Abstract
Intranasal (IN) administration is a promising approach for rapid-onset delivery of medications and to circumvent their first-pass elimination when taken orally. Metoclopramide Hydrochloride (MET HCl) is a potent antiemetic, effective even for preventing emesis induced by cancer chemotherapy. The feasibility of developing an efficacious intranasal formulation of metoclopramide has been undertaken in this study. Formulations were modulated so as to have gelation at physiological ion content after intranasal administration. Gelation was determined by physical appearance. The mucoadhesive force in terms of detachment stress, determined using sheep nasal mucosal membrane, increased with increasing concentration of carbopol. The results of in vitro drug permeation studies across sheep nasal mucosa indicate that effective permeation could be significantly increased by using in situ gelling formulation with carbopol concentration 0.15% or greater. Histological examination did not detect any damage during in vitro permeation studies. Finally, the bioavailability study in rabbits revealed that the absolute bioavailability of MET HCl was significantly increased from 40.67% in the case of the oral drug solution to 54.61% in the case of the nasal in situ gel. This study points to the potential of mucoadhesive nasal in situ gel in terms of ease of administration, accuracy of dosing, prolonged nasal residence and improved drug bioavailability.
Introduction
The past decade has been marked by a steadily growing interest using the nasal route for systemic delivery. Compounds ranging in size from simple, traditional drug molecules of ∼ 200–300 Da to complex proteins with molecular weight exceeding 100,000 have been investigated. The nasal delivery appears to be desirable alternative to the parenteral medication because of the existence of a rich vasculature and a highly permeable structure within the nasal membranes (CitationBehl et al., 1998). The well perfused nasal mucosa provides an excellent site for rapid absorption of centrally acting drugs such as midazolam for the control of epileptic seizures, morphine, and ketamine for the treatment of pain and dihydroergotamine for the relief of migraine (CitationIllum, 2003). In addition, nasal application circumvents first pass elimination and/or degradation in GI tract, and may be employed routinely without any pain (CitationHussain, 1998). Furthermore, studies demonstrated the existence of direct transport from the nasal cavity to the cerebrospinal fluid and proceeding to the brain. From a pharmacokinetic standpoint, after intranasal administration absorption is rapid due to the existence of a rich vasculature and a highly permeable structure within the nasal membrane which should provide faster onset of action as compared to per oral administration. Hence, intranasal delivery could be especially important in the management of crisis situations such as severe nausea and vomiting (CitationChein, 1992).
Metoclopramide Hydrochloride (MET HCl) is a potent antiemetic, effective in the treatment of nausea and vomiting associated with cancer therapy, pregnancy, migraine, etc. Oral bioavailability of MET HCl is highly variable, showing values between 32–98% due to extensive pre-systemic metabolism (CitationHarrington et al., 1983). Oral forms of MET HCl often get vomited out before systemic absorption. Parenteral or rectal administration results in low patient compliance. In this regard, the intranasal delivery seems to be an attractive alternative. The major disadvantage associated with nasal drug delivery is rapid mucociliary clearance (MCC) that limits the time available for drug absorption from applied dosage form (CitationUgwoke et al., 2001). The half-life of clearance for both liquid and powder formulation that are not mucoadhesive is in the order of 15–20 min (CitationIllum, 1999). Therefore, a plausible strategy is to decrease MCC by the use of gel/mucoadhesive formulations to prolong the residence time at the nasal absorption site and thereby facilitate the uptake of drug. Ordinary gels are difficult to administer and an accurate drug dose cannot be measured. Mucoadhesive powders are not highly favored products. They can cause irritation on the nasal mucosa and give a gritty feel to the tissue, besides the difficulty and the cost of manufacturing powders with specified morphology. A nasal mucoadhesive in situ gel appears very attractive since it is fluid like prior to nasal administration and thus can be easily be instilled as a drop, allowing accurate drug dosing.
Gellan gum is an anionic heteropolysaccharide produced by aerobic fermentation of the bacterium sphingomonas eloda (formerly known as pseudomonas eloda) (CitationKedzierevicz et al., 1999). The chemical structure made up of repeating units of tetrasaccharide is composed of β-D-glucose, β-D-glucoronic acid, α-L rhamnose residues in the molar ration of 2:1:1. Because of its ability to form strong clear gels at physiological ion concentration, it can provide a longer contact time for drug transport across the nasal membrane before the formulation is cleared by mucociliary clearance mechanism. These features, along with bioadhesivity, biodegradability, biocompatibility and absence of toxicity of this polymer, attracted widespread interest in gellan gum as an ion responsive gelling agent. The aim of the present study is to develop a MET HCl, mucoadhesive nasal in situ gel using an ion-sensitive polymer. The optimized nasal in situ gel with favorable gelation, rheological behavior, release and mucosal permeation ability was selected for further bioavailability study.
Materials and methods
Materials
Metoclopramide Hydrochloride was a kind gift from IPCA Laboratory (India Rep Office, Andheri, India), and Gellan Gum (Deacetylated) a gift sample from CPKelco division of the Monsanto Company (Mumbai, India). Carbopl 934P NF was a gift sample from Lubrizol Inc. (Colaba, India). All chemicals were of analytical grade and used without any purification.
Methods
Preparation of in situ gels
Gellan gum was weighed and dispersed in distilled water. The dispersions were then stirred for 20 min at 100°C in a water bath and then cooled to room temperature. Metoclopramide hydrochloride (2.5% w/v) was added during a cooling process. Carbopol 934P (as shown in ) was added slowly with stirring. Appropriate quantities of mannitol and benzalkonium chloride were also added simultaneously. Final pH of formulations was adjusted to between 4.5–5.5; by using 0.1N HCl. display compositions of all the formulations. Formulations were filled in 10-mL amber colored glass vials, capped with rubber closures, and sealed with aluminum caps. In their final pack, the formulations were terminally sterilized by autoclaving at 121°C and 15 Pa for 20 min. Sterilized formulations were stored in a refrigerator (4–8°C) until use.
Table 1. Composition of in situ gels.
Evaluation of prepared in situ gels
Gelation studies
Gellan gum is known to undergo gelation in the presence of cations. Gelation is the process by which the liquid phase makes a transition into gel. A 10-mL transparent vial containing a magnetic bar and each formulation (2 mL) was placed on a magnetic stirrer. The Simulated Nasal Fluid, SNF (aqueous solution containing 8.77 mg/ml NaCl, 2.98 mg/ml KCl and 0.59 mg/ml CaCl2 per liter), having the cationic composition of nasal secretions, was added slowly while stirring. The gelation point was determined when the magnetic bar stopped moving due to gelation. The consistency of formed gel was checked and graded, as indicated in .
Table 2. Degree of gelation, gel strength, and viscosity determination.
Viscosity measurements and rheological behavior studies.
Viscosities of formulations before and after gelation were measured by a Brookfield DV-E viscometer using Spindle number 3 at 100 rpm shear rate. Rheological behavior of resultant gels was evaluated by treating viscosity reading using the following pair of equations (CitationMitchka, 1982).
where τi = shear stress, Kατ = conversion factor (0.279 for spindle number 3), and α = torque dial.
where γi = shear rate, n = flow index of fluid (computed from slope of log τi vs log N), knγ = conversion factor (taken on the basis of obtained values of n), and Ni = rotational speed.
Gel strength determination
It is expressed in terms of time (in seconds) required by a 35 g piston for penetration of 5 cm distance, through the 50 g gel formulation. This test was performed using ‘Gel strength apparatus’ modified at the laboratory as mentioned by CitationYong et al. (2001). Gellan carbopol solution (50 g) was placed in a 100 ml-measuring cylinder and gelation was induced by SNF. The apparatus for measuring gel strength (weight: 35 g) was then placed onto the gel. The gel strength was measured as the time (in seconds) required to move the apparatus 5 cm down through the gel. In cases that took more than 10 min to drop the apparatus into the gel, various weights were placed on top of the apparatus and gel strength was described by the minimal weights that pushed the apparatus 5 cm down through the gel.
Mucoadhesive strength measurement.
The mucoadhesive potential of each formulation was determined by measuring the force required to detach the formulation from nasal mucosal tissue using a modified method described by CitationMurthy et al. (2006). In brief, nasal tissues were carefully removed from the nasal cavity of sheep obtained from the local slaughter house. Tissues were immediately used after separation. At the time of testing, a section of nasal tissue was secured (keeping the mucosal side out) to the upper probe using a cyanoacrylate adhesive. The upper probe was attached to a pre-calibrated force displacement transducer SS12LA (BIOPAC Systems Inc., USA) connected to the Student’s Physiograph apparatus. The surface area of each exposed mucosal membrane was 4.2 cm2. At room temperature, fixed amount of samples of each formulation were placed on the lower probe. The probes were equilibrated and maintained at 37°C. The probe with nasal tissue was lowered until the tissue contacted the surface of the sample. Immediately, a force of 0.1 N was applied for 2 min to ensure intimate contact between the tissues and the samples. The probe was then moved upwards at a constant speed of 0.15 mm/s. The bioadhesive force, expressed as the detachment stress in dyne/cm2, was determined from the minimal weights that detached the tissues from the surface of each formulation using the following equation.
where m is the weight added to the balance in grams; g is the acceleration due to gravity (taken as 980 cm/s2); and A is surface area of mucosal tissue in cm2.
Optimization of initial contact time.
The effect of varying contact time (1, 2, 3, 5, and 10 min) was investigated for some of the gel preparations to optimize initial contact time. Formulations were allowed to be in contact with nasal mucosa for calculating contact time (1, 2, 3, 5, and 10 min) and bioadhesive force was determined as described above. Contact time that resulted in maximum strength was selected as optimum contact time required for adequate adhesion.
In vitro release study
In vitro drug release studies were performed using a Franz diffusion cell with dialysis membrane (cut-off Mw = 12,000). The receptor compartment contained 16 ml phosphate buffer solution (PBS), pH 6.6, that was within the pH range in nasal cavity and maintained at 37 ± 0.5°C. The donor compartment contained 3 ml of simulated nasal fluid. After an equilibration of membrane, formulation equivalent to 2.5 mg of metoclopramide hydrochloride was placed in the donor compartment. At set time intervals, 0.5 ml samples were withdrawn from the receiver compartment and concentration of metoclopramide hydrochloride was determined spectrophotometrically at 273 nm (Shimadzu, UV-1700, Japan). The buffer contained in the receiving compartment was replaced with an equal quantity to maintain a constant volume (CitationCerchiara et al., 2005).
Analysis of drug release data.
The drug release from the dosage form is affected by the polymer type and other formulation parameters. For determining the drug release mechanism from gel matrices; the release data from in vitro release studies was analyzed by the commonly used exponential Korsemeyer-Peppas equation as follows:
where Mt/M is the fraction of drug released at time t, k is the release rate constant, and n is the diffusion exponent indicating the release mechanism.
When n is equal to 0.5, the drug release is with a fickian diffusion mechanism (Higuchi model). If 0.5 < n > 1 this indicates anomalous or non-fickian release, while if n = 1 this indicates zero order release (CitationPeppas, 1985).
In vitro permeation studies.
Fresh nasal tissues were carefully removed from the nasal cavity of sheep obtained from the local slaughterhouse. Tissue samples were placed in Franz type diffusion cells displaying a permeation area of 0.785 cm2. The receiver compartment containing 16 ml of PBS pH 6.6 was maintained at 37 ± 0.5°C. After a pre-incubation time of 20 min, pure drug solution and formulation equivalent to 2.5 mg of Metoclopramide hydrochloride was placed in the donor chamber containing 3 ml of SNF. At pre-determined time interval samples were withdrawn from the receiver compartment, replacing the sampled volume with PBS pH 6.6 after each sampling, for a period of 4 h. The samples withdrawn were filtered and used for analysis. The amount of permeated drug was determined using a UV-visible spectrophotometer at 273 nm.
Permeability coefficient (p) was calculated by the following formula:
where, dQ/dt is the flux or permeability rate (mg/h), C0 is the initial concentration in the donor compartment, and A is the effective surface area of nasal mucosa.
Histological examination of nasal mucosa
Histopathological evaluation of tissue incubated in PBS (pH 6.6) after collection was compared with tissue incubated in the diffusion chamber with gel formulations. Tissue was fixed in 10% buffered formalin (pH 7.2), routinely processed and embedded in paraffin. Paraffin sections (7 μm) were cut on glass slides and stained with hematoxylin and eosin (HE). Sections were examined under a light microscope, to detect any damage to the tissue during in vitro permeation study.
In vivo study
The animal experiment was carried out in compliance with the protocol of the Institutional animal ethical committee (Registration No: 651/02/C/CPCSEA under CPCSEA, India).
Six New Zealand white rabbits with mean weight of 2.5 ± 0.3 kg were used. The rabbits were accommodated to the dosing for 1 month before the study to prevent withdrawal and defense reaction that may lead to inaccurate dosing. The rabbits were kept in a single cage and fasted for 12 h before the study with free access to water during the experiments. A cannula was inserted into the marginal ear vein for blood sampling and flushed with heparinized normal saline solution.
Study design.
New Zealand white rabbits were selected as an experimental model because they provide a well controlled animal model for screening the nasal absorption potential of nasal formulations (CitationDoneti et al., 1995, CitationDesai et al., 1998). In a cross-over study with 1 week apart as a wash out period, 400 µl nasal in situ gel (equivalent to 10 mg MET HCl) was deposited into the both nostrils (200 µl into each nostril). The animals also received 5 ml of oral drug solution (equivalent to 10 mg MET HCl) by an oral tube as well as i.v.bolus of Reglan® (10 mg/2 ml) injected into their marginal ear vein.
Sample collection and analysis
After administration of the different formulations, blood samples (1.5 ml) were collected at time intervals of 5, 10, 20, 45, 60, 90, 180, and 240 min from the marginal ear vein of the rabbits. Blood samples were allowed to clot and then centrifuged at 3000 rpm for 15 min. The obtained serum samples were deep frozen at −20°C pending HPLC analysis. At the time of analysis, 1 ml of methyl paraben (IS) solution in methanol (10 µg/ ml) and 1 ml acetonitrile were added to 1 ml of each thawed serum sample. The treated samples were vortex mixed for 2 min, centrifuged at 3000 rpm for 15 min, filtered through a nylon membrane filter (0.45 µm), and degassed by ultrasonication for 10 min thereafter; 20 µl were injected into a HPLC column (C-18, Reverse phase column, Eclipse XDB, 5 µm, 4.6 mm × 150 mm, Agilent 1200 series, Singapore). The mobile phase consisted of methyl alcohol:water (65:35) v/v and acetic acid to adjust pH at 3.5. The mobile phase was delivered into the HPLC apparatus at flow rate of 1 ml/min. (Quaternary Pump, Model G1354A, Agilent 1200 series). The detection wavelength was 254 nm (Ultra Violet variable wavelength detector, Model G1315D, Agilent 1200 series Diode Array Detector) (CitationRadwan, 1998).
Data treatment and statistical analysis
Results from HPLC analysis were plotted as plasma drug concentration vs time. Non-compartment pharmacokinetic parameters including Tmax, Cmax, and AUC were estimated by Kinetica 5.0® computer program. The AUC values for each curve were calculated from tome zero to the last data point using the trapezoidal rule with extrapolation to infinity. The AUC0–∞ values obtained from curve were used to calculate the absolute bioavailability.
Results
Preparation of in situ gels
Gellan gum, an anionic polysaccharide, forms gels in the presence of cations, hence carbapol 934P, anionic mucoadhesive polymer was added to improve mucoadhesion. Mannitol and benzalkonium chloride were used as tonicity adjusting agent and preservative, respectively.
Gelation studies
Gelation studies were carried out using SNF. In these studies the gelling capacity (speed and extent of gelation) for all formulations were determined. Thus, the in situ formed gel should preserve its integrity without dissolving or eroding so as to localize the drug at absorption site for extended duration.
Gelation characteristics was assessed on an ordinal scale ranging between – and ++++, as shown in . After easy instillation into the nasal cavity the liquid polymeric solutions should undergo a rapid sol-to-gel transition by means of ionic gelation. The composition of nasal electrolyte was rich in cationic content. Our objective was to develop a nasal ion sensitive gel with anionic polymers. It would behave as a liquid at room temperature for ease of administration and accurate measurement of the dose but sets into a gel with increased residence time at the lower physiological cations contents. All gellan gum formulations showed instantaneous gelation, depending upon the polymer concentration. Formulation GM1, GM2 showed weakest gelation while GM5 showed very stiff gelation, hence excluded from further studies. Formulations GM3 and GM4 showing good gelation were further considered for combination with carbopol. Carbopol did not affect the gelation in concentration range 0.05–0.2% w/v. Further increases in carbopol concentration lead to the formation of a stiff gel which could be undesirable for nasal delivery. Hence, 0.2% w/v carbopol with 0.3% w/v gellan GM3C3 was selected as optimized formulation.
Viscosity measurements and rheological behavior studies
Apparent viscosity values were measured for liquid formulations and gel using Brookfield viscometer DV-E with spindle no 3 at 100 rpm. The results showed a marked increase in viscosity in all formulations after sol-to-gel transition. The apparent viscosity of both solutions and gel formulations was found to be proportionate to the increase in polymer concentration. All gels exhibited non-Newtonian flow and exhibited shear thinning behavior (). It was observed that use of mucoadhesive polymer did not alter the shear thinning behavior of gellan gum in situ gels. Concerning the viscosity of the formulated in situ gels, polymer concentration substantially contributed to the enhancement of viscosity. The gels studied exhibited shear thinning behavior after sustained shearing, i.e. as the shear rate increased the measured viscosity decreased. The difference in gel properties measured in vitro will influence the efficacy of their spreading and retention in vivo. Shear thinning behavior will increase spread ability of gel (CitationOwen et al., 2000). This is advantageous as it increases the solutions tendency to stay in place after development.
Figure 1. Rheological behavior of gel formulation (GM3C3).
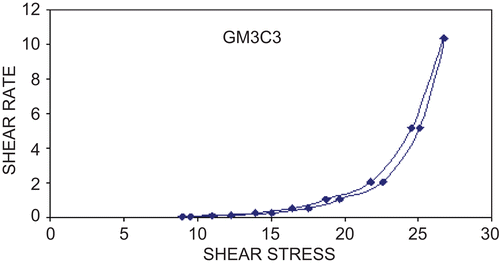
Gel strength
In the development of a nasal in situ gelling system, the gel strength is important in finding the condition, which allows easy administration as droplets and delays the post-nasal drip or anterior leakage. The gel strength was found to be affected by concentrations of gelling and bioadhesive polymers. Optimal in situ gel must have suitable gel strength so as to be administered easily and can be retained at nasal mucosa without leakage after administration. In the Gellan gum gels carbopol was found to increase the gel strength. The gel strength values between 25–50 s were considered sufficient. The gel strength less than 25 s may not retain its integrity and may erode rapidly while gels having strength greater than 50 s are too stiff and may cause discomfort to the mucosal surfaces.
The GM3C2 and GM3C5 formulation series showed the gel strength values in the range 35–40 s () which are acceptable.
In vitro mucoadhesion
All formulations were subjected to in vitro mucoadhesion studies by the method reported by CitationMurthy et al. (2006). displays the comparison of mucoadhesion force of formulations. The mucoadhesion force is an important parameter for in situ gelling nasal formulations, since it prolongs the nasal clearance of gels and increases its residence time in nasal cavity. The reinforcement of the mucoadhesive forces in the nasal in situ gels by the use of mucoadhesive polymers could be explained by the fact that secondary bond forming groups (hydroxy, ethoxy, and amine) are the principle source of mucoadhesion. The bioadhesive force is known to be dependent on the nature and concentration of bioadhesive polymers. The stronger the bioadhesive force the more is the nasal residence time. However, the strong mucoadhesion to nasal mucosal membrane might not actually be advantageous. The rapid mucus turnover is one of the most important protective mechanisms in the nose, and if the dosage form is extensively bound to the mucus it might be cleared from the mucosa at the same speed as that of the mucus.
Figure 2. Mucoadhesion studies of all formulations.
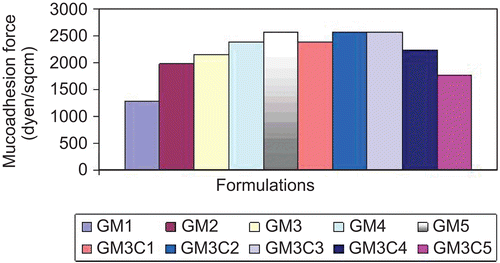
Combination of carbopol with 0.3% w/v gellan gum solution showed better mucoadhesion as compared to that in 0.4% Gellan gum formulation series; 0.2% w/v carbopol showed the highest mucoadhesion. Further increase in the carbopol 934P concentration reduced the mucoadhesive force.
Two minutes of initial contact time gave optimum mucoadhesive strength. Further increase in contact time did not affect the mucoadhesive strength, whereas decreased contact time resulted in increased mucoadhesive strength resulting from sufficient time for entanglement of polymer chains with mucin. Assessment of the mucoadhesive strength in terms of detachment stress showed that the gellan gum formulation possessed adhesive properties that increased with the addition of carbopol 934 concentration. Earlier work with carbopol polymers has clearly indicated that it is the availability of carboxyl groups that determines bioadhesion. Carboxylic groups that gradually undergo hydrogen bonding with sugar residues in oligosaccharide chains in the mucus membrane, resulting in the formation of a strengthened network between polymer and mucus membrane. Thus, carbopol with high density of hydrogen bonding groups would be able to interact more strongly with mucin glycoprotein. Carbopol may also adopt more favorable macromolecular confirmation with increased accessibility of its functional groups for hydrogen bonding.
In vitro drug release study
In vitro drug release studies of formulations were performed using the Franz diffusion cell using dialysis membrane, phosphate buffer of pH 6.6 was used as diffusion media. Releases profiles of formulations are elaborated in Initial rates of release of drug were very rapid due to incomplete gel formation, but as the time progressed the release rate decreased due to complete gel formation. With increase in concentration of carbopol the release rates were found to decrease gradually.
Figure 3. Percent cumulative drug release from formulations.
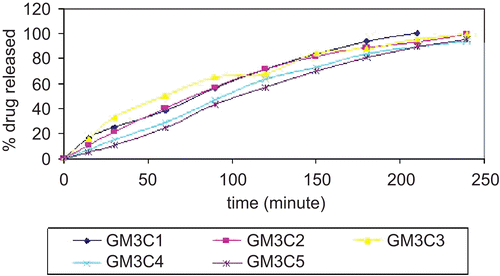
The release profiles exhibited an inflection point, which indicated the gel formation in the donor compartment of the diffusion cell. During gel formation, a portion of drug might be loaded into the gel matrix, thus the cross-linking of polymer reduces the drug release rate. The initial rapid release of metoclopromide might be due to formation of pre-hydrated matrix containing water filled pores due to the presence of aqueous vehicle. The results showed that the formed gels had the ability to extend the release of metoclopromide for the duration of ∼ 3 h. In vitro release study indicated that the release of drug varied according to the type and concentration of polymers. The results further showed that the amount of the drug released in the first hour decreased with the increasing polymer concentration, and this pattern continued for the entire duration of study. Formulations of gellan gum showed the complete drug release up to duration of 180 min. Release profiles were overlapping to that of aqueous solution of metoclopromide.
Mathematical treatment to in vitro release data
On the basis of gelation properties, gel strength, viscosity, mucoadhesion, percent drug content, in vitro diffusion profile, the optimum in situ gelling formulation of gellan gum, and carbopol 934P combination ‘GM3C3’ was selected and subjected for further studies.
The graph between cumulative drug release and square root of time showed an almost linear relationship after the initial period. It was not possible to correlate the release in the early stages of drug release due to incomplete gel formation. After the complete gel formation, the release profiles were found to be linear with square root of time and followed Higuchi’s equation. The drug transport mechanism of the same formulation was determined by using the Korsmeyer-Peppas exponential equation. [(Mt/M) = ktn]. Form the plot of log (Mt/M) vs log of time, the kinetic parameters n and k were calculated. The slope of the plot was observed to be 0.9148, indicating the anomalous (non-fickian) release kinetic, i.e. release of Metoclopramide HCl followed erosion-diffusion mechanism (Peppas & Shalin, Citation1989).
Ex vivo permeation
Formulation GM3C3 was further subjected to ex vivo permeation studies using the sheep nasal mucosa. The percent drug permeated after 4 h was found to be 89.5%. The permeability coefficient (P) was also calculated and found to be 0.5681 cm/h.
Histological examination of nasal mucosa
The successful use of the mucoadhesive nasal delivery system is not only limited to their bioadhesive efficacy but equally important is their safety. Hence it was important to investigate the safety of the optimized in situ gel formulation (GM3C3). The histological study revealed the safety of formulation on sheep nasal mucosa. Photomicrographs of sheep nasal mucosa after the permeation studies were observed for histopathological changes in comparison with the untreated mucosa. Results are displayed in . The section of mucosa treated with formulation GM3C3 showed very slight degeneration of nasal epithelium along with slight erosion. There was increased vascularity in basal membrane and the superficial part of the sub-mucosa as compared with untreated mucosa. This might be the result of the mucoadhesive and permeability enhancing properties of carbopol in the formulation. There was no sign of remarkable destructive effect of formulations on the treated nasal mucosa.
Figure 4. Photomicrographs of sheep nasal mucosa subjected to histological examination.
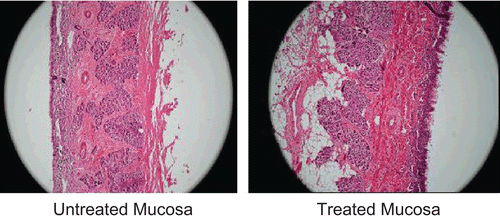
In vivo study
In the present study the rabbits remained conscious through the experiment, thereby providing functional mucociliary transport function. Usual anesthetizing agent could affect the in vivo absorption of drug (CitationMayor & Illum, 1997), therefore we avoided anesthesia.
The pharmacokinetic parameters were determined for the optimized formulation (GM3C3) as compared to the peroral solution. The mean serum drug concentration time profile after administration of the I.V. and oral solutions as well as the nasal in situ gel of metoclopramide are illustrated in (mean serum concentration vs time). From this profile it is clear that higher serum drug levels are achieved in the case of nasal in situ gel as compared to the oral drug solution. shows the pharmacokinetic and bioavailability data for the different formulations. The Cmax values were 3.35 ± 0.22 μg/ml for the nasal in situ gel while it was 2.49 ± 0.13 μg/ml for the oral solution; however this difference was significant at p < 0.05. Moreover the AUC0–∞ values were 693.17, 282.17, and 378.92 μg/ml min for the I.V solution, oral solution, and the nasal in situ gel, respectively. These values corresponds to absolute bioavailability (Fabs) of 40.67 ± 2.21% and 54.61 ± 6.33% for oral and nasal in situ gel, respectively, which was statistically significant (p < 0.05).
Table 3. Comparative pharmacokinetic parameters of metoclopramide hydrochloride following administration of intravenous and oral solution and nasal in situ gel in rabbits.
Figure 5. Serum concentration– time profile following administration of intravenous, oral solution and nasal in situ gel of MET HCl in rabbits.
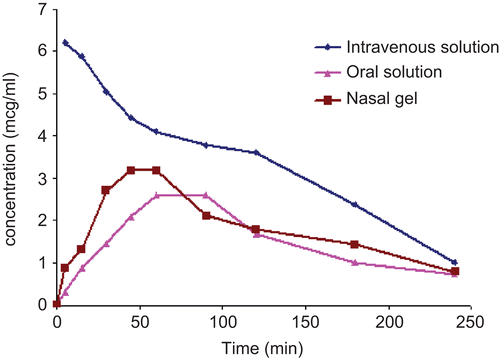
The pharmacokinetics of the optimized formulation (GM3C3) was studied in rabbits. Results revealed that higher serum MET HCl levels were achieved in the case of the nasal in situ gel as compared to the oral drug solution as indicated by the Cmax and AUC values. The superior absolute bioavailability of the nasal in situ gel (54.61%) compared to the oral solution (40.67%) clearly indicated higher absorption of MET HCl when administered intranasally. MET HCl is a class I drug according to the biopharmaceutical classification system (CitationAmidon et al., 1995); the significantly higher bioavailability of its intranasal gel as compared to its oral solution could therefore be attributed avoiding the first pass metabolism associated with peroral drug administration. Similar results were reported by CitationHussain et al. (1980) working on propranolol. Furthermore, CitationMurthy et al. (2006) demonstrated a significant increase in the in vitro sumatriptan permeation across sheep nasal mucosa by using poloxomer in situ gelling formuation containing 0.3% carbapol.
Consistently, the situ gel developed in the present study was able to retain the drug in nasal cavity for a time long enough to be absorbed nasally. Meanwhile, the nasal mucosal permeation enhancement effect of both gellan gum and carbopol, ingredients of the in situ gel, cannot be ruled out. Gellan gum might enhance permeation across nasal mucosa by increasing contact time after gelation. As far as carbopol is concerned, the enhancement of permeation could be attributed to carbopol mediated tight junctional modulation.
Conclusion
The nasal delivery system of MET HCl formulated in this study was feasible for nasal administration, and was expected to rapidly exert its antiemetic effect. Moreover, the optimized in situ gel demonstrated an enhanced bioavailability of MET HCl as compared to the oral drug solution The most prominent advantage of the in situ gel is that it is fluid-like prior to contact with the nasal mucosa; this feature is satisfactory for convenience of administration for patients, maintaining accuracy of drug dosing, and avoidance of the bitter taste of the antiemetic drug.
Acknowledgements
The authors are grateful to Principal, R.C. Patel Institute of Pharmaceutical Education and Research, Shirpur, India for providing facilities to carry out present research work.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Amidon, G.L., Lennernas, H., Shah, V.P., Crison, J.R. (1995). A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 12:413–20.
- Behl, C.R., Pimplaskar, H.K., Sileno, J., Romeo, V.D. (1998). Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev. 29:89–116.
- Cerchiara, T., Luppi, B., Chidichimo, G., Bigicci, M., Zecchi, V. (2005). Chitosan and poly (methyl vinyl ether-co-maleic anhydride) microparticles as nasal sustained delivery systems. Eur J Pharm Biopharm. 61:195–200.
- Chien, Y.W. (1992). Nasal drug delivery systems. In: Swacrbrick, J., ed. Novel drug delivery systems. New York: Marcel Dekker.
- Desai, S.D., Balchandra, J. (1998). In vitro evaluation of pluronic F127-based controlled release ocular drug delivery system for pilocarpine. J Pharm Sci. 87:226–230.
- Doneti, P., Zia, H., Needham, T.E. (1995). In vivo evaluation of spray formulation of human insulin for nasal delivery. Int J Pharm. 122:91–105.
- Harrington, R.A., Hamilton, C.W., Brogden, R.N., Linkewich, J.A. (1983). Heel RC. Metoclopramide: an updated review of its pharmacological properties and clinical use. Drugs. 25:451–94.
- Hussain, A., Poster, T., Hirai, S., Kashihara, T., Jones, M. (1980). Nasal absorption of propranolol in humans. J Pharm Sci. 69:1240–3.
- Hussain, A.A. (1998). Intranasal drug delivery. Adv Drug Deliv Rev. 29:39–49.
- Illum L. (1999). Bioadhesive formulations for nasal peptide delivery. In:Mathiowitz E, Chickering DE, Lehr DE, eds. Bioadhesive drug delivery systems. New York, NY: Marcel Dekker. 507–539.
- Illum, L. (2003). Nasal drug delivery—possibilities, problems and solutions. Contr Rel. 87:187–98.
- Kedzierevicz, F., Lombry, C., Rios, R., Hoffman, M., Manicent, P. (1999). Effect of the formulation on the in-vitro release of propranolol from gellan beads. Int J Pharm. 178:129–36.
- Mayor, S.H., Illum, L. (1997). Investigation of the effect of anesthesia on nasal absorption of insulin in rats. Int J Pharm. 149:123–9.
- Mitchka, P. (1982). Simple conversion of Brookfield R.V.T readings into viscosity functions. Rheol Acta. 21:207–9.
- Murthy, R.S.R., Majithiya, R.J., Ghosh, P.K. (2006). Thermoreversible-mucoadhesive gel for nasal delivery of Sumatriptan. AAPS PharmSciTech. 7:E1–7.
- Owen, D.H., Peters, J.J., Katz, D.F. (2000). Rheological properties of contraceptive gels. Contraception. 62:321–6.
- Peppas, N.A. (1985). Analysis of fickian and non fickian drug release from polymers. Pharm Acta Helv. 60:110–1.
- Peppas, N.A., Shalin, J.J. 1989. A simple equation for the description of solute release III. Coupling of diffusion and relaxation. Int J Pharm. 57:169–72.
- Radwan, M.A. (1998). Determination of metoclopramide in serum by HPLC assay and its application in pharmacokinetic study in rat. Anal Lett. 31:2397–410.
- Ugwoke, M.I., Verbeke, N., Kinjet, R. (2001). The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J Pharm Pharmacol. 53:3–22.
- Yong, C.S., Choi, J.S., Rhee, J.D. (2001). Effect of sodium chloride on the gelation, gel strength and bioadhesive force of poloxamer gels. Int J Pharm. 275:195–205.