Abstract
A novel multilamellar vesicular delivery system was developed for the controlled release application. Multilamellar vesicles were prepared by thin film hydration and converted into proliposomes by freeze-drying. A model drug metoclopramide, a highly hydrophilic drug, was successfully encapsulated into proliposomes. The proliposomes produced were non-sticky, free-flowing powders. The proliposomes were formulated into a unit dosage form by combining with various excipients. The effect of different compositions such as type and concentration of phospholipid or hydrophilic polymer was investigared to optimize the formulation. The formation of multilamellar vesicles was confirmed by observing the process of hydration of proliposomes under an optical microscope. The spherical shape of vesicles was confirmed by transmission electron microscopy (TEM) and mean particle sizes were in the range of 1.3–2.5 µm, as measured by dynamic light scattering technique. Differential scanning calorimetry (DSC) study of formulations was conducted to understand the crystalline nature of drug in the vesicles. The results indicated a molecular level dispersion of drug into proliposomes with encapsulation efficiency up to 43%. Critical formulation parameters were identified to obtain a near zero order in vitro release pattern. Proliposomal formulations produced were suitable as multiparticulate drug delivery systems for the controlled release of a highly hydrophilic molecule.
Introduction
The encapsulation of drugs in liposomes is valuable in reducing toxicity and improving the therapeutic effectiveness of a wide variety of drugs, products such as AmBisome® or Doxil® are successful at clinically achieving these goals for parenteral administration of amphotericin B and doxorubicin, respectively (CitationSoppimath & Betageri, 2008). The drug release from Doxil® liposomes was reported to be slow and follows first order kinetics, demonstrating controlled release of encapsulated drug (CitationElbayoumi & Torchilin, 2008). Drug release from liposomes depends on the lipophilicity of drug molecule and lipid composition other than the type of liposomes. It was reported that multilamellar vesicles (MLV) have shown barrier property for the release of encapsulated water-soluble drug, e.g. atenolol, and retarded the drug release compared to drug encapsulated in small unilamellar vesicles (SUV) (CitationBetageri & Parsons, 1992). Drug release from MLV has two main points of interests. First, phospholipid bilayers may simulate some fundamental properties of biological membranes and constitute a model system to investigate passive drug transport. Second, there is a possibility of application of these systems for sustained or controlled drug delivery application, mainly for non-parenteral route of administration (CitationBetageri & Parsons, 1992).
Liposomal formulations offer numerous advantages andare extensively studied for parenteral drug delivery and topical application in the cosmetic industry. In recent years, a greater understanding of gastrointestinal (GI) physiology, uptake mechanisms, and promotion of the oral delivery of colloidal dosage forms such as microparticles, nanoparticles, and vesicles have led to a number of novel concepts for oral drug delivery, especially for vaccines (CitationChen et al., 1996; CitationDiBiase & Morrel, 1997; CitationFlorence, 1997; CitationRogers & Anderson, 1998). Oral controlled release multiple unit dosage forms such as microparticles, beads, and pellets are becoming more popular than conventional single unit dosage forms due to their added advantages (CitationAgnihotri & Aminabhavi, 2004). Once the multiparticles are released from the tablet or capsule, uniform distribution of these multi-unit dosage forms along the GIT could result in more reproducible drug absorption and reduced risk of local irritations (CitationGaleone et al., 1981). Also multiparticulate unit dosage forms allow more flexibility in modulating the drug release and can be easily filled into hard gelatin capsules or compressed into tablets for the ease of administration (CitationAgnihotri & Aminabhavi, 2004). The formulation of multiparticulates into tablets has the advantage of preventing tampering, as hard gelatin capsules have been tampered (Tylenol®) (CitationBodmeier, 1997).
Oral vesicular formulations have been criticized because of their instability, leakage, and potential destruction in gastric fluids. The use of proliposomes represents an alternative to conventional liposomal formulations (CitationKumar et al., 2001; CitationBrocks & Betageri 2002; CitationDeshmukh et al., 2008). Proliposomes are a dry, free-flowing powder which forms a MLV suspension when it comes in contact with water (CitationPayne et al., 1986). The stability problems associated with conventional liposomes, including aggregation, susceptibility to hydrolysis, and oxidation, may be avoided by using proliposomes. Since these formulations exist as a dry powder and upon hydration transform into MLV makes them more suitable for developing sustained release formulations for oral delivery.
Metoclopramide is a potent antiemetic and prokinetic agent commonly used for the management of gastrointestinal disorders (CitationReynolds, 1993). It is highly water-soluble (1 g in 0.7 g of water) (CitationPitre & Stradi, 1982). It is rapidly absorbed after oral administration and undergoes significant first-pass metabolism (CitationTeng et al., 1977; CitationUSP DI, 1998). It has a short biological half-life and is usually administered in a dose of 10–15 mg four times daily in order to maintain effective concentrations throughout the day. In long-term therapy, fluctuation in the plasma concentrations with high concentration peaks are common for drugs with rapid absorption and elimination (CitationUSP DI, 1998). The secondary effects of metoclopramide on the central nervous system in the form of extrapyramidal symptoms, will surface if plasma levels markedly exceed therapeutic levels (Ganza-Gonzalez et al., 1999). In such instances controlled release formulations might be expected to ameliorate such problems.
Proliposomes have been extensively investigated in our laboratory to increase the permeation of highly hydrophilic compounds and to increase the solubility of poorly water-soluble drugs (CitationBetageri, 2004; Citation2005). However, this is the first time we are reporting our investigation on the effect of tableting on proliposomes. In the present investigation we have encapsulated metoclopramide into proliposomes with various phospholipid compositions and formulated into multiparticulate unit dosage forms. Proliposomes were characterized for particles size, encapsulation efficiency, DSC, and TEM. Formation of vesicles from proliposomes was also studied. Investigation was also extended to evaluate drug release properties of proliposomes in the presence of hydrophilic polymeric matrices.
Materials and methods
Materials
Metoclopramide, Cholesterol, and Chitosan (CS) (Medium mol. weight, 75–85% deacetylated) were obtained from Sigma-Aldrich Inc (St. Louis, MO). Dimyristoyl-phosphocholine (DMPC) and dimyristoyl-phosphoglycerol (DMPG) were obtained from Genzyme Corporation (Cambridge, MA). Dipalmitoyl-phosphocholine (DPPC) was procured from Avanti Polar Lipids (Alabaster, AL). Egg-phosphatidylcholine (EPC) was obtained from NOF Corporation (Japan). Hydroxypropyl cellulose (HPC) (Klucel HXF) was obtained from Hercules Inc (Wilmington, DE). Carbopol 71G (CP) was procured from BFGoodrich Co (Cleveland, OH). Gelatin (GL) was procured from Spectrum chemicals (Gardena, CA). Microcrystalline cellulose (Avicel PH 101) was obtained from FMC Corporation (Newark, DE). Stearylamine was obtained from MP Biomedicals LLC (Solon, OH). All other chemicals and reagents used in the study were of analytical or HPLC grade and used without further purification.
Methods
Preparation of pro-liposomes
Liposomes were prepared by thin film hydration; briefly, metoclopramide, phospholipid (DMPC, DMPG, DPPC, or EPC), and cholesterol in varying ratios were dissolved in ethanol. The solvent was dried under vacuum by using a rotavapor (Buchi, Switzerland) to make a thin film. This thin film was hydrated with phosphate buffer saline (PBS) pH 7.4 at a temperature slightly higher than the phase transition temperature of the phospholipid (at 25°C for DMPC, DMPG, or EPC and 42°C for DPPC) for 10 min followed by bath sonication for 10 min at room temperature. Liposomes were transformed to proliposomes by freeze-drying the liposomal suspension for 24 h.
Encapsulation efficiency
The proliposomes were hydrated with PBS pH 7.4 by bath sonication for 5 min. To separate the non-encapsulated drug from liposomes, a Centrifree® ultrafiltration technique was used (CitationDipali et al., 1996). Each Centrifree® unit consists of a sample reservoir underneath which is a filter membrane and a filter cup. The liposomal suspension was placed in sample reservoir and the unit was centrifuged at 2000 rpm for 30 min. The liposomes, along with encapsulated drug, remained on top of the membrane, and the free drug associated with supernatant was separated in the bottom filter cup. The concentration of metoclopramide was analyzed spectrophotometrically at the λmax value of 273 nm (CitationPitre & Stradi, 1982). A calibration curve was constructed over the concentration range of 1–30 μg/mL (r2 = 0.999, n = 6). To avoid the interference from liposome carriers, the supernatant of the placebo liposomes was used as the blank. Encapsulation efficiency was calculated as:
where total drug is the theoretical amount of drug used in the formulation and free drug is the amount of drug in the supernatant.
Particle size measurement
Particle size and size distributions were measured by using dynamic light scattering technique (NICOMP model 370 submicron particle sizer, Santa Barbara, CA). Intensity-weighted Gaussian analysis was used for unimodal distribution. The run time stopped automatically when a fitting error of 1 or when a Chi-squared value of less than 1 was achieved. The proliposomes were hydrated and the liposomal suspension was diluted suitably and particle size and size distribution were recorded.
Preparation of pro-liposomal tablets
Proliposomes were blended uniformly with hydrophilic polymer (HPC, CP, CS, or GL) and microcrystalline cellulose (MCC). Amount of MCC used was changed with relative changes in the concentration of phospholipid or polymer to keep the tablet weight constant. Tablets with 9 mm diameter were compressed using a 10-station rotary compression machine. Various formulations were prepared by varying the concentration or type of phospholipid or polymer. Effect of different phospholipids, phospholipid concentration, different polymers, polymer concentration, and charge of phospholipid and polymer was investigared. The composition of various formulations is summarized in .
Table 1. The composition of various formulations.
Since chitosan is fibrous in nature and lacks good flow properties and compressibility (CitationSabnis et al., 1997), an additional step of freeze-drying chitosan along with MCC was introduced to improve these properties. Briefly, chitosan was dissolved in 2% aqueous acetic acid to obtain a clear solution and then MCC (one part chitosan and four parts MCC) was dispersed under magnetic stirring to obtain a uniform suspension. The suspension was subjected to freeze-drying for 48 h and the freeze-dried material was used for tableting of proliposomes.
Drug content in tablets (assay)
Three tablets from each formulation were crushed into powder in a mortar and then the powder equivalent to the weight of one tablet was shaken in 250 mL of 10% Triton X-100 aqueous solution for 30 min (Precision horizontal shaker, VA) followed by sonication for 30 min (Branson 5510 bath sonicator, CT). The resulting suspension was filtered using 0.22 µm syringe filters. Then the clear solution was diluted suitably and the concentration of metoclopramide was analyzed spectrophotometrically at the λmax value of 273 nm. A 10% Triton X-100 aqueous solution diluted similarly as that of sample was used as blank.
Differential scanning calorimetric (DSC) analyses
Differential scanning calorimetry (DSC) was performed to understand the physical state of the drug in the liposomes. DSC was performed on pure drug, physical mixture of drug, and phospholipid, as well as drug-loaded proliposomes. Samples were analyzed using Shimadzu DSC-60 (Kyoto, Japan) in nitrogen environment with a heating rate of 10°C/min.
Formation of vesicles from proliposomes
Formation of vesicles from free-flowing proliposomal powder was confirmed by observing the process of hydration of proliposomes with water under an optical microscope (Nikon, Eclipse E400, Japan) equipped with a digital camera. Proliposomes were taken on a glass slide and few drops of nanopure water were added and the hydration process was followed by taking a series of time-lapse photographs. The proliposomal hydration process was viewed at 400× magnification.
Transmission electron microscopic (TEM) studies
The morphology of vesicles was observed under a FEI Tecnai G2 Spirit transmission electron microscope (Eindhoven, Netherlands). Proliposomes were hydrated with water and sonicated to produce a liposomal suspension. A drop of liposomal suspension was dried on a carbon-coated grid and stained with aqueous solution of phosphotungstic acid. After drying the specimen was viewed under the microscope at an accelerating voltage of 100 kV.
In vitro drug release
Drug release studies were performed on fully automated dissolution equipment coupled with the UV system (Carry 50, Varian, USA) using USP Type II apparatus at the stirring speed of 50 rpm. Each tablet was placed in 600 mL of PBS (pH 7.4) maintained at 37°C. The instrument automatically measures the concentration of drug released at the particular time interval by a UV spectrophotometer coupled with flow through cells attached to the instrument and then replaces the solution back into the dissolution bowl. Concentration of metoclopramide was analyzed spectrophotometrically at the λmax value of 273 nm. These studies were performed in triplicate for each sample and average values were used for data analysis.
During in vitro release studies, the formation and liberation of liposomes from the tablet was confirmed by observing a drop of dissolution media (at 4 h) under an optical microscope at 400× magnification.
Results and discussion
Preparation and characterization of proliposomes
Proliposomes prepared were white, non-sticky powders, which were successfully formulated into tablet unit dosage form with good tableting characteristics. The encapsulation efficiency, drug content, and particle size of proliposomes produced using different phospholipids are summarized in . The encapsulation efficiency of proliposomes was found to be in the range of 18–43%. It is evident from the results that phospholipid concentration was critical for encapsulation efficiency. Proliposomes produced using DMPC with 1:0.5, 1:1, and 1:1.5 (drug:phospholipid) ratio had encapsulation efficiency of 18, 42, and 43%. Whereas those produced with DMPG, DPPC, and EPC had 40, 37, and 34% encapsulation, respectively. The mean particle size of proliposomes was between 1.3–2.5 µm. Proliposomes with EPC produced particle size of 1.3 µm and that with DPPC produced particle size of 2.5 µm. This could be attributed to the differences in phase transition temperature of phospholipids. Above the phase transition temperature, the lipid bilayer is in fluid state and easily gets hydrated to form particles which need less energy to form smaller particles compared to when processed at temperatures below transition temperature. On a population basis, the particle size distribution was found to be unimodal with the narrow size distributions. The drug content of the final tableted formulations was found to be in the range of 98–99%. Tablets with hardness in the range of 5–8 Kp and friability less than 1% were prepared.
Table 2. Results of percent encapsulation efficiency, drug content, and particle size.
Most of the phospholipid membranes of liposomes are not sufficiently rigid; they often leak encapsulated drugs during processing and storage (CitationKulkarni et al., 1995). Cholesterol, when added to lipid bilayer, fills in the gaps created by imperfect packing of other lipid bilayers and modulates the membrane fluidity, elasticity, and permeability. Thus, cholesterol improves the retention (CitationBetageri, 1993) of hydrophilic drugs by decreasing bilayer permeability. Incorporation of cholesterol also modulates the phase transition temperature of phospholipids, imparts rigidity to lipid bilayers, and improves the physical stability. Cholesterol content will also modify the hydration property of the lipid due to its effect on membrane fluidity. As the present investigation is designed to develop proliposomes for oral drug delivery application, rehydration of the proliposomes into liposomes in the presence of biological fluid at body temperature is essential for success of these systems (CitationBetageri & Kulkarni, 1999). Thus, cholesterol was used in all the proliposomal formulations. As encapsulation efficiency is influenced by fluidity and rigidity of lipid membrane we have optimized the ratio of cholesterol to balance these properties (phospholipid:cholesterol ratio of 1:0.5).
DSC studies
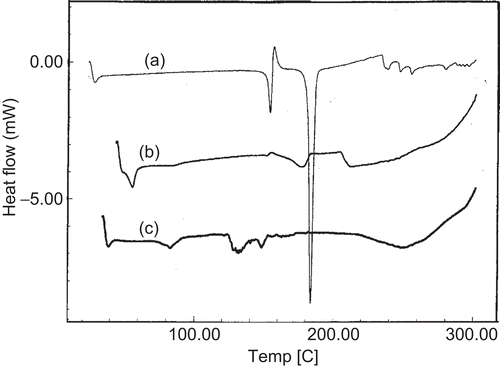
The DSC thermograms for pure drug, drug, and phospholipid (DMPC) physical mixture and drug-loaded proliposomes are presented in . Pure drug has shown a sharp endothermic peak at 184°C indicating the melting endothermic transition with enthalpy (ΔH) of −112.45 J/g. In the case of physical mixture of drug and phospholipid, a broad endothermic peak is observed at 177°C with ΔH of −25.27 J/g. While in the case of drug-loaded proliposomes these peaks have disappeared. It is evident from the results that, in the physical mixture, ΔH has reduced drastically and the peak shifts to the lower temperature. In the physical mixture, phospholipid could be dominating the transitions and hence a broad peak is observed with lower ΔH value. In the case of drug-loaded proliposomes, absence of the peak indicates that the drug is dispersed in amorphous form. It was also reported in the literature that presence of liposomal structure in the glassy state will prevent the liposomal fusion as well as solute leakage in the solid state; hence, based on the results, we can predict that proliposomes might be stable with respect to membrane fusion and drug leakage in solid state (CitationSun et al., 1996).
Figure 1. DSC thermograms of (a) pure drug, (b) physical mixture of phospholipid (DMPC) + drug, and (c) drug-loaded proliposomes (DMPC).
Formation of vesicles from proliposomes
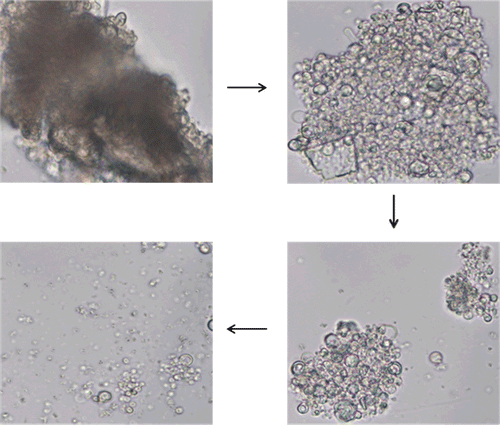
Hydration of proliposomes into vesicles progressed quickly upon contact with water. The series of time-lapse photographs of hydration of proliposomes are shown in . Approximately within 4 min the hydration was complete and proliposomes were converted into vesicles. The process of formation of vesicles occurs by progressive hydration of phospholipid present on the surface of proliposomes and slowly liposomes detach from the cluster of proliposomes. From this observation it could be assumed that proliposomes would progressively and rapidly convert into vesicles upon contact with physiological fluids present in the body after oral administration. Discrete vesicular structures were observed in the photographs taken after the lapse of 4 min, which also reveal the multilamellar nature of the vesicles formed.
Figure 2. Series of time-lapse photomicrographs depicting the process of hydration of proliposomes (DMPC) at 400× magnification (time lapse of 1 min between each photograph).
Transmission electron micrographs
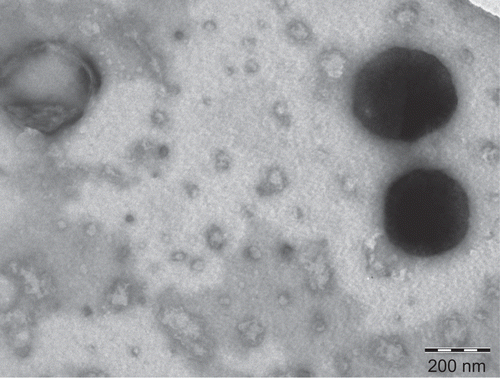
Transmission electron micrographs (TEM) confirmed the formation of vesicles from proliposomes. A TEM image of proliposomes derived vesicles is shown in are spherical in shape. The figure is representative of the proliposomes of formulation using DMPC, we observed a similar trend in the proliposomes of formulations using other phospholipids. It is quite interesting to notice that the particle size obtained from TEM is significantly smaller as compared to particle size measured by dynamic light scattering technique. This could be due to the shrinkage of the vesicles, as the aqueous dispersion of vesicles is dried during the sample preparation for TEM.
Figure 3. TEM of liposomes derived from proliposomes (DMPC).
In vitro drug release
Various proliposomal formulations were developed into matrix tablet dosage forms by combining with hydrophilic polymer using conventional tablet manufacturing process. Tableted proliposomal formulations were evaluated for in vitro drug rerelease in pH 7.4 PBS. The formation and liberation of vesicles from tableted proliposomes was confirmed by observing the dissolution media under an optical microscope. represents the vesicles formed during the in vitro release study. Once the formulation is in contact with release medium, the proliposomes get hydrated to form multilamellar vesicles and facilitate the drug release. As the in vitro release proceeds, initially the formulation imbibes the dissolution medium to form a swollen hydrogel matrix followed by hydration of proliposomes and formation of multilamellar vesicles. Interestingly, it was observed that during the initial hours of the release study the concentration of vesicles observed were less, and approximately during the middle of the study the concentration of vesicles start rising (as per optical microscopic observation of the unfiltered dissolution samples). This indicates the existence of a lag phase for the release of vesicles from the dosage from.
Figure 4. Photomicrograph of liposomes formed during in vitro release (4 h sample) of formulation F2 at 400× magnification.

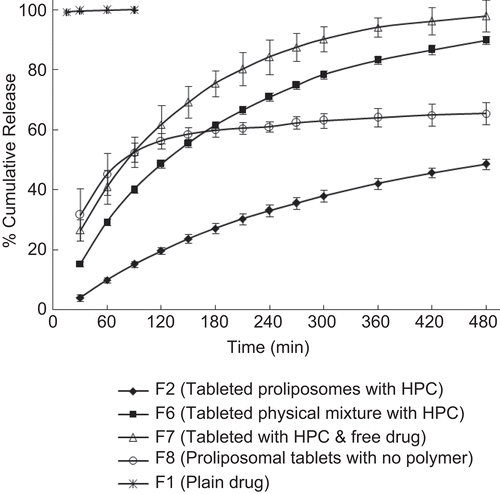
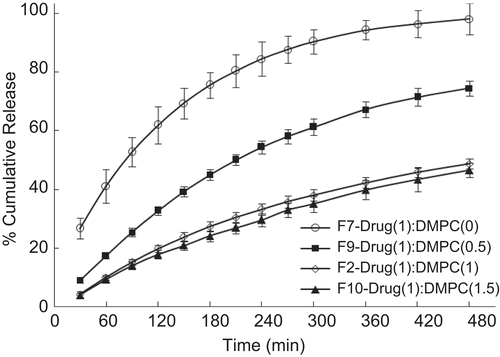
The effect of encapsulation of drug into proliposomes and tableting of proliposomes by incorporating hydrophilic polymer on the in vitro release is displayed in . It is observed that metoclopramide release from proliposomal tablets (containing DMPC proliposomes and HPC polymer) was only 49% over a period of 8 h with no initial burst release. Whereas formulations without proliposomes (conventional HPC matrix tablet) exhibited a burst release of 27% in the first 30 min and 98% of drug was released over a period of 8 h. In the case of proliposomal tablets formulated without the hydrophilic polymer (HPC), 66% of drug was released over a period of 8 h with a burst release of 32% in the first 30 min. Physical mixture of drug, phospholipid, and polymer showed 90% drug release over a period of 8 h. The tablet containing plain drug (no proliposomes and polymer) released almost 100% in 30 min.
Figure 5. Evaluation of the effect of encapsulation of drug into proliposomes and incorporation of hydrophilic polymer on the in-vitro release profile of drug. Each data point represents mean ± standard deviation (n = 3).
These results indicate that when proliposomes are compressed directly with diluents such as microcrystalline cellulose, the hydration process of proliposomes is incomplete, as evidenced by slow and incomplete drug rerelease with an initial burst release. This might have resulted owing to the formation of the proliposome-MCC matrix due to compression pressure and MCC being water insoluble might have hindered hydration of the proliposomal system. This prompted us to attempt hydrophilic polymers such as HPC as excipient to facilitate the hydration process. The results show that incorporation of small amounts of HPC have not only reduced the initial burst release but also extended the drug release over a period of time. This indicates that a proper combination of proliposomes and pharmaceutical excipients are necessary to yield tailorable release profiles from the tableted proliposomes.
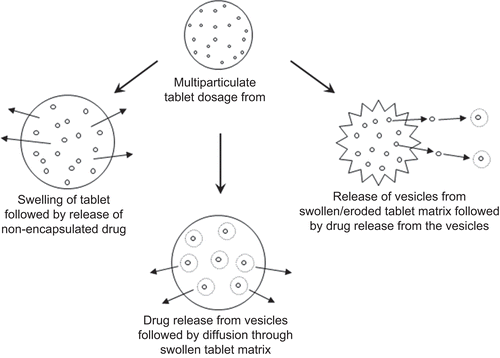
This phenomenon might be attributed to the fact that the drug release could be controlled by three possible mechanisms, i.e. (i) water permeation, hydrophilic polymer swelling, non-encapsulated drug dissolution, and diffusion through swollen matrix, (ii) hydration of proliposomes into multilamellar vesicles followed by subsequent drug release from vesicles and diffusion of drug through swollen matrix, and (iii) liberation of vesicles from swollen or eroded matrices into the release medium followed by drug release from vesicles. These mechanisms could be schematically summarized as in . Combining the proliposomal technology with release controlling polymer not only helps in reducing the burst release and controlling the release throughout the GI transit, but also reduces the amount of release controlling polymer required. This makes such systems suitable for delivering the molecules with high water solubility and dose.
Figure 6. Illustration of possible mechanism of drug release from the proposed proliposomal multiparticulate unit dosage forms.
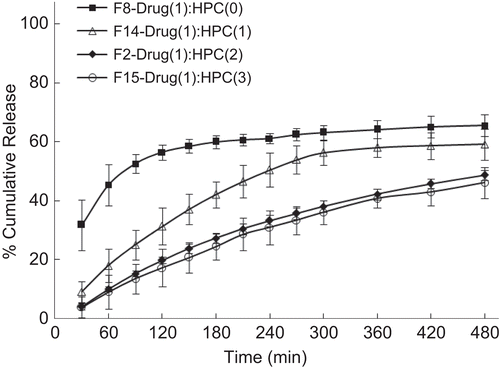
Once the great potential of controlled release offered by combining proliposomes with conventional polymer matrix was realized, various formulations were prepared to study the effect of different variables and to optimize the formulation. The effect of different phospholipids is displayed in . Proliposomal tablets prepared using DMPG exhibited a slower drug release as compared to other phospholipids studied. It can be seen that DMPC proliposomal tablets released only 49% over a period of 8 h in contrast to EPC proliposomes, which released 88%, whereas DMPG and DPPC proliposomes released 65 and 84%, respectively. All the phospholipids studied were effective in controlling the burst release. It is a well known fact that the presence of cholesterol modulates the membrane properties of phospholipids. Even though the Tg of DPPC is above the body temperature, cholesterol helps in easy hydration at body temperature. The possible mechanism of drug release is diffusion. The effect of various concentrations of phospholipid (DMPC) is displayed in . It clearly demonstrates that increasing the concentration of phospholipid reduces the rate of drug release. However, there is no significant difference observed between drug:phospholipid ratios of 1:1 and 1:1.5. It could be concluded that a drug:phospholipid ratio of 1:1 is optimum for controlling the release.
Figure 7. Effect of different phospholipid formulations on the in-vitro release profile of drug. Each data point represents mean ± standard deviation (n = 3).
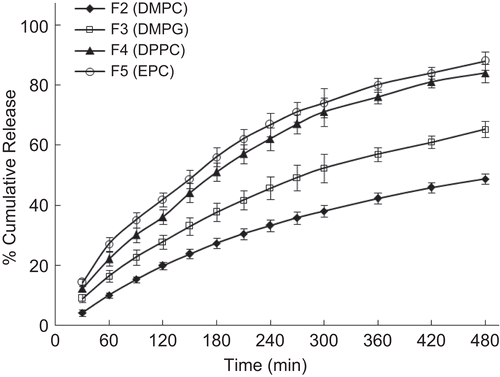
Figure 8. Effect of phospholipid concentration on the in-vitro release profile of drug. Each data point represents mean ± standard deviation (n = 3).
compares the release profiles of formulations containing various hydrophilic polymers. It is evident from the figure that HPC had slower release compared to carbopol, chitosan, and gelatin. All the polymers studied were effective in controlling the burst effect. In the case of formulations with gelatin, release was almost 2-fold higher compared to HPC. Effect of polymer concentration on release profile is displayed in . It is observed that release is inversely proportional to the polymer concentration; as expected the release rates are slower for formulations containing higher amounts of polymer. The proliposomes in the polymer matrix might be acting as inert fillers by occupying the free volume spaces of the swollen hydrogel. Proliposomes within the polymer matrix acts as a plasticizer and, thus, increase the free volume within the network. This, in turn, will create a more tortuous path for water molecules to permeate through, but the degree of tortuosity depends upon the volume fraction of the filler (CitationPeppas, 1980).
Figure 9. Effect of different polymer formulations on the in-vitro release profile of drug. Each data point represents mean ± standard deviation (n = 3).
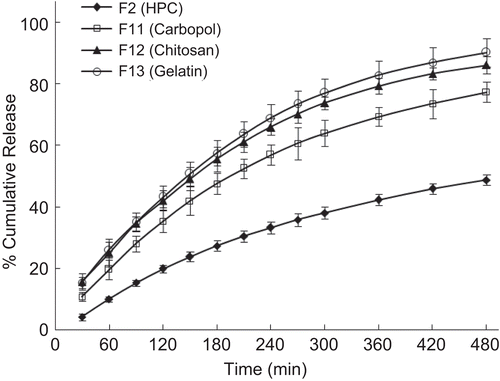
Figure 10. Effect of polymer concentration on the in-vitro release profile of drug. Each data point represents mean ± standard deviation (n = 3).
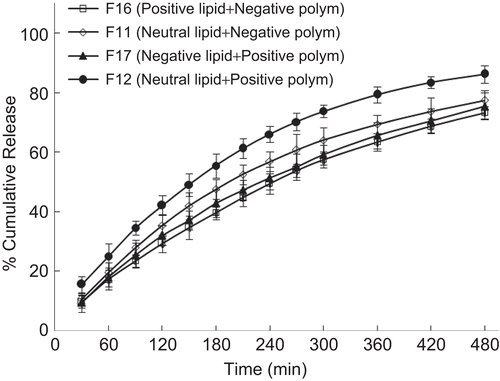
The presence of charge often alters the properties of vesicular structures, as seen in many studies (CitationHenriksen et al., 1994; CitationThongborissute et al., 2006). In order to evaluate the interaction of oppositely charged phospholipid and polymer on drug release profile, positive charge was induced by incorporating stearylamine along with DMPC and negative charge was induced by using DMPG. In the case of polymers, carbopol and chitosan were used to induce negative and positive charges, respectively in the matrix. Also, control formulations were prepared by using neutral lipid (DMPC). displays the release profiles of formulations. It can be seen that charged formulations displayed a slightly slower release profile compared to control formulations. This possible slow release could be due to the mild electrostatic interaction between the oppositely charged phospholipid and polymer which hinders the release of drug. This demonstrates that reported proliposomal systems could be possibly further designed to meet the required drug release profile by using charged proliposomes and polymeric matrix.
Figure 11. Effect of charge of phospholipid and polymer on the in-vitro release profile of drug. Each data point represents mean ± standard deviation (n = 3).
Results of drug release were also analyzed using the empirical equation (CitationRitger & Peppas, 1987) to understand the mechanism of drug release. The initial 60% drug release data (i.e. linear region of the plots) were fitted to equation (2) to estimate the exponential coefficient, n:
By applying the least-squares estimation method to the release data at 95% confidence level the values of n have been determined. These data, along with the values of correlation coefficient r2, are presented in . These values ranged between 0.23–0.94. It is quite interesting to note that wide variations of release mechanisms (CitationBaker & Lonsdale, 1974) are prevailing depending upon the variation in the composition of formulations. In the case of formulations prepared without the polymer (formulation F8) the n-value is 0.23, indicating a Fickian (n ≤ 0.45) trend. In the case of formulation without the proliposomes (formulation F7) the n-value is 0.66, indicating anomalous (0.45< n< 0.85) behavior. Whereas, in the formulation containing both proliposomes (DMPC) and polymer (HPC) the n-value is 0.94, indicating a case II transport (n ≥ 0.89), i.e. a near zero order release pattern. All the other formulations indicated anomalous behavior.
Table 3. Results of exponential coefficient (n) and correlation coefficient (r2) calculated from equation (2).
Conclusions
A highly hydrophilic drug, metoclopramid,e was successfully encapsulated into multilamellar vesicles to form proliposomes for possible oral controlled release applications. Multilamellar vesicle structures are formed spontaneously from proliposomes and also after formulating into single unit dosage form. Type and concentration of phospholipid and hydrophilic polymer have shown varying effects on the drug release from the tableted proliposomes. Proliposomes, when formulated into unit dosage form using a hydrophilic polymer, reduce the burst release and further control the drug release. The combination of phospholipid and hydrophilic polymer is highly effective in modulating the release rate. An exceptionally low amount of phospholipid and polymer are needed to produce this effect, reducing the size of the dosage form; thereby making it cost effective technology. Encapsulation of drug modifies the mechanism of drug release and by combining proliposomes and hydrophilic polymers a near zero order release can be achieved. Results are convincing from this study that proliposomal technology can be exploited to deliver hydrophilic molecules for a prolonged period of time. Although the present investigation is focused on water-soluble molecules, the proposed delivery systems could be extended to drug molecules with diversified physicochemical properties to modulate release profile by carefully selecting a combination of both phospholipid and polymer.
Acknowledgements
The authors thank Ms Nirupama (Semler Research Center, Bangalore, India) for helping in performing DSC and TEM experiments. Part of this paper was presented at the AAPS Annual Meeting and Exposition, Atlanta, GA, USA, November 16–20 (2008).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Agnihotri, S.A., Aminabhavi, T.M. (2004). Formulation and evaluation of novel tableted chitosan microparticles for the controlled release of clozapine. J Microencapsul. 21:709–18.
- Baker, R.W., Lonsdale, H.K. (1974). Controlled release: mechanisms and rates. In: Tanquarry, A.C., Lacey, R.E., eds. Controlled release of biologically active agents. New York: Plenum Press, 15–71.
- Betageri, G.V. (1993). Liposomal encapsulation and stability of dideoxyinosine triphosphae. Drug Dev Ind Pharm. 19:531–9.
- Betageri, G.V. (2004). Enteric-coated proliposomal formulations for poorly water soluble drugs. U.S. Patent. No. 6,759,058.
- Betageri, G.V. (2005). Proliposomal drug delivery system. U.S. Patent. No. 6,849,269.
- Betageri, G.V., Kulkarni, S.B. (1999). Preparation of liposomes. In: Arshady, R., ed. Microspheres, microcapsules and liposomes, Volume 1. London: Citus Books, 490–521.
- Betageri, G.V., Parsons, D.L. (1992). Drug encapsulation and release from multilamellar and unilamellar liposomes. Int J Pharm. 81:235–41.
- Bodmeier, R. (1997). Tableting of coated pellets. Eur J Pharm Biopharm. 43:1–8.
- Brocks, D.R., Betageri, G.V. (2002). Enhanced oral absorption of halofantrine enantiomers after encapsulation in a proliposomal formulation. J Pharm Pharmacol. 54:1049–53.
- Chen, H.M., Torchilin, V., Langer, R. (1996). Polymerized liposomes as potential oral vaccine carriers: Stability and bioavailability. J Contr Rel. 42:263–72.
- Deshmukh, D.D., Ravis, W.R., Betageri, G.V. (2008). Improved delivery of cromolyn from oral proliposomal beads. Int J Pharm. 358:128–36.
- DiBiase, M.D., Morrel, E.M. (1997). Oral delivery of microencapsulated proteins. In: Sanders, L.M., Wayne Hendren, R., eds. Protein delivery: Physical systems. New York: Plenum Press, 255–88.
- Dipali, S.R., Kulkarni, S.B., Betageri, G.V. (1996). Comparative study of separation of non-encapsulated drug from unilamellar liposomes by various methods. J Pharm Pharmacol. 48:1112–5.
- Elbayoumi, T.A., Torchilin, V.P. (2008). Tumor-specific antibody-mediated targeted delivery of Doxil® reduces the manifestation of auricular erythema side effect in mice. Int J Pharm. 357:272–9.
- Florence, A.T. (1997). The oral absorption of micro- and nanoparticulates: neither exceptional nor unusual. Pharm Res. 14:259–66.
- Galeone, M., Nizzola, L., Cacioli, D., Mosie, G. (1981). In-vitro demonstration of delivery mechanism from sustained-release pellets. Curr Therap Res. 29:217–34.
- Henriksen, I., Smistad, G., Karlsen, J. (1994). Interactions between liposomes and chitosan. Int J Pharm. 101:227–36.
- Kulkarni, S.B., Betageri, G.V., Singh, M. (1995). Factors affecting microencapsulation of drugs in liposomes. J Microencapsul. 12:229–46.
- Kumar, R., Gupta, R.B., Betageri, G.V. (2001). Formulation, characterization and in-vitro release of glyburide from proliposomal beads. Drug Deliv. 8:25–7.
- Payne, N.I., Timmins, P., Ambrose, C.V., Ward, M.D., Ridgway, F. (1986). Proliposomes: a novel solution to an old problem. J Pharm Sci. 75:325–9.
- Peppas, N.A. (1980). Mathematical modeling of diffusion process in drug delivery polymeric systems. In: Smolen, V.F., ed. Bioavailability and the pharmacokinetic control of drug response. New York: Wiley.
- Pitre, D., Stradi, R. (1982). Analytical profiles of drug substances. In: Florey, K., ed. Vol. 16. Orlando, FL: Academic Press, 327.
- Reynolds, J.R., ed. (1993). Martindale: The extra pharmacopoeia, 30th Ed. London: Pharmaceutical Press, 892–4.
- Ritger, P.L., Peppas, N.A. (1987). A simple equation for description of solute release. II. Fickian and anomalous release from swellable devices. J Contr Rel. 5:37–42.
- Rogers, J.A., Anderson, K.E. (1998). The potential of liposomes in oral drug delivery. Crit Rev Ther Drug Carrier Sys. 15:465–524.
- Sabnis, S., Rege, P., Block, L.H. (1997). Use of chitosan in compressed tablets of diclofenac sodium: inhibition of drug release in an acidic environment. Pharm Dev Technol. 2:243–55.
- Soppimath, K.S., Betageri, G.V. (2008). Nanostructures for cancer therapy. In: Gonsalves, K.E., Halberstadt, C.G., Laurencin, C.T., Nair, L.S., eds. Biomedical nanostructures. New Jersey: John Wiley & Sons, 409–38.
- Sun, W.Q., Leopold, A.C., Crowe, L.M., Crowe, J.H. (1996). Stability of dry liposomes in sugar glasses. Biophys J. 70:1769–76.
- Teng, L., Bruce, R.B., Dunning, L.K. (1977). Metaclopramide metabolism and determination by high-pressure liquid chromatography. J Pharm Sci. 66:1615–8.
- Thongborissute, J., Takeuchi, H., Yamamoto, H., Kawashima, Y. (2006). Properties of liposomes coated with hydrophobically modified chitosan in oral liposomal drug delivery. Pharmazie. 61:106–11.
- USP DI. (1998). Drug information for the health care professional. Vol. I, 18th Ed. United States Pharmacopoeial Convention, Rockville, 2009–13.