Abstract
The aim of this study was to formulate and evaluate in vitro, ceftriaxone sodium lipospheres dispersions for oral administration. Ceftriaxone sodium lipospheres were prepared by melt-emulsification using 30%w/w Phospholipon® 90H in Softisan® 154 as the lipid matrix containing increasing quantities of PEG 4000 (10, 20, 30, and 40%w/w). Characterization based on particle size, particle morphology, encapsulation efficiency, loading capacity and pH were carried out on the lipospheres. Microbiological studies of the ceftriaxone sodium-loaded lipospheres were performed using Escherichia coli as the model organism. In vitro permeation of ceftriaxone sodium from the lipospheres through artificial membrane (0.22 μm pore size) was carried out using Franz cell and simulated intestinal fluid (SIF) without pancreatin as acceptor medium. Photomicrographs revealed spherical particles within a micrometer range with minimal growth after 1 month (Maximum size = 64.76 ± 3.81 μm). Microbiological studies indicated that lipospheres formulated with 20%w/w of PEG 4000 containing 2%w/w or 3%w/w of ceftriaxone sodium gave significantly (p < 0.05) higher inhibition zone diameter than those with 30%w/w or 40%w/w of PEG 4000. The result also indicated that lipospheres with 10%w/w PEG 4000 resulted in significantly higher encapsulation efficiency (p < 0.05) while those with 30%w/w gave the least, while the loading capacity values ranged from 3.22 mg of ceftriaxone sodium/100 mg of lipid to 6.36 mg of ceftriaxone sodium/100 mg of lipid. Permeation coefficient values varied and ranged from 8.55 × 10−7 cm/s to 2.08 × 10−6 cm/s depending on the concentration of PEG 4000. The result of this study gave insight that the issue of ceftriaxone stability in oral formulation could be adequately addressed by tactical engineering of lipid drug delivery systems such as lipospheres.
Introduction
The method by which a drug is delivered can have a significant effect on its efficacy. The very slow progress in the efficacy of the treatment of severe diseases has suggested a growing need for a multidisciplinary approach to the delivery of therapeutics to targets in tissues (CitationCostas, 2006). There is growing interest and investment in the use of lipid-based systems in drug discovery and product development to effectively overcome physical and biological barriers related to poor aqueous solubility and stability, membrane permeability, drug efflux, and availability (CitationWestesen & Siekmann, 1998). However, pharmaceutical development is one of the most challenging aspects of pharmaceutical and biopharmaceutical innovations. It involves navigating the complexities of pharmaceutical sciences and manufacturing, while achieving regulatory compliance. An array of lipid systems such as emulsions, micellar solutions, liposomes, lipid nanoparticles, structured lipid carriers, self-emulsifying lipid formulations, solid dispersions, dry emulsions, solid–liquid compacts, and drug–lipid conjugates is available to pharmaceutical formulators. Among the various lipid systems, lipospheres have been developed to address some issues such as stability and low payload capacity of some lipid systems. Lipospheres drug carriers can modify the bioavailability of encapsulated drug (CitationBekerman et al., 2004) and have been used to modify sunscreen formulation (CitationIannucceli et al., 2008) and release characteristics of some drugs, e.g. allopurinol (CitationEl-Gibaly & Abdel-Ghaffar, 2005). They can therefore be used to improve the therapeutic index of drugs by increasing their efficacy and/or reducing their toxicity if the delivery systems are carefully designed. Lipospheres can help overcome the delivery problems of new classes of active molecules such as peptides, proteins, genes, and oligonucleotides, and may also extend the therapeutic potential of established drugs (CitationDidier, 2000; CitationRabasco & Gonzalez, 2000; CitationZhang et al., 2008).
The rapid growth in the use of lipid-based drug delivery systems is primarily due to the diversity and versatility of pharmaceutical grade lipid excipients and drug formulations, and their compatibility with liquid, semi-solid, and solid dosage forms (CitationCraig, 2004; CitationAttama & Nkemnele, 2005). Lipospheres are solid water-insoluble microparticles formed of a solid hydrophobic core having a layer of a phospholipid embedded on the surface of the core containing a biologically active agent in the core. The average particle diameter is between 0.3–250 μm (CitationDomb, 1993). Lipospheres have several advantages over other lipid delivery systems such as emulsions, vesicles, and liposomes, in terms of stability, low cost of reagents, ease of manufacture, high dispersibility in aqueous medium, and a release rate for the entrapped substance that is controlled by the phospholipid coating and the carrier. In a liposphere, there is no equilibrium of substances in and out of the vehicle as in an emulsion system. Lipospheres also have a lower risk of reaction of substance to be delivered with vehicle than in emulsion systems because the vehicle is a solid inert material. Moreover, the release rate of a substance from the lipospheres can be manipulated by altering either or both inner solid vehicle or the outer phospholipids (CitationDomb, 1993). The pharmaceutical uses include providing extended release of active agent including drugs such as vaccines and anesthetics; in oral formulations for release into the lower portions of the gastrointestinal tract; in oral formulation to mask the taste or odour of the substance and as a component in lotions and sprays for topical use (CitationWestesen & Siekmann, 1998). Oxytetracycline (OTC) has been encapsulated in a liposphere drug delivery system composed of solid triglycerides, phospholipids, buffer solution, and preservatives, to prolong the duration of action the drug (CitationDomb, 1995).
This study aimed at evaluating ceftriaxone sodium-loaded lipospheres formulated with Softisan 154®, PEG 4000, and a phospholipid (Phospholipon® 90H) for oral delivery of ceftriaxone sodium. Softisan® 154 (hydrogenated palm oil) is a hard fat based on triglyceride blends of natural, saturated even-numbered vegetable fatty acids, with chain length of C10 to C18. It is free of any anti-oxidant and stabilizer (Sasol, Citation2008). Polyethylene glycol (PEG)-conjugated lipids are commonly employed for steric stabilization of liposomes and modification of drugs and formulations (CitationHamidi et al., 2006; CitationRyan et al., 2008). Ceftriaxone belongs to the third spectrum cephalosporins which are broad spectrum β-lactam antibiotics. Like other third generation cephalosporins, it has broad spectrum activity against Gram positive and Gram negative bacteria and is highly stable to most β-lactamases. The drug is stable under ordinary conditions but reconstituted solution retains the physical and chemical stability for 6 h at room temperature or for 24 h in the refrigerator at 2–8°C (CitationRocephin®, Roche Switzerland, drug information). It is usually formulated as reconstitutable powder for injection.
Materials and methods
Materials
The following materials were used as procured from their local suppliers without further purification: Softisan® 154 (CitationSasol GmbH, Germany), Phospholipon® 90H (Phospholipid GmbH, Köln Germany), sorbitol (Across Organics, Germany), thiomersal (Synochem, Germany), PEG 4000 (Sigma, UK), ceftriaxone sodium (Avicef®, Turkey), nutrient agar (Antec Diagnostic Products, UK), and monobasic potassium phosphate, sodium hydroxide, and hydrochloric acid (BDH, UK). All other materials used were of analytical grade. Distilled water was obtained from our laboratory.
Preparation of lipid matrix
The lipid matrix was prepared using Softisan® 154 and Phospholipon® 90H (P90H). A 15 g quantity of P90H was carefully weighed and added to 35 g of Softisan 154® (i.e. 30% of P90H in Softisan® 154) in a crucible and the mixture heated on a water bath to melt. The molten lipids were then stirred thoroughly until solidification to ensure adequate mixing.
Preparation of drug loaded and unloaded lipospheres
The lipid base, i.e. 30%w/w of P90H in Softisan® 154 (from above) and PEG 4000 (10%w/w) were carefully weighed, transferred into a 250 ml beaker and heated to melting on a water bath, and ceftriaxone sodium (1%w/w) was introduced into the melted lipid and stirred thoroughly. Thiomersal and sorbitol were carefully weighed and dissolved in distilled water at 75°C and immediately transferred into the lipid phase containing the drug at the same temperature. The mixture was then homogenized with a mixer (Silverson, UK) for 4 min and allowed to recrystallize at room temperature. The above procedure was repeated using increasing quantities of PEG 4000 to yield 20, 30, and 40%w/w, respectively, each time using 2%w/w and 3%w/w of the drug. The formula corresponding to lipid matrix (PEG in 30%w/w P90H in Softisan® 154) 10%w/w, ceftriaxone sodium 1, 2, or 3%w/w (Batches A, B, and C, respectively), thiomersal 0.005%w/w, sorbitol 4%w/w, and sufficient distilled water to make 100%w/w, as presented in , was used in the formulation of loaded lipospheres. Lipospheres containing no drug (unloaded lipospheres), which served as the control, were also prepared to contain 10, 20, 30, and 40%w/w of PEG 4000 (Batch D).
Table 1. Quantities of starting materials for the formulation of different batches of unloaded and ceftriaxone sodium-loaded lipospheres.
Particle size analysis and morphological characterization of lipospheres
The particle size of the lipospheres was determined by computerized image analysis of at least 50 lipospheres on a photomicroscope (Lieca, Germany). Each of the batches was mounted on a slide and observed under a light microscope. With the aid of the software in the microscope, the projected perimeter diameters of the particles corresponding to the particle sizes of the lipospheres were determined and average calculated. The particle morphologies were also observed and photomicrographs taken. All these were done in a time-dependent manner (24 h, 1 week, and 1 month).
pH analysis
With the aid of a pH meter (Digital pH Meter, Labtech), the pH of the different batches of the liposphere formulations including those of the control were measured. This was also carried out in a time-dependent manner (24 h, 1 week, and 1 month). Triplicate determinations were done in each measurement.
Encapsulation efficiency (EE%)
The EE% of each formulation was determined after 1 month of preparation. In each case, a 6 ml volume of each batch was placed in a centrifuge tube and centrifuged (Chem. Lab. Instrument, UK) for 30 min at an optimized speed of 1500 rpm to obtain two phases (i.e. aqueous and lipid phases). A 1 ml volume of the aqueous phase was thereafter diluted 1000-fold using distilled water. The absorbance of these solutions was determined in a UV-spectrophotometer (UNICO UV-2102 PC, USA) at a wavelength of 239 nm, and the encapsulation efficiency calculated using equation (1) (CitationMorkhade & Joshi, 2007). The regression equation of Beer’s plot for ceftriaxone sodium in distilled water was: A = 0.717C + 0.028 (R2= 0.9890), where A is the absorbance of the solution and C is ceftriaxone sodium concentration.
Loading capacity (LC)
LC expresses the ratio between the entrapped API and the total weight of the lipids. It is determined as follows:
where Wlis the weight of lipid added in the formulation, Wais the weight of API added to the formulation, and Wsis the amount of API determined in supernatant after separation of the lipid and aqueous phase.
Release study as a function of IZD
The plate agar diffusion method was used for this study conducted after 1 month of lipospheres preparation. This method depends on the diffusion of antibiotics from holes on the surface of the microbial seeded agar. The plate was seeded with Escherichia coli. Using a sterile cork borer, four cups were aseptically bored at equal distances from each other. Using a sterile applicator, one drop of the formulation of a batch was applied in the holes. The plates were incubated at 37 ± 1°C for 24 h and the diameter of each inhibition zone was measured and the average determined (CitationHassan et al., 2003).
In vitro drug permeation studies
A Franz diffusion cell (CitationFranz, 1975) was used for the permeation studies. In each case, a finite volume of the formulation was placed in the donor compartment of the Franz diffusion cell separated from the receptor compartment by an artificial membrane (pore size 0.22 μm). The receptor compartment was filled with SIF (pH 7.4) and maintained at a temperature of 37 ± 1°C by means of a thermostatically controlled water bath, with agitation provided by a magnetic stirring bar at 50 rpm. Aliquot was removed and replaced by an equal volume of the receptor phase at different time interval up to 6 h and the samples collected analyzed for drug content spectrophotometrically using a spectrophotometer (UNICO UV-2102 PC, USA) at 241 nm. The drug content at each time point was calculated by reference to Beer’s calibration. The regression equation of Beer’s plot for ceftriaxone sodium in SIF (pH = 7.4) was: A = 0.565C – 0.000 (R2= 0.9990), where A is the absorbance of the solution and C is ceftriaxone sodium concentration.
The permeation parameters of ceftriaxone sodium from the lipospheres were calculated by plotting the amounts of drug permeated through the membrane (μg/cm2) vs time (min). The steady-state flux (J) values across the membrane were evaluated from the linear ascents of the permeation graphs by means of the relationship:
where Q indicates the quantity of substance crossing the artificial membrane; A is the area of the membrane exposed; and t is the time of exposure. The permeation coefficient P in each case was calculated from equation (3).
where C0 represents the initial drug concentration in the donor compartment.
Data and statistical analysis
All experiments were performed in replicates (n= 3) for validity of statistical analysis. Results were expressed as mean ± SD. ANOVA and Student t-tests were performed on the data sets generated using Origin® for Windows and SigmaPlot® 11. Differences were considered significant for p values < 0.05. Permeation calculations were performed with a special Microsoft excel program.
Results and discussion
Characterization of lipospheres
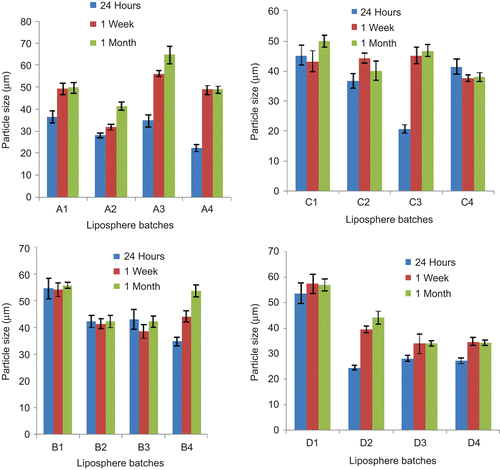
shows the photomicrographs of the representative batches of lipospheres containing 30%w/w PEG 4000 and 1%w/w ceftriaxone sodium (A3), 30%w/w PEG 4000 and 2%w/w ceftriaxone sodium (B3), 30%w/w PEG 4000 and 3%w/w ceftriaxone sodium (C3), and 30%w/w PEG 4000 and no ceftriaxone sodium (D3) after 1 month. From the micrographs it could be seen that the lipospheres formulated were spherical in shape. It should be noted that the micrograph presents the particles in two dimensions and particles viewed edge-on may not appear spherical. The result of particle size analysis of the various batches of lipospheres which were carried out after 24 h, 1 week, and 1 month are presented in . Particle size may be a function of either one or more of the following: formulation excipients, degree of homogenization, homogenization pressure, rate of particle size growth, crystalline habit of the particle, etc. From the result after 24 h, it was observed that sub-batches B1, C1, and D1 had the largest particle size. The reverse was the case for the lipospheres formulated with the highest quantity (40%w/w) of PEG 4000 except for batch C4 containing 3%w/w ceftriaxone sodium. In the unloaded lipospheres, increase in PEG 4000 reduced the particle size more than the loaded lipospheres. This shows that drug loading did significantly (p < 0.05) affect the size of the lipospheres. There was higher particle growth between 24 h and 1 month for lipospheres from batches A3, A4, C3, and D2, containing varying quantities of PEG 4000 and the active pharmaceutical ingredient (API) (). Increase in particle size is usually as a result of aggregation and subsequent growth by Ostwald ripening or sintering. Because of the apparent particle size stability obtained after 1 month, these systems could be adjudged to be relatively stable. Microscopic studies show that many disperse systems, e.g. emulsions flocculate a few seconds after preparation (CitationWashington, 1996). Many particle size analyses require a diluted sample, and this dilution in many cases causes flocs to break-up. In this study, dilution of sample was not done and the particle sizes obtained reflect the true sizes of the particles at the time of analysis. Phospholipids alone or in combination with mobile surfactants have been shown to produce very stable disperse systems (CitationAttama et al., 2007). Oral multiparticulate systems show several advantages in comparison with single unit dosage forms, such as more predictable gastric emptying, gastric emptying less dependent on the state of nutrition, high degree of dispersion in the digestive tract, lesser risk of dose dumping, and reduced local irritation (CitationShivakumar et al., 2007). Oral administration of drugs is still preferred to injections by many patients.
Figure 1. Photomicrographs of representative batches of lipospheres containing 30%w/w PEG 4000 after 1 month: A3, B3, C3, and D3 contain 1, 2, 3, and 0%w/w of ceftriaxone sodium, respectively. Bar represents 50 μm.

Figure 2. Time-resolved particle size analysis of the lipospheres containing 10, 20, 30, and 40%w/w PEG 4000 (n= 3): A1–A4, B1–B4, C1–C4, and D1–D4 contain 1, 2, 3, and 0%w/w ceftriaxone sodium, respectively.
pH analysis
The pH of the different batches of lipospheres were measured 24 h, 1 week, and 1 month after preparation to ascertain the variation of pH with time, which could be a function of degradation of the API or excipients. A priori stable API may be affected by degradation of excipients with storage through generation of unfavorable pH (increase or decrease) or reactive species for the API. It is necessary to know the pH of maximum of stability of an API or its stability profile and utilize that in designing a suitable formulation for the API. This will inform the formulation scientist whether to include a stabilizer or not in the new formulation. shows the pH values of the various batches of the ceftriaxone sodium-loaded and unloaded lipospheres. The pH varied from between 6.55 ± 0.12 and 7.96 ± 0.09 within 24 h of preparation to between 3.94 ± 0.23 and 7.56 ± 0.27 within 1 month of preparation, with lipospheres containing 30%w/w PEG 4000 (A3) having the highest reduction in pH after 1 month (pH = 3.94 ± 0.23). From , it is clear that there was a gradual decrease in pH value of lipospheres containing ceftriaxone sodium (API) as well as for the unloaded lipospheres. A 12%w/v solution of ceftriaxone sodium powder has a pH range of 6–8 (CitationBP, 2001), while the pH of the ceftriaxone sodium lipospheres were within the value given for a 1%w/w aqueous solution of ceftriaxone sodium (pH ≈ 6.7). From the overall values, lipospheres loaded with 1%w/w ceftriaxone sodium containing 10%w/w, 20%w/w, 30%w/w, and 40%w/w PEG 4000 (Batches A1–4) had the highest significant (p < 0.05) reduction. The slight decline in the pH values from alkalinity through neutrality to acidity in most of the lipospheres was not attributed to drug degradation since there was also a fall in the pH of the unloaded lipospheres, Ceftriaxone sodium-loaded lipospheres produced high activity after 1 month of storage. Degradation of lipid excipients to free fatty acids may be implicated in the fall to acidic pH. One has to be aware that particle surface pH in some systems may be two-to-three units different from the pH of the bulk because of double layer forces of attraction/adsorption of other ions in the continuous phase (CitationLasic & Templeton, 1996).
Table 2. Result of some physical parameters of the lipospheres.
Encapsulation efficiency (EE%) and loading capacity (LC)
The role of the formulated lipospheres is to deliver the API to the target tissues intact. Thus, the ability of the lipospheres to accommodate active molecules is an important property. It can be expressed by the entrapment efficiency (EE%) and loading capacity. EE% defines the ratio between the weight of entrapped API and the total weight of API added to the dispersion, while LC expresses the ratio between the entrapped API and the total weight of the lipids (Doktorovova & Souto, Citation2009). shows the EE% and the LC obtained for the various batches of lipospheres after 1 month of storage. Both EE% and LC are dependent on several parameters, such as the lipophilic properties of the API, the screening of the most appropriate lipid composition/ratio and surfactant combination, as well as the production procedure used. LC ranging from 3.22 mg of ceftriaxone sodium/100 mg of lipid to 6.36 mg of ceftriaxone sodium/100 mg of lipid were obtained (). These values were somewhat low because ceftriaxone sodium is very soluble in an aqueous system, and the formulation was not lyophilized to obtain dry lipospheres.
As could be seen in , the EE% ranged from 29.52 ± 2.78% for batch C3 lipospheres to 60.09 ± 3.55% for batch A1 lipospheres. The varied EE% may be as a result of API and matrix physicochemical and material characteristics. All the same, higher EE% values were obtained for batch A lipospheres compared with other batches. There was no significant difference (p < 0.05) in the EE% obtained for batches B2, B3, and B4 lipospheres. The EE% decreased as the amount of the polymer (PEG 4000) in the lipid matrix (30%w/w of phospholipon® 90H in Softisan® 154) increased. Some trends were observed in the case of sub-batches 4 (i.e. A4, B4, and C4) which had the highest amount of PEG 4000 (40%w/w) in the lipid matrix yet had a greater efficiency than sub-batches 3 (i.e. A3, B3 and C3). From , A4 (with 40%w/w PEG 4000) had a greater efficiency than A2 and A3 which had 20%w/w and 30%w/w, respectively, of PEG 4000 in the lipid matrix. Also B4 (with 40%w/w of PEG 4000 in lipid matrix) had a greater efficiency than B3 (with 30% of PEG 4000 in lipid matrix). A similar phenomenon was also observed in Batch C. The reason for this is uncertain, but may be related to increase in crystallinity of the lipospheres with increase in PEG 4000 concentration that resulted in difficulty in encapsulation of ceftriaxone sodium. However, this has to be proven by crystallinity studies. Although there was reduction in the EE% with increase in PEG 4000 concentration, there appears to be a limiting concentration of PEG 4000 (30%w/w) beyond which increase in EE% resulted (). This behavior may be due to formation of other structures in the systems, which could favor drug encapsulation. For instance, when added in high concentration, PEG-lipids induce formation of mixed micelles (CitationRyan et al., 2008).
Release studies as a function of inhibition zone diameter (IZD)
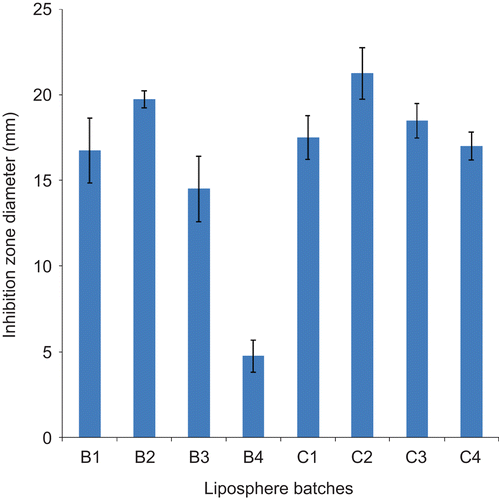
This test was performed to establish that ceftriaxone sodium did not lose activity during formulation and after short-term storage, and was done 1 month after preparation. The result of drug release studies using the agar plate diffusion method is presented in . The determination of IZD using agar plate method is based on the diffusion of an antibiotic agent or formulation thereof through a solidified nutrient agar. From the result presented in , the liposphere formulations gave these zones of inhibition in decreasing order of magnitude: B2 > B1 > B3 > B4 for batch B lipospheres, and C2 > C3 > C1 > C4 for batch C lipospheres. A very small zone of inhibition was observed for liposphere batches formulated with 10, 20, 30, and 40%w/w PEG 4000 and 1%w/w ceftriaxone sodium (A1, A2, A3, and A4), probably because the concentration of drug contained in this batch was low to yield concentrations equal to or above the minimum inhibitory concentration (MIC) to cause significant inhibition. Also batch D, which served as the control, had no inhibition since it contained no drug.
Figure 3. Ceftriaxone release from drug-loaded lipospheres as a function of inhibition zone diameter using E. coli as test organism obtained after 1 month of storage (n= 3): B1–B4 contain 2%w/w ceftriaxone sodium; and C1–C4 contain 3%w/w ceftriaxone sodium.
In vitro permeation studies
Permeation studies carried out on the various batches of lipospheres resulted in different permeation profiles shown in –. There was controlled permeation of ceftriaxone from the lipospheres without burst effect in all the formulations. The amount of ceftriaxone permeated showed capacity limited permeation because the amount depended on the initial drug load, with higher drug loading producing higher ultimate amount permeated. However, within a batch the ultimate amount permeated was not proportional to the amount of PEG 4000 added. In batches B and C lipospheres, sub-batches B3 and C3 containing 30%w/w of PEG 4000 had the highest ultimate amount permeated after 6 h ( and ). For each permeation study the linear ascent of the curve was used to determine the flux J (CitationWinkler & Müller-Goymann, 2002). The flux J of ceftriaxone sodium (μg/cm2s) permeating the artificial membrane in the Franz cell is given by the mass (μg) of the permeant in the receptor solvent (as determined at each sampling time t, by UV analysis) and the membrane diffusion area (cm2). The flux data may be expressed by Fick’s second law of diffusion, which takes into account the initial donor concentration, its partition coefficient between the donor solution and the membrane, the diffusion coefficient of the permeant in the artificial membrane, and the thickness of the membrane. The permeation coefficient P was calculated as the quotient of the flux J, and the initial concentration C, of each sample introduced into the donor compartment of the Franz cell (CitationAzzi et al., 2006). The following permeation coefficient values were obtained: 2.08 × 10−6 cm/s, 1.63 × 10−6 cm/s, 1.41 × 10−6 cm/s, and 1.54 × 10−6 cm/s, respectively, for liposphere batches A1, A2, A3, and A4 containing, respectively, 10, 20, 30, and 40%w/w PEG 4000 and 1%w/w ceftriaxone sodium each; 1.19 × 10−6 cm/s, 1.66 × 10−6 cm/s, 1.88 × 10−6 cm/s, and 1.82 × 10−6 cm/s, respectively, for liposphere batches B1, B2, B3, and B4 containing, respectively, 10, 20, 30 and 40%w/w PEG 4000 and 2%w/w ceftriaxone sodium each; and 8.55 × 10−7 cm/s, 1.66 × 10−6 cm/s, 1.99 × 10−6 cm/s, and 1.92 × 10−6 cm/s, respectively, for liposphere batches C1, C2, C3, and C4 containing, respectievly, 10, 20, 30, and 40%w/w PEG 4000 and 3%w/w ceftriaxone sodium each. These values were within the range obtained for some lipophilic and lipophobic drugs (CitationFriedrich et al., 2005; CitationAttama et al., 2008). Sub-batches B3 and C3 produced the highest p-values within their batches. They show prolonged release of the incorporated drug consistent with findings in earlier studies (CitationDomb, 1995; CitationBekerman et al., 2004; CitationShivakumar et al., 2007).
Figure 4. Permeation profile of ceftriaxone sodium from lipospheres loaded with 1%w/w of drug in SIF (n= 3). A1–A4 contain 10, 20, 30, and 40%w/w PEG 4000, respectively.
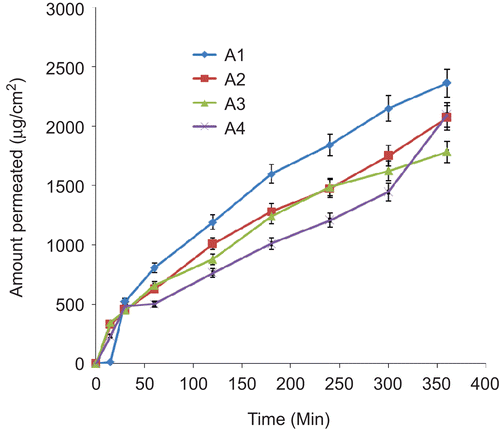
Figure 5. Permeation profile of ceftriaxone sodium from lipospheres loaded with 2%w/w of drug in SIF (n= 3). B1–B4 contain 10, 20, 30, and 40%w/w PEG 4000, respectively.
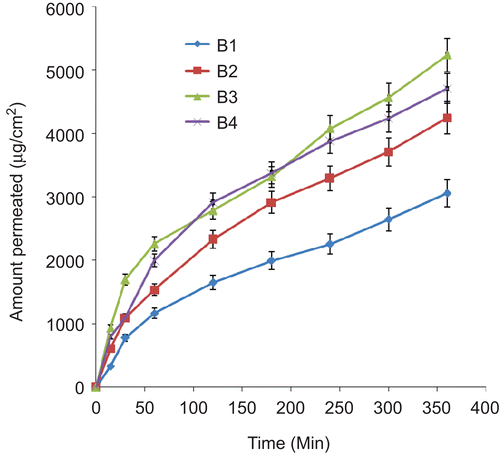
Figure 6. Permeation profile of ceftriaxone sodium from lipospheres loaded with 3%w/w of drug in SIF (n= 3). C1–C4 contain 10, 20, 30, and 40%w/w PEG 4000, respectively.
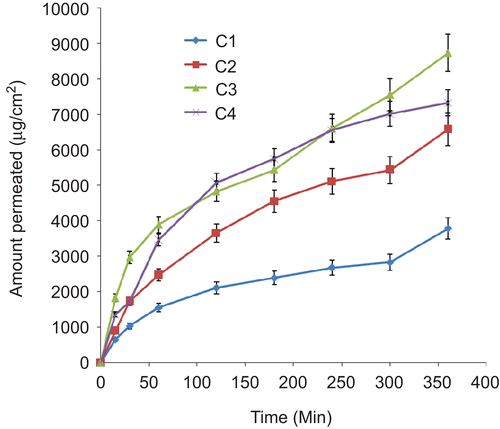
We are concerned primarily with the linear steady-state portion of the permeation plot where an intrinsic equilibrium state of ceftriaxone sodium diffusion is occurring under the test constraint employed in the study. The effect of drug concentration as well as the amount of polymer (PEG 4000) incorporated into the lipid matrix on the permeation coefficient can be obtained from the permeation data. The effect of PEG 4000 concentration on the permeation coefficient can be represented in decreasing order of magnitude of the permeation coefficients: batch A lipospheres: A1 > A4 > A2 > A3, batch B lipospheres: B3 > B4 > B2> B1; and batch C lipospheres: C3 > C4 > C2 > C1. On the other hand, the effect of drug concentration can equally be represented in decreasing order of permeation coefficient: A1 > B1 > C1, C2 > B2 > A2, C3 > B3 > A3, and C4 > B4 > A4. The drug concentration effect on the permeation coefficients tended to be predominantly linear as the batches with highest amount of ceftriaxone sodium gave the highest permeation coefficient. Looking at the effect of PEG 4000 on the permeation coefficients, it was observed that lipospheres formulated with 30%w/w of the polymer had the highest permeation coefficient P, followed by those of 40%w/w, 20%w/w, and 10%w/w, in that order for batches B and C lipospheres. However, a different pattern was observed for batch A lipospheres. The reason for this is uncertain, but may be related to the low amount of API added. Ceftriaxone has been shown to produce good results when used in treatment. For instance, in a recent study, liposome-encapsulated ciprofloxacin was compared with conventional ciprofloxacin and ceftriaxone in a rat model of pneumococcal pneumonia (CitationEllbogen et al., 2003). When the antibiotics were given once daily, ceftriaxone was more effective than either form of ciprofloxacin. However, in this study novel formulation of ceftriaxone sodium was studied in vitro for possible oral delivery of the API.
Conclusions
In designing a drug delivery device such as those based on lipids or combinations thereof, the choice as well as the combination ratio of lipids and other excipients should be considered based on the desired release pattern, route of administration, stability, and other physicochemical considerations. In this research work, the effect of drug concentration on the permeation characteristics was directly related. It was observed that the most concentrated formulation had the highest permeation coefficient. One can suggest that sub-batches 3 (for batches B and C) containing 30%w/w of PEG 4000 in the lipid matrix and 2%w/w and 3%w/w ceftriaxone sodium, respectively, offer the most promising combination as regards permeability, and could be further investigated for oral delivery of ceftriaxone sodium. However, sub-batches 2 (for batches B and C) had the highest activity against E. coli in the microbiological studies. The fall in pH of the unloaded lipospheres and ceftriaxone-loaded lipospheres was attributed to the changes in the excipients used in formulation, but needs further investigation to completely distinguish it from changes in the integrity of the API.
Acknowledgements
We wish to thank Phospholipid GmbH, Köln, Germany for providing the sample of Phospholipon® 90H used in this study.
Declaration of interest: The authors state no conflicts of interest and have received no funding for the research or in the preparation of this manuscript.
References
- Attama, A.A., Nkemnele, M.O. (2005). In vitro evaluation of drug release from self micro-emulsifying drug delivery systems using a novel biodegradable homolipid from Capra hircus. Int J Pharm. 304:4–10.
- Attama, A.A., Reichl, S., Müller-Goymann, C.C. (2008). Diclofenac sodium delivery to the eye: in vitro evaluation of novel solid lipid nanoparticle formulation using human cornea construct. Int J Pharm. 355:307–13.
- Attama, A.A., Schicke, B.C., Paepenmüller, T., Müller-Goymann, C.C. (2007). Solid lipid nanodispersions containing mixed lipid core and a polar heterolipid: characterization. Eur J Pharm Biopharm. 67:48–57.
- Azzi, C., Zhang, J., Purdon, H.C., Chapman, J.N., Nitcheva, D., Herbert, R.J., Smith, W.E. (2006). Permeation and reservoir formulation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and benzo[a]pyrene(B[a]P) across porcine esophageal tissue in the presence of ethanol and methanol. Carcinogenesis 27:137–45.
- Bekerman, T., Golenser, J., Domb, A. (2004). Cyclosporin nanoparticulate lipospheres for oral administration. J Pharm Sci. 93: 1264–70.
- British Pharmacopoeia. (2001). London: Her Majesty’s Stationery Office. p. 275.
- Costas, K. (2006). Recent advances in novel drug delivery system. Azjono J Nanotechnol Online. 2:1–11.
- Craig, D.Q.M. (2004). Lipid matrices for sustained release—an academic overview. Bull Tech Gattefossé. 97:9–19.
- Didier, L.M. (2000). Rationale and application of lipids as pro-drug carriers. Eur J Pharm Sci. 11:243–55.
- Doktorovova, S., Souto, E.B. 2009. Nanostructured lipid carrier-based hydrogel formulations for drug delivery: a comprehensive review. Exp Opin Drug Deliv. 6: 165–76.
- Domb, A.J. (1993). Lipospheres for controlled delivery of substances. Available online at: freepatentonline.com/5188837.html. Accessed October 10, 2008.
- Domb, A.J. (1995). Long acting injectable oxytetracycline-liposphere formulation. Int J Pharm. 124:271–8.
- El-Gibaly I, Abdel-Ghaffar SK. (2005). Effect of hexacosanol on the characteristics of novel sustained-release allopurinol solid lipospheres (SLS): factorial design application and product evaluation. Int J Pharm. 294:33–51.
- Ellbogen, M.H., Olsen, K.M., Gentry-Nielsen, M.J., Preheim, L.C. (2003). Efficacy of liposome-encapsulated ciprofloxacin compared with ciprofloxacin and ceftriaxone in a rat model of pneumococcal pneumonia. J Antimicrob Chemother. 51:83–91.
- Franz, T.J. (1975). Percutaneous absorption- on the relevance of in vitro data. J Invest Dermatol. 64:190–5.
- Friedrich, I., Reichl, S., Müller-Goymann, C.C. (2005). Drug release and permeation studies of nanosuspensions based on solidified reverse micellar solutions (SRMS). Int J Pharm. 305:167–75.
- Hamidi, M., Azadi, A., Rafiei, P. (2006). Pharmacokinetic consequences of PEGylation. Drug Deliv. 13:399–409.
- Hassan, M.A., Mohammed, F.A., Sabour, E.A. (2003). Formulation and evaluation of ciprofloxacin hydrochloride and norfloxacin gel. STP Pharma Sci. 13:195–201.
- Iannuccelli, V., Coppi, G., Sergi, S., Mezzena, M., Scalia, S. (2008). In vivo and in vitro skin permeation of butyl methoxydibenzoylmethane from lipospheres. Skin Pharmacol Physiol. 21:30–8.
- Lasic, D.D., Templeton, N.S. (1996). Liposomes in gene therapy. Adv Drug Deliv Rev. 20:221–66.
- Morkhade, D.M., Joshi, S.B. (2007). Evaluation of gum dammar as a novel microencapsulating material for ibuprofen and diltiazem hydrochloride. Indian J Pharm Sci. 67:263–8.
- Rabasco, A., Gonzalez, R. (2000). Lipids in pharmaceutical and cosmetics preparation. Coden Gracan. 15:114.
- Rocephin®, Drug package insert. Switzerland: Roche.
- Ryan, S.M., Mantovani, G., Wang, X., Haddleton, D.M., Brayden, D.J. (2008). Advances in PEGylation of important biotech molecules: delivery aspects. Expert Opin Drug Deliv. 5:1–13.
- Sasol. 2008. Softisan® 154, Product information. Available online at: http://www.sasolos.com/products/pdf/excipients_pharmaceuticals.pdf, accessed October 10, 2008.
- Shivakumar, H.N., Patel, P.B., Desai, B.G., Ashok, P., Arulmozhi, S. (2007). Design and statistical optimization of glipizide loaded lipospheres using response surface methodology. Acta Pharm. 57:269–85.
- Washington, C. (1996). Stability of lipid emulsions for drug delivery. Adv Drug Deliv Rev.20:131–45.
- Westesen, K., Siekmann, B. (1998). Solid lipid nanoparticles of bioactive agent and method for the manufacture and use thereof. United States Patent 5785 976.
- Winkler A., Müller-Goymann C.C. (2002). Comparative permeation studies for different formulations enriched with 5-aminolevulinic acid and its n-butyl ester through excised human stratum corneum. Arch Pharm Med Chem. 333:67.
- Zhang, X., Gan, Y., Nie, S., Pan, W. (2008). PEGylated nanostructured lipid carriers loaded with 10-hydroxycamptothecin: an efficient carrier with enhanced anti-tumour effect against lung cancer. J Pharm Pharmacol. 60:1077–87.