Abstract
The present study was aimed to evaluate the anti-tumor efficacy and systemic toxicity of chitosan-based plumbagin microspheres in comparison to free plumbagin. The optimized formulation had a mean particle size of 106.35 μm with an encapsulation efficiency of 80.12%. Pharmacokinetic studies showed a 22.2-fold increase in elimination half-life (t1/2) of plumbagin from chitosan microspheres as compared to free plumbagin. Administration of plumbagin microspheres resulted in a significant tumor growth inhibition and reduced systemic toxicity. These results suggest that chitosan-based microspheres could be a promising strategy for the systemic delivery of anti-cancer agents like plumbagin.
Introduction
Conventional dosage forms for cancer therapy suffer from poor bioavailability due to low aqueous solubility, shorter biological half-life, rapid metabolism, and elimination, requiring frequent administration of the drug which may be associated with debilitating effects. Thus, development of controlled drug delivery systems to optimize the pharmaceutical action of a drug while reducing its toxic side effects in vivo remains a challenging task. In this regard, various biodegradable polymeric controlled release dosage forms have been developed to overcome the shortcomings of conventional dosage forms with enhanced anti-cancer efficacy. Chitosan, a biodegradable polymer obtained by alkaline deacetylation of chitin, has been utilized for encapsulating many anti-cancer agents (CitationThanoo et al., 1992; CitationJameela et al., 1998; CitationSingh & Udupa, 1998; CitationSinha et al., 2004; CitationPatashnik et al., 2005; Prabaharan & Mano, Citation2005a). It is believed that chitosan forms colloidal particles and entraps drug molecules by chemical cross-linking or complexation (Prabaharan & Mano, Citation2005b) allowing the control of release rate of the active drug, thus maintaining the therapeutic dosage for a prolonged period of time with minimum side-effects.
Plumbagin (2-methyl, 5-hydroxy,1,4-naphthoquinone), a naphthoquinone from Plumbago zeylanica L., has shown promising anti-cancer activity both in vitro against a broad spectra of human tumor cells such as ME-180 human cervical cancer, KBM-5 human chronic myeloid leukemia, U937 human histiocytic lymphoma, U266 human multiple myeloma, H1299 lung adenoma, MCF7 breast adenocarcinoma, A549 human non-small cell lung cancer, BG1 ovarian adenocarcinoma, PC3 prostate cancer (CitationSrinivas et al., 2004; CitationHsu et al., 2006; CitationSandur et al., 2006; CitationPowolny & Singh, 2008; CitationThasni et al., 2008) and against tumor cells growing in vivo in mouse model (CitationKrishnaswamy & Purushothaman, 1980; CitationPurushothaman et al., 1985; CitationSugie et al., 1998; CitationHsu et al., 2006; CitationKuo et al., 2006). Our previous studies as well as others on mouse tumor cells grown in vitro and in vivo have shown anti-tumor and radiosensitizing properties (CitationDevi et al., 1994; CitationPrasad et al., 1996; CitationNair et al., 2008). Plumbagin, being a quinone moiety, is reported to be highly toxic (CitationSolomon et al., 1993) and acts as spindle poison by inhibiting cell mitosis at low concentrations and at higher concentrations it exhibits radiomimetic and cytotoxic effects (CitationSingh & Udupa, 1997). Literature suggests that the structure of plumbagin is similar to vitamin K and therefore the chronic administration of plumbagin leads to prolonged bleeding time by altering platelet adhesiveness and coagulation. (CitationVijayakumar et al., 2006). Also, reports suggest that plumbagin exhibits shorter biological half-life with rapid elimination in vivo (CitationChandrasekaran & Nagarajan, 1981), which may undermine its therapeutic potential. Various approaches have been attempted by different routes of administration viz., intraperitoneal, subcutaneous, and intravenous in order to improve the anti-cancer potential and reduce the toxicity associated with plumbagin viz. niosomes and albumin microspheres, PLGA based microparticles, and thermosensitive liposomes (CitationNaresh et al., 1996; CitationKini et al., 1997; CitationSingh and Udupa, 1997; CitationTiwari et al., 2002). It has been reported that the formulation of plumbagin as niosomes and albumin microspheres administered via i.p. route resulted in slight reduction in the toxicity, however these approaches did not improve the anti-cancer efficacy of plumbagin. PLGA-based formulations are expensive and necessitate the use of toxic organic solvents which limit their usage. Although thermosensitive liposomes may offer significant therapeutic advantages in terms of improvement in anti-cancer efficacy and reduction in toxicity, in the case of deep-seated tumors this approach may not be clinically feasible as it requires technical expertise for delivering the heat precisely.
Chitosan, compared to synthetic biodegradable polymers, is less expensive and being hydrophilic it eliminates the use of toxic organic solvents to dissolve the polymer during formulation development. In addition, it is a suitable carrier for even low molecular weight compounds (CitationDass & Choong, 2008), and has been used as an absorption enhancer for poorly-absorbable drugs (CitationSchipper et al., 1997). It is also reported that chitosan possesses inherent anti-cancer activity (CitationMurata et al., 1989; CitationChen et al., 1997; CitationLin et al., 2007; CitationQi et al., 2007; CitationDass & Choong, 2008), which makes it more ideal for encapsulation of anti-cancer agents. Hence, in the present study, an attempt has been made to develop chitosan-based plumbagin-loaded microspheres using glutaraldehyde as cross-linking agent. The prepared formulations were evaluated for in vitro release characteristics, pharmacokinetics, and pharmacodynamic, as well as systemic toxicity.
Materials and methods
Materials
Plumbagin, 2,3-dicholoro-1,4-naphthoquinone, chitosan, minimum essential medium (MEM), fetal bovine serum (FBS), and dialysis membrane (12,000 KD) were purchased from Sigma Chemicals (St. Louis, MO, USA). Methanol and all the other reagents used for HPLC analysis were of HPLC grade and procured from Merck (Mumbai, India).
Animal tumor model and cell line
The murine B16F1 melanoma cell line was procured from National Center for Cell Sciences (NCCS) (Pune, India) and maintained in our laboratory. The cells were routinely grown in 25-cm2 T-flasks (Falcon, Becton Dickinson, USA) with loosened caps, containing minimum essential medium (MEM) supplemented with fetal bovine serum (FBS) and other required components at 37°C in a humidified atmosphere (5% CO2 in 95% air). C57BL/6J mice were procured from the National Institute of Nutrition (Hyderabad, India). The animal care and handling was carried out according to the guidelines issued by the World Health Organization (Geneva, Switzerland) and the INSA (Indian National Science Academy, New Delhi, India). Mice of either sex, 8–12 weeks old, weighing 20–25 g, were selected for experimentation from an inbred colony maintained under the controlled conditions of temperature (23 ± 2°C), humidity (50 ± 5%) and light (14 and 10 h of light and dark, respectively). All the animal experiments were carried out with prior approval from the institutional animal ethics committee (Kasturba Medical College, Manipal, India).
Formulation development
Preparation of chitosan microspheres
Different concentrations of chitosan (1, 1.5, 2.0, and 3.0% w/v) were prepared by stirring chitosan in 2% v/v acetic acid in a beaker using a magnetic stirrer. The drug was dispersed in chitosan solution and was added drop-wise to a beaker containing liquid paraffin oil and span-80 as surfactant under constant stirring at 600 rpm (Remi, Mumbai, India) for 30 min resulting in the formation of w/o emulsion. Glutaraldehyde (25% v/v) as a cross-linking agent was added in varying amounts depending upon the cross-linking density required and stirring was continued for 4 h at 1500 rpm to form microspheres. The formed microspheres were washed 2–3-times with petroleum ether (60–80°C) to remove the adhered liquid paraffin as well as glutaraldehyde, and separated through vacuum filtration (CitationThanoo et al., 1992; CitationAkbuga & Bergisadi, 1996; Citation1999). The microspheres thus formed were finally dried in a vacuum desiccator for 24 h. The composition of various formulations is depicted in . The effect of drug-to-chitosan ratio and glutaraldehyde concentration on entrapment efficiency and release rate were studied.
Table 1. Composition of plumbagin-loaded chitosan microspheres.
Differential scanning calorimetry (DSC)
Differential scanning calorimetry was performed by using DSC-60 (Shimadzu, Tokyo, Japan). The instrument comprised of a calorimeter (DSC 60), flow controller (FCL 60), Thermal analyzer (TA 60), and operating software TA 60 from Shimadzu Corporation (Japan). The samples were placed in aluminium pans and were crimped, followed by heating under nitrogen flow (30 ml/min) at a scanning rate of 5°C/min from 25–200°C. Aluminium pan containing same quantity of indium was used as reference. The heat flow as a function of temperature was measured for both the drug and formulated microspheres.
Characterization of microspheres
Determination of encapsulation efficiency
The drug content in the microspheres was measured using a UV-Visible spectrophotometer (UV-1601PC, Shimadzu, Tokyo, Japan). Twenty milligrams of dried microspheres was crushed and suspended in methanol to extract the drug from microspheres. After 24 h, the suspension was filtered through 0.45 μM membrane filter and the concentration of the drug in the filtrate was assayed spectrophotometrically at 264 nm for drug content against methanol as blank. Corresponding drug concentrations in the samples were calculated from the standard calibration plot.
The percentage encapsulation efficiency was calculated using following equation:
Particle size analysis
Particle size was determined by Malvern particle size analyzer (Malvern Instruments, UK). Briefly, 10 mg of dry microspheres were first suspended in 100 ml of distilled water (containing 1% Tween 80) and subjected to sonication for 30 s (CitationZhu et al., 2005). The suspension was then placed in the laser particle counter. The mean diameter and the particle size distribution was then determined.
Morphological characterization of microspheres
The shape and surface morphology of the microspheres was examined using scanning electron microscopy (SEM) (JSM-T20, Tokyo, Japan). An appropriate sample of microspheres was mounted on metal stubs, using double-sided adhesive tape. Samples were gold coated and observed for morphology, at acceleration voltage of 15 KV.
In vitro release study
The drug release experiment was carried out using diffusion cell apparatus. One end of the diffusion cell was enclosed with dialysissac membrane (Sigma Aldrich, St. Louis, MO) previously soaked in phosphate buffer saline (PBS) pH 7.4 for 24 h. About 100 mg microspheres were suspended in 5 ml of PBS into the diffusion cell and immersed into the diffusing media containing 50 ml of PBS solution (pH 7.4) acting as a receptor compartment kept under stirring at 500 rpm. At set time intervals, 5 ml sample was withdrawn, passed through 0.45 μM membrane filter and assayed for drug using a UV-spectrophotometer at 264 nm. The removed samples were replaced with an equal quantity of fresh PBS in order to maintain sink conditions. The in vitro release studies were performed in triplicate for each prepared formulation.
Analysis of release mechanism
To study the release rate kinetics and mechanism of the drug release from the optimized formulation, F6, the in vitro drug release data was treated with mathematical equation such as zero order (percent release vs t), first order (log percent release vs t) kinetics (CitationNicholas, 1987), Higuchi’s diffusion equation (CitationHiguchi, 1963) (Q = Kt1/2), and Korsmeyer equation (CitationKorsmeyer et al., 1983) (Mt/M∞ = Ktn), where Mt is the amount of drug released at time t and M∞ is the amount released at time infinity, thus the Mt/M∞ is the fraction of drug released at time t, k is the kinetic constant, and n is the diffusional exponent, a measure of the primary mechanism of drug release. R2 values were calculated for the linear curves obtained by regression analysis of the above plots.
Pharmacokinetic evaluation in tumor-bearing mice
In order to monitor the plasma levels of plumbagin in C57BL/6J mice bearing solid tumor B16F1 melanoma, free or formulated chitosan microspheres were injected via intramuscular route at an optimal dose of 6 mg/kg body weight on the basis of earlier study (CitationTiwari et al., 2002). At pre-determined time intervals the blood was withdrawn from the retro-orbital vein using heparinized capillaries (at each point, blood was collected from four mice); plasma was separated by centrifugation (3 min, 13,000 rpm) and stored in vials at −20°C until HPLC analysis.
Bioanalytical method validation
The method was evaluated for linearity using freshly prepared spiked plasma samples in the concentration range of 0.1–10 μg/ml. A calibration curve for plumbagin was constructed prior to the experiments and the acceptance criteria for the calibration curve was with a correlation value of 0.995 or more. The intra-batch accuracy and precision were determined by analysis of six replicates each of low, medium, and high concentration quality control samples; while inter-batch accuracy and precision were determined by the analysis of these quality control samples on three separate occasions. The quality control samples were prepared at concentrations of 0.3 μg/ml (low), 5.0 μg/ml (medium), and 8.0 μg/ml (high) for plumbagin. The overall precision of the method was expressed as relative standard deviation (RSD) and accuracy of the method expressed as percent to true value.
Recovery of mice plasma
The stock solution of plumbagin was added to mice plasma to yield final concentrations of 0.3, 5.0, and 8.0 μg/ ml. Also, plumbagin stock solution was diluted to 0.3, 5.0, and 8.0 μg/ml in methanol. Both the plasma samples and the diluted solutions were processed as described in the following section and the ratio of peak area (plasma sample/diluted solution) for plumbagin was used to calculate the recovery in mice plasma.
Sample preparation for HPLC analysis
HPLC analysis was performed using Waters 2695 separations module with 2487 dual wavelength absorbance detector plus auto sampler. A reverse-phased Grace Vydac (Grace) C18 silica gel column (250 mm × 4.6 mm, 5 μm) was used at room temperature and the detector wavelength was set at 264 nm. Mixture of methanol:glacial acetic acid (0.1% v/v) (68:32) was used as the mobile phase at a flow rate of 1 ml/min. The drug was extracted from the plasma as described earlier (CitationHsieh et al., 2006) with minor modifications. Briefly, 100 μl of plasma was mixed with 100 μl of 0.1 N HCl and 2 ml of ethyl acetate into a clean RIA vial, vortexed for 15 min and centrifuged at 10,000 rpm for 10 min. The resultant upper organic layer was transferred to glass tubes and evaporated to dryness under nitrogen flow. The dried residue was reconstituted with 150 μl of the mobile phase and 100 μl aliquot of the clear solution was injected to the HPLC system. 2, 3-dichloro-1, 4-naphthoquinone (10 μl of 10 μg/ml) was used as an internal standard. Pharmacokinetic parameters were calculated using the pharmacokinetic software PK Solutions 2.0 (Montrose, CO).
Evaluation of antitumor activity
The mouse melanoma B16F1 cell line was used to induce solid tumors by intradermal administration of 5 × 105 viable tumor cells suspended in 50 μl of minimum essential medium on the dorsal side of the C57BL/6J mice. Once the tumor became palpable, diameters in three perpendicular planes was measured as explained earlier (CitationDevi & Rao, 1993) and tumor volume (V) was calculated from the formula:
where D1; D2; D3 are tumor diameters along three perpendicular planes.
The free plumbagin as well as microspheres (F6) were evaluated for anti-tumor efficacy in mice bearing solid tumors (n = 8/group). The microspheres equivalent to a dose of 6 mg/kg plumbagin were suspended in a vehicle (consisting of aqueous solution of 0.5% w/v sodium carboxymethyl cellulose, viscosity of 9 cps, 0.9% w/v sodium chloride). Once the tumor size reached 100 ± 10 mm3, the animals were injected (50 μl) using 21-gauge needle intramuscularly (i.m.) with free plumbagin or formulated at a dose of 6 mg/kg on the first, third, and seventh days. After the treatment, the tumor response was assessed by volume doubling time (VDT) and growth delay (GD), and the animals were observed further for survival. The actuarial survival curves were plotted using Kaplan-Meier analysis. Some of the tumor-bearing animals which were under stress due to excess tumor burden were terminated from the study due to humane considerations and were considered as censored in the survival analysis.
Hematological toxicity
The hematological toxicity of the free drug and of the plumbagin microspheres was evaluated using blood collected by retro-orbital vein using heparinized propylene tubes. Blood parameters such as total RBC and WBC counts, hemoglobin, hematocrit values, and platelet measurements were obtained by the using automated blood cell counter PCE-210 Erma (Japan).
Statistical analysis
The data obtained from the experiments was analysed by GraphPAD InStat Software (California, USA). Statistical analysis was performed using Kaplan-Meier analysis for survival, the Student’s t-test, One-way ANOVA followed by Tukey post-hoc test for comparison between the groups. For survival studies, a statistical significance (p < 0.05) of the mean values between the treatment groups were analyzed using Mantel-Cox Log Rank test. A p-value below 0.05 was considered to be significant.
Results and discussion
Preparation and characterization of microspheres
The aim of the present study was to develop and evaluate the chitosan-based microspheres of plumbagin for its anti-tumor efficacy and reduced systemic toxicity. Plumbagin-loaded chitosan microspheres were prepared by emulsion cross-linking method using glutaraldehyde (GA) as a cross-linking agent. Chitosan as a biodegradable polymeric drug carrier in sustained release technology offers potential advantages for the prolonged release of low molecular weight compounds. Molecules such as 5-flurouracil (CitationAkbuga & Bergisadi, 1996) and cisplatin (CitationAkbuga & Bergisadi, 1999) have been successfully incorporated into chitosan microspheres. In addition, cross-linking of the chitosan can potentially influence the release pattern of the loaded drug. Glutaraldehyde is reported as the best cross-linking agent for various classes of drugs (CitationDesai & Park, 2005). Hence, in the present study, glutaraldehyde was used as a cross-linking agent. The concentration of plumbagin was kept constant in all the prepared formulations. The particle size and size distribution of microspheres was recorded by laser light diffraction technique. The mean particle size of the optimized F6 formulation was found to be 106.35 μm. The SEM images revealed that chitosan microspheres (F6) were spherical with porous outer surface, as shown in . The roughness on the surface of the microspheres may be attributed to the removal of the drug crystals present on the surface during washing.
Figure 1. Scanning electron micrograph of plumbagin-loaded chitosan microspheres (a) ×35 and (b) ×100.

The encapsulation efficiency varied from 30–80% by varying the concentration of chitosan and the volume of cross-linking agent added during formulation, as shown in . The studies indicated an improved encapsulation of the drug from 30.12% to 65.12% when the concentration of the chitosan was increased from 1 to 1.5% w/v (F1 and F2). However, further increase in the chitosan concentration did not improve the drug encapsulation (F3 and F4). When the volume of liquid paraffin oil, continuous phase, (F5) was increased, entrapment efficiency was increased to 68.74%. This efficiency was further increased to 80.12% in F6 by increasing the volume of cross-linking agent. The microspheres prepared with high concentration of chitosan (F7 and F8) resulted in decreased entrapment efficiency with slight aggregation of the particles. It could be due to saturation of the polymer in the formulation or may be due to insufficient amount of cross-linking agent. Since highly concentrated solutions made the addition procedure difficult, the optimum concentration of chitosan was chosen as 1.5% w/v.
Table 2. Characteristics of plumbagin-loaded chitosan microspheres.
A preliminary study conducted for optimum selection of chitosan concentration revealed that the formulated microspheres with chitosan concentration less than 1% showed coagulation and concentration above 3% w/v made the addition process difficult with decreased entrapment efficiency, and also resulted in particle aggregation due to greater viscosity of the polymeric solution. Hence, chitosan concentration of 1–3% by weight was chosen for the optimization experiments. Also, the stirring rate was optimized in the pilot batches and the stirring speed was kept constant at 1500 rpm for the preparation of microspheres due to uniform particle size distribution. The increase in the stirring speed above 1500 rpm resulted in decreased encapsulation efficiency and microspheres of smaller particle size were observed, whereas below 1000 rpm the size of the formed microspheres was bigger and irregular in shape.
Compatibility studies using differential scanning calorimetric analysis
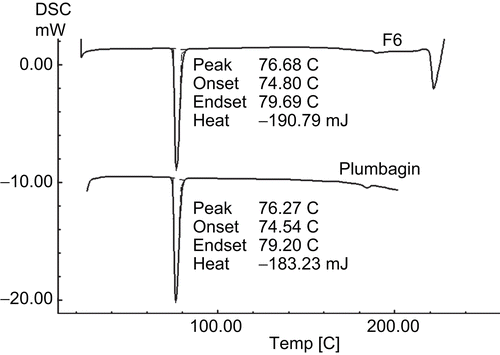
The possible interaction between the drug and the excipients was studied by DSC. The DSC thermograms of pure plumbagin and the optimized F6 formulation are shown in . Pure plumbagin showed a sharp endotherm at 76.27°C, while the formulated plumbagin (F6) showed at 76.68°C, corresponding to its melting point/transition temperature, suggesting no interaction between the drug and other excipients used in this formulation.
Figure 2. Differential scanning calorimetric thermograms of pure plumbagin and plumbagin-loaded chitosan microspheres of formulation F6.
In vitro release study
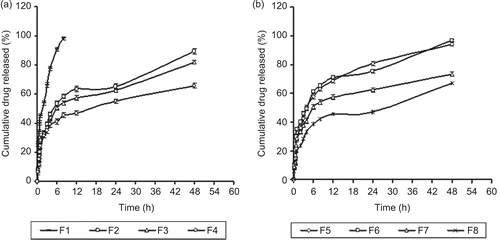
The effect of chitosan concentration on the in vitro release profile was studied and the release profiles of plumbagin from the formulated chitosan microspheres are shown in . The results of release profile of formulation F1 (1% chitosan w/v) showed a faster release of 98.98 ± 1.2 in 12 h, whereas F2–F4 showed 89.32 ± 1.58, 81.9 ± 1.29, and 65.89 ± 1.59 at 48 h, respectively, where a constant volume (2 ml) of cross-linking agent was used. However, no difference in the release rate was observed in the formulations F5 and F6, which showed 94.94 ± 1.25 and 96.72 ± 1.3 release at 48 h when the glutaraldehyde volume was increased from 2 to 3 ml. The release profiles of F7 and F8 were found to be 73.47 ± 1.39 and 67.21 ± 1.58 at 48 h. It is observed that the increase in chitosan concentration to 3% w/v (F4 and F8) showed a retarded release rate of 67.21 ± 1.39 at 48 h. This may be attributed to both the low solubility and the higher viscosity of the gel layer formed around the drug particles at higher concentrations of chitosan on contact with the dissolution medium and also it might be due to increase in the thickness of the outer layer of microspheres (CitationNishioka et al., 1989). In the present study, increase in the volume of cross-linking agent (F5 and F6) did not result in significant difference in the release properties of plumbagin-loaded microspheres. These results were similar to the earlier reports (CitationThanoo et al., 1992) where there was no significant difference observed in the release properties of cisplatin-loaded microspheres cross-linked with different volumes of glutaraldehyde aqueous solution.
Figure 3. Release profile of plumbagin from chitosan based microspheres as a function of drug loading (n = 3) (a) Formulations F1–F4, (b) Formulations F5–F8.
Analysis of release mechanism
The curvilinear nature of the cumulative percent drug release vs time (hour) plot () suggests that the formulation F6 did not followed zero order release kinetics (R2 = 0.635). The F6 release data, when treated using Higuchi model, showed a regression value R2 = 0.8704, while with the dissolution data, when plotted in accordance with the Korsemeyer-Peppas method, the calculated n-value of 0.611 with R2 = 0.9729 suggest that the F6 formulation release mechanism followed anomalous (non-fickian) transport. It means that water diffusion and also the polymer relaxation have an essential role in the drug release. The correlation coefficient of 0.9492 obtained when the release data fitted to first order kinetics suggest that the drug release was matrix drug load dependent.
The formulation F6 with optimized release characteristics and particle size distribution was further evaluated for its pharmacokinetic and pharmacodynamic profile.
Bioanalytical method validation
Linearity
The peak area ratio of plumbagin to I.S. in mice plasma was linear with respect to the analyte concentration over the range of 0.1–10 μg/ml. The correlation coefficient (r2) for plumbagin was greater than 0.996 over the concentrations used. The LLOQ was established as 0.1 μg/ml, the lowest concentration on the calibration curve that could quantify plumbagin with acceptable precision and accuracy.
Precision and accuracy
The results shown in indicate that this method is reproducible for replicate analysis of plumbagin in mice plasma. The mean precision defined by RSD ranged from 3.29–10.14%, whereas the mean accuracy ranged between 87.30–97.91%.
Table 3. Precision and accuracy of the HPLC method for determining plumbagin concentrations in mice plasma.
Recovery
As per the earlier reported methods, ethyl acetate, a widely used extracting solvent because of its high polarity and volatility, was chosen for liquid–liquid extraction method.
The use of ethyl acetate as extraction solvent resulted in ∼ 62–69% recovery of plumbagin from mice plasma.
Pharmacokinetics
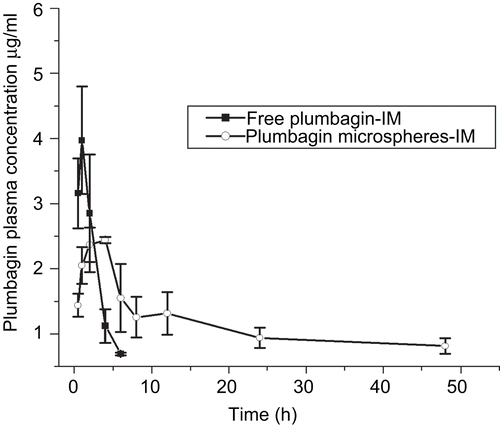
The plasma concentration time profile of free plumbagin and the optimized formulation (F6) are shown as . The plasma drug concentration for free plumbagin was detectable only up to 6 h, which may be due to the faster clearance leading to very short biological half-life (CitationChandrasekaran & Nagarajan, 1981). On the other hand, plasma concentrations of plumbagin from the microspheres were detectable up to 48 h, with a drastic increase in elimination half-life (22.2-fold increase). Also, there was a significant alteration in the pharmacokinetic parameters when plumbagin was formulated as microspheres with 9.28-fold increase in area under concentration-time curve AUC(0–∝), 2.50-fold increase in Tmax, and 24.94-fold increase in the mean residence time. This may be attributed to the sustained release of plumbagin from chitosan microspheres up to 48 h, which correlates well with the in vitro release data. The detection of plumbagin from microspheres was not possible at 72 h interval as the drug might have got completely eliminated or it might be below the limits of detection.
Figure 4. Pharmacokinetic profiles of free plumbagin and encapsulated chitosan microspheres after intramuscular injection in tumor-bearing C57BL/6J mice at a single dose of 6 mg/kg. The data represents the mean ± SD (n = 4).
Pharmacodynamics
Results obtained from the treatment of mice bearing B16F1 solid tumor is summarized in . Administration of both free plumbagin and optimized plumbagin-loaded chitosan microspheres (F6) resulted in significant inhibition of tumor growth, whereas the tumors in the untreated animals grew rapidly and reached to a mean size of 5.7 cm3 within 14 days. As can be seen in , treatment with plumbagin microspheres resulted in delayed tumor growth (p < 0.05) when compared with free plumbagin at the same dose level. However, the initial volume doubling time (VDT) values did not differ significantly. The observed increase in GD value () with the microspheres (F6) could be explained due to the slow release of the plumbagin from the formulation up to 48 h leading to the prolonged systemic circulation of the drug as evidenced in pharmacokinetic data. Plumbagin, being a quinone moiety, is said to exhibit its anti-cancer activity by generation of reactive oxygen species (ROS), and has been found to inhibit the activity of topoisomerase II (Topo II) through the stabilization of the Topo II-DNA cleavage complex. The ROS have been recognized as key molecules, which selectively can modify the proteins and hence regulate the cellular signaling including apoptosis (CitationKawiak et al., 2007). However, multiple administration of free plumbagin or its formulation did not exhibit complete regression of the tumor, as the total quantity of the plumbagin released might not have been enough to completely inhibit the growth kinetics of tumor cells.
Figure 5. Tumor growth inhibition by intramuscular injection of free plumbagin and optimized encapsulated chitosan microspheres formulation (F6) in tumor-bearing C57BL/6J mice. Murine B16F1 melanoma cells (5 × 105) were inoculated subcutaneously on the dorsal side of the mice and the experiment commenced once the tumor reached 100 ± 10 mm3. The animals were administered intramuscularly with vehicle (0.5% w/v sodium carboxymethyl cellulose in saline), free plumbagin, and F6 formulation on 1st, 3rd and 7th day at a dose of 6 mg/kg body weight. The data represents the mean of eight animals ± SD. Significant levels: * p < 0.001 compared with vehicle-treated group, a p < 0.05 compared to free plumbagin-treated group, b p < 0.001 compared to free plumbagin-treated group.
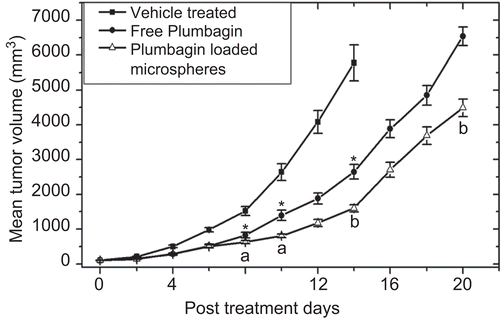
Table 4. Anti-tumor efficacy of plumbagin-loaded chitosan microspheres in C57BL/6J mice bearing melanoma B16F1 solid tumor.
Table 5. Pharmacokinetic parameters of plumbagin encapsulated as microspheres after intramuscular injection in tumor-bearing mice at a dose of 6 mg/kg (n = 4).
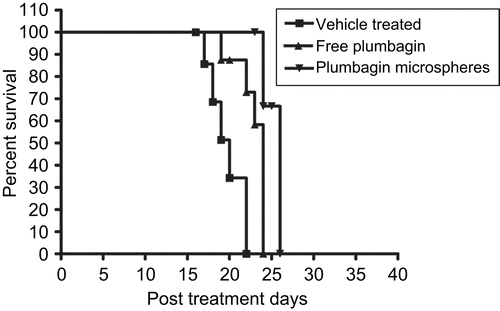
Survival of the mice carrying B16F1 tumors in response to the above treatments was also determined. The results are summarized as Kaplan-Meier plots in . The median survival in days for vehicle treated control, free plumbagin, and formulated plumbagin microspheres were found to be 20, 24, and 26 days, respectively. Also, the formulated plumbagin microspheres showed an increase in life span percent (ILS %) of 30% against 20% with that of the free plumbagin alone.
Figure 6. Effects of free plumbagin and plumbagin-loaded microspheres (F6) on the survival of C57BL/6J mice inoculated with B16F1 cells. The treatment groups and schedules were as mentioned in .
Hematological toxicity parameters
The administration of plumbagin in multiple doses has shown little effect on the total RBC count. However, there was an increase in the percentage of granulocytes in both the treated groups (free plumbagin as well as formulated microspheres F6) when compared with the respective vehicle treated controls on day 1 and day 15. We observed that animals treated with plumbagin microspheres had improved mean platelet counts compared to free plumbagin treated group (). The reason behind the reduced systemic toxicity for microspheres (F6) could be attributed to the slow release of the plumbagin from the microspheres over a period of 48 h in comparison to the free plumbagin, which is released in ∼ 6 h duration as evidenced in pharmacokinetics study. Also, the survival data further supports the reduction in the systemic toxicity with the plumbagin-loaded microspheres with increased life span (ILS) of 10% in comparison to free plumbagin.
Table 6. Effect of free plumbagin and plumbagin-loaded chitosan microspheres for F6 formulation on hematological parameters on 1st and 15th day of post-treatment (n = 4).
Conclusions
Microspheres are known to reduce the toxicity profile of a number of chemotherapeutic agents. The optimized chitosan microspheres (F6) prepared were well formed and exhibited good drug loading efficiency. Our results concluded that the intramuscular administration of the chitosan microspheres improved the anti-tumor efficacy of plumbagin with reduced systemic toxicity. Thus, the present investigation demonstrates the ability of biodegradable polymer chitosan in providing a promising delivery system such as microspheres which is safe and cost effective in the delivery of anti-cancer agents like plumbagin for better therapeutic efficacy and reduced toxicity.
Acknowledgement
The authors are thankful to Dr K. Satyamoorthy, Director, Manipal Life Sciences Centre, Manipal University, Manipal for providing the facilities and encouragement during this study.
Declaration of interest
The financial support from Indian Council of Medical Research (ICMR), New Delhi, India (IRIS No: 2005-00150), to carry out this study is acknowledged.The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Akbuga, J., Bergisadi, N. (1996). 5-Fluorouracil-loaded chitosan microspheres: preparation and release characteristics. J Microencapsul 13:161–8.
- Akbuga, J., Bergisadi, N. (1999). Effect of formulation variables on cis-platin loaded chitosan microsphere properties, J Microencapsul 16:697–703.
- Chandrasekaran, B., Nagarajan, B. (1981). Metabolism of echitamine and plumbagin in rats. J Biosci 3:395–400.
- Chen, W.R., Adams, R.L., Carubelli, R., Nordquist, R.E. (1997). Laser-photosensitizer assisted immunotherapy: a novel modality for cancer treatment. Cancer Lett 115:25–30.
- Dass, C.R., Choong, P.F. (2008). The use of chitosan formulations in cancer therapy. J Microencapsul 25:275–9.
- Desai, K.G., Park, H.J. (2005). Preparation of cross-linked chitosan microspheres by spray drying: effect of cross-linking agent on the properties of spray dried microspheres. J Microencapsul 22:377–95.
- Devi, P.U., Rao, B.S. (1993). Response of mouse sarcoma-180 to bleomycin in combination with radiation and hyperthermia. Strahlenther Onkol 169:601–7.
- Devi, P.U., Solomon, F.E., Sharada, A.C. (1994). In vivo tumor inhibitory and radiosensitizing effects of an Indian medicinal plant, Plumbago rosea on experimental mouse tumors. Indian J Exp Biol 32:523–8.
- Higuchi, T. (1963). Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci 52:1145–9.
- Hsieh, Y.J., Lin, L.C., Tsai, T.H. (2006). Measurement and pharmacokinetic study of plumbagin in a conscious freely moving rat using liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 844:1–5.
- Hsu, Y.L., Cho, C.Y., Kuo, P.L., Huang, Y.T., Lin, C.C. (2006). Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J Pharmacol Exp Ther 318:484–94.
- Jameela, S.R., Kumary, T.V., Lal, A.V., Jayakrishnan, A. (1998). Progesterone-loaded chitosan microspheres: a long acting biodegradable controlled delivery system. J Contr Rel 52:17–24.
- Kawiak, A., Piosik, J., Stasilojc, G. (2007). Induction of apoptosis by plumbagin through reactive oxygen species-mediated inhibition of topoisomerase II. Toxicol Appl Pharmacol 223:267–76.
- Kini, D.P., Pandey, S., Shenoy, B.D., Singh, U.V., Udupa, N., Umadevi, P., Kamath, R., Nagarajkumari Ramanarayan, K. (1997). Antitumor and antifertility activities of plumbagin controlled release formulations. Indian J Exp Biol 35:374–9.
- Korsmeyer, R.W., Gurny, R., Doelker, E., Buri, P., Peppas, N.A. (1983). Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm 15:25–35.
- Krishnaswamy, M., Purushothaman, K.K. (1980). Plumbagin: a study of its anticancer, antibacterial & antifungal properties. Indian J Exp Biol 18:876–7.
- Kuo, P.L., Hsu, Y.L., Cho, C.Y. (2006). Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol Cancer Ther 5:3209–21.
- Lin, S.Y., Chan, H.Y., Shen, F.H., Chen, M.H., Wang, Y.J., Yu, C.K. (2007). Chitosan prevents the development of AOM-induced aberrant crypt foci in mice and suppressed the proliferation of AGS cells by inhibiting DNA synthesis. J Cell Biochem 100:1573–80.
- Murata, J., Saiki, I., Nishimura, S., Nishi, N., Tokura, S., Azuma, I. (1989). Inhibitory effect of chitin heparinoids on the lung metastasis of B16-BL6 melanoma. Jpn J Cancer Res 80:866–72.
- Nair, S., Nair, R.R., Srinivas, P., Srinivas, G., Pillai, M.R. (2008). Radiosensitizing effects of plumbagin in cervical cancer cells is through modulation of apoptotic pathway. Mol Carcinog 47:22–33.
- Naresh, R.A., Udupa, N., Devi, P.U. (1996). Niosomal plumbagin with reduced toxicity and improved anticancer activity in BALB/C mice. J Pharm Pharmacol 48:1128–32.
- Nicholas, G.L. (1987). Sustained release dosage forms. In: Lachman, L., Lieberman, H.A., Kanig, J.L., eds. The theory and practice of industrial pharmacy. New York: Marcel Dekker, 433–7.
- Nishioka, Y., Kyotani, S., Masui, H., Okamura, M., Miyazaki, M., Okazaki, K., Ohnishi, S., Yamamoto, Y., Ito, K. (1989). Preparation and release characteristics of cisplatin albumin microspheres containing chitin and treated with chitosan. Chem Pharm Bull (Tokyo). 37:3074–7.
- Patashnik, S., Rabinovich, L., Golomb, G. (2005). Preparation and evaluation of chitosan microspheres containing bisphosphonates. J Drug Target 4:371–80.
- Powolny, A.A., Singh, S.V. (2008). Plumbagin-induced apoptosis in human prostate cancer cells is associated with modulation of cellular redox status and generation of reactive oxygen species. Pharm Res 25:2171–80.
- Prabaharan, M., Mano, J.F. (2005a). Chitosan-based particles as controlled drug delivery systems. Drug Deliv 12:41–57.
- Prabaharan, M., Mano, J.F. (2005b). Hydroxypropyl chitosan bearing beta-cyclodextrin cavities: synthesis and slow release of its inclusion complex with a model hydrophobic drug. Macromol Biosci 5:965–73.
- Prasad, V.S., Devi, P.U., Rao, B.S., Kamath, R. (1996). Radiosensitizing effect of plumbagin on mouse melanoma cells grown in vitro. Indian J Exp Biol 34:857–8.
- Purushothaman, K.K., Mohana, K., Susan, T. (1985). Biological profile of plumbagin. Bull Med Ethno-bot Res 6:177–88.
- Qi, L., Xu, Z., Chen, M. (2007). In vitro and in vivo suppression of hepatocellular carcinoma growth by chitosan nanoparticles. Eur J Cancer 43:184–93.
- Sandur, S.K., Ichikawa, H., Sethi, G., Ahn, K.S., Aggarwal, B.B. (2006). Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem 281:17023–33.
- Schipper, N.G., Olsson, S., Hoogstraate, J.A., DeBoer, A.G., Varum, K.M., Artursson, P. (1997). Chitosans as absorption enhancers for poorly absorbable drugs 2: mechanism of absorption enhancement, Pharm Res 14:923–9.
- Singh, U.V., Udupa, N. (1997). Reduced toxicity and enhanced antitumor efficacy of betacyclodextrin plumbagin inclusion complex in mice bearing Ehrlich ascites carcinoma. Indian J Physiol Pharmacol 41:171–5.
- Singh, U.V., Udupa, N. (1998). Methotrexate loaded chitosan and chitin microspheres—in vitro characterization and pharmacokinetics in mice bearing Ehrlich ascites carcinoma. J Microencapsul 15:581–94.
- Sinha, V.R., Singla, A.K., Wadhawan, S. (2004). Chitosan microspheres as a potential carrier for drugs. Int J Pharm 274:1–33.
- Solomon, F.E., Sharada, A.C., Devi, P.U. (1993). Toxic effects of crude root extract of Plumbago rosea (Rakta chitraka) on mice and rats. J Ethnopharmacol 38:79–84.
- Srinivas, P., Gopinath, G., Banerji, A., Dinakar, A., Srinivas, G. (2004). Plumbagin induces reactive oxygen species, which mediate apoptosis in human cervical cancer cells. Mol Carcinog 40:201–11.
- Sugie, S., Okamoto, K., Rahman, K.M., Tanaka, T., Kawai, K., Yamahara, J., Mori, H. (1998). Inhibitory effects of plumbagin and juglone on azoxymethane-induced intestinal carcinogenesis in rats. Cancer Lett 127:177–83.
- Thanoo, B.C., Sunny, M.C., Jayakrishnan, A. (1992). Cross-linked chitosan microspheres: preparation and evaluation as a matrix for the controlled release of pharmaceuticals. J Pharm Pharmacol. 44:283–6.
- Thasni, K.A., Rakesh, S., Rojini, G., Ratheeshkumar, T., Srinivas, G., Priya, S. (2008). Estrogen-dependent cell signaling and apoptosis in BRCA1-blocked BG1 ovarian cancer cells in response to plumbagin and other chemotherapeutic agents. Ann Oncol 19:696–705.
- Tiwari, S.B., Pai, R.M., Udupa, N. (2002). Temperature sensitive liposomes of plumbagin: characterization and in vivo evaluation in mice bearing melanoma B16F1. J Drug Target 10:585–91.
- Vijayakumar, R., Senthilvelan, M., Ravindran, R., Devi, R.S. (2006). Plumbago zeylanica action on blood coagulation profile with and without blood volume reduction. Vascul Pharmacol 45:86–90.
- Zhu, K.J., Li, Y., Jiang, H.L., Yasuda, H., Ichimaru, A., Yamamoto, K., Lecomte, P., Jerome, R. (2005). Preparation, characterization and in vitro release properties of ibuprofen-loaded microspheres based on polylactide, poly(ϵ-caprolactone) and their copolymers. J Microencapsul 22:25–36.