the drug into nanoparticles by electrostatic forces, and (ii) the negative zeta potential of the nanoparticles and the specific excipients included in Novorapid® which could destabilize the medium, leading to the leak of insulin.
and illustrate the in vitro release profile of each formulation, in pH 7.4 phosphate buffer containing 0.1% (v/v) of Tween® 80, by representing the percentage of Actrapid® and Novorapid® release with respect to the amount of drug encapsulated, respectively. They were characterized by an initial burst during which insulin was rapidly released over 1 h followed by a plateau characterized by an incomplete release of drug up to 24 h. Although the entire encapsulated drug was not released within 24 h, the dissolution test was limited to this time since the goal of this research work was to prepare an oral delivery system which has to take into account the intestinal residence time. When nanoparticles are prepared by w/o/w method, water-soluble drugs and proteins show a significant tendency to migrate to the aqueous dissolution medium, thereby concentrating at the surface of the particles and generally involving a burst effect (CitationJameela et al., 1997). All formulations prepared with Eudragit® RS or with blends of Eudragit® RS and PCL released 55–70% of encapsulated drug, whereas nanoparticles prepared with PCL alone exhibited less drug release (17–35%). No significant difference could be observed between formulations containing Eudragit® RS, whatever its concentration. The higher drug release observed with Eudragit® RS alone or in combination with PCL, compared with PCL alone may be explained by the high hydrophobicity of PCL that reduced the wettability of the nanoparticles as well as the diffusion of the drug to the dissolution medium. In addition, Ha et al. (1997) showed that the degradation of PCL was very slow in an aqueous medium because of its semi-crystallinity and hydrophobicity. This indicates that in vitro drug release was a result of diffusion and not a degradation of the polymer. This is also compatible with the short duration time of the release test which was limited to 24 h. Nevertheless, in vivo, the release of insulin from such nanoparticles could be increased since the biological environment (i.e. enzymes) may partially degrade PCL. Although no significant difference was observed between formulations of Novorapid® and Actrapid® prepared with Eudragit® RS alone or in combination with PCL, the higher release was from nanoparticles prepared with the 25/75 blend, proving that electrostatic interactions between Eudragit® RS and insulin did not interfere with the release of the drug.
As a first step, even if all formulations were able to release insulin, only one formulation was tested in vivo as a preliminary test to evaluate the ability of insulin-loaded nanoparticles to decrease glycemia in rats. Previous works based on heparin-loaded nanoparticles (CitationHoffart et al., 2006) have demonstrated that the blend 50/50 permitted the increase of anti-Xa activity after oral delivery. Moreover, a previous work has already studied Actrapid®-loaded nanoparticles in diabetic rats (CitationDamgé et al., 2007). So, Novorapid®-loaded nanoparticles based on the same blend of Eudragit® RS and PCL (50/50) were chosen to be orally administered to rats. The dose of 50 IU/kg was higher than the classical dose used for subcutaneous injection of insulin in the treatment of diabetes. Due to lower absorption figures associated with the oral route, it was deemed necessary to administer a higher dose for the oral route than for the parenteral one. As shown in , the decrease of glycemia started 2 h after oral administration. This lag time could be due to the time required for nanoparticles to reach the site of the gastro-intestinal tract where nanoparticles or free insulin released from nanoparticles could be absorbed. The release of insulin from nanoparticles is first based on the diffusion of the drug through the polymers matrix which also takes time. The encapsulation of insulin into polymeric nanoparticles allowed insulin to be protected against degradation by proteolytic enzymes (i.e. trypsin, chymotrypsin), as previously shown by CitationDamgé et al. (1997) with poly(alkylcyanoacrylate) nanospheres. Indeed, non-encapsulated insulin does not affect glycemia after oral delivery. The quaternary ammonium groups of Eudragit® RS confer a global positive zeta potential to nanoparticles which can interact with the negative charges of intestinal mucus (due to the presence of sialic acid in its composition). These interactions could be responsible for mucoadhesion of nanoparticles on the surface of the intestinal barrier, allowing a closer intimacy of contact between drug and mucus membrane at the absorption sites, thus enhancing the permeability as well as reducing the local degradation of the drug. Insulin-loaded nanoparticles and/or insulin released from nanoparticles could be taken up by three potential mechanisms: (i) uptake via a paracellular pathway, (ii) transcytosis or receptor-mediated transcytosis and transport via the epithelial cells of the intestinal mucosa, and (iii) lymphatic uptake via the M-cells of the Peyer’s patches mostly abundant in the ileum (CitationDamgé et al., 2008). Mucoadhesion of cationic nanoparticles made of Eudragit® RS to mucin layer on Caco-2 cells has already been demonstrated by CitationLamprecht et al. (2006). The mechanism was governed by non-specific particle adhesion to the epithelial barrier followed by a replacement of the drug on the particle surface by mucin. The free drug was then taken up into or transported through the cells. This mechanism is in line with a transport phenomenon described for enhanced particle deposition close to biological barriers, leading to a highly increased concentration of the entrapped drug at the apical side being responsible for enhanced absorption by a drug gradient concentration towards the basolateral compartment.
We have demonstrated the ability of nanoparticles based on different blends of PCL and Eudragit® RS to encapsulate and to release in vitro two types of insulin. It seems that the ratio of polymers did not influence the encapsulation efficiency of the drug, except for Novorapid®-loaded nanoparticles based only on PCL. Regarding the in vitro kinetic profiles, nanoparticles composed of PCL alone displayed a lower release when compared with all formulations based on the blend of polymers or Eudragit® RS alone. The effect of oral administration of Novorapid®-loaded nanoparticles (50/50) on glycemia of diabetic rats has proven that insulin was still active after encapsulation and was absorbed by the gastro-intestinal tract. Compared with subcutaneous administration of insulin, such nanoparticles allowed a hypoglycemic effect from 2–8 h after oral delivery to be observed. Insulin nanoparticles could represent an interesting dosage form to increase the compliance and the comfort of diabetic patients.
Acknowledgements
Declaration of interest: The authors declare no conflict of interest.
References
- Benita, S., Levy, M.Y. (1993). Submicron emulsions as colloidal drug carriers for intravenous administration: comprehensive physicochemical characterization. J Pharm Sci. 82, 1069–79.
- Brange, J., Langkjaer, L. (1997). Insulin formulation and delivery. Pharm Biotechnol. 10, 343–409.
- Carlson, M.G., Campbell, P.J. (1993). Intensive insulin therapy and weight gain in IDDM. Diabetes. 42, 1700–7.
- Chen, H., Langer, R. (1998). Oral particulate delivery: status and future trends. Adv Drug Deliv Rev. 34, 339–50.
- Chernysheva, Y., Babak, V., Kildeeva, N., Boury, F., Benoit, J.P., Ubrich, N., Maincent, P. (2003). Effect of the type of hydrophobic polymers on the size of nanoparticles obtained by emulsification-solvent evaporation. Mendeleev Commun. 13, 65–8.
- Damgé, C., Maincent, P., Ubrich, N. (2007). Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J Contr Rel. 117, 163–70.
- Damgé, C., Michel, C., Aprahamian, M., Couvreur, P. (1988). New approach for oral administration of insulin with polyalkylcyanoacrylate nanocapsules as drug carrier. Diabetes. 37, 246–51.
- Damgé, C., Reis, C.P., Maincent, P. (2008). Nanoparticle strategies for the oral delivery of insulin. Expert Opin Drug Deliv. 5, 45–68.
- Damgé, C., Vranckx, H., Balschmidt, P., Couvreur, P. (1997). Poly(alkyl cyanoacrylate) nanospheres for oral administration of insulin. J Pharm Sci. 86, 1403–9.
- Hoffart, V., Lamprecht, A., Maincent, P., Lecompte, T., Vigneron, C., Ubrich, N. (2006). Oral bioavailability of a low molecular weight heparin using a polymeric delivery system. J Contr Rel. 113, 38–42.
- Hoffart, V., Ubrich, N., Lamprecht, A., Bachelier, K., Vigneron, C., Lecompte, T., Hoffman, M., Maincent, P. (2003). Microencapsulation of low molecular weight heparin into polymeric particles designed with biodegradable and nonbiodegradable polycationic polymers. Drug Deliv. 10, 1–7.
- Jameela, S.R., Suma, N., Jayakrishnan, A. (1997). Protein release from poly(epsilon-caprolactone) microspheres prepared by melt encapsulation and solvent evaporation techniques: a comparative study. J Biomater Sci Polym Ed. 8, 457–66.
- Jiao, Y., Ubrich, N., Hoffart, V., Marchand-Arvier, M., Vigneron, C., Hoffman, M., Maincent, P. (2002a). Preparation and characterization of heparin-loaded polymeric microparticles. Drug Dev Ind Pharm. 28, 1033–41.
- Jiao, Y., Ubrich, N., Marchand-Arvier, M., Vigneron, C., Hoffman, M., Lecompte, T., Maincent, P. (2002b). In vitro and in vivo evaluation of oral heparin-loaded polymeric nanoparticles in rabbits. Circulation. 105, 230–5.
- Kennedy, F.P. (1991). Recent developments in insulin delivery techniques. Current status and future potential. Drugs. 42, 213–27.
- Lamprecht, A., Koenig, P., Ubrich, N., Maincent, P., Neumann, D. (2006). Low molecular weight heparin nanoparticles: mucoadhesion and behaviour in Caco-2 cells. Nanotechnology. 17, 3673–80.
- Lewis, G.F., Zinman, B., Groenewoud, Y., Vranic, M., Giacca, A. (1996). Hepatic glucose production is regulated both by direct hepatic and extrahepatic effects of insulin in humans. Diabetes. 45, 454–62.
- Monaco, L., Geffken, G., Silverstein, J.H. (1996). Accuracy of injection site identification among children with insulin dependent diabetes mellitus: a comparison of traditional and new visual aids. Clin Pediatr (Phila). 35, 191–7.
- Owens, D.R., Zinman, B., Bolli, G. (2003). Alternative routes of insulin delivery. Diabet Med. 20, 886–98.
- Pauletti, G.M., Gangwar, S., Knipp, G.T., Nerurkar, M.M., Okumu, F.W., Tamura, K., Siahaan, T.J., Borchardt, R.T. (1996). Structural requirements for intestinal absorption of peptide drugs. J Contr Rel. 41, 3–17.
- Peppas, N.A., Kavimandan, N.J. (2006). Nanoscale analysis of protein and peptide absorption: insulin absorption using complexation and pH-sensitive hydrogels as delivery vehicles. Eur J Pharm Sci. 29, 183–97.