Abstract
The objective of this study was to enhance the solubility, dissolution rate, and oral bioavailability of a very poorly water-soluble anti-fungal agent, ketoconazole (KET), by inclusion complexation with a highly-soluble cyclodextrin derivative, hydroxypropyl-β cyclodextrin (HP-β-CD). Two groups of tablets containing KET alone and KET:HP-β-CD (1:2) kneaded product (KP) including magnesium stearate and lactopress (anhydrous and spray-dried) as excipients were prepared by direct compression method. After the characterization studies, the in vitro dissolution studies of these tablets in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) were carried out. To evaluate the in vivo bioavailability, the tablets were administered orally to rabbits and drug levels in serum were determined by HPLC. Tablets containing the cyclodextrin complex showed a higher in vitro dissolution rate and bioavailability compared to the tablets containing KET alone.
Introduction
Ketoconazole (KET) is a broad-spectrum anti-fungal imidazole derivative which interferes with the synthesis of ergosterol and therefore alters the permeability of the cell membrane of sensitive fungi (CitationBennett, 2001; CitationKarasulu et al., 2002). It is administered topically or orally and practically insoluble in water. The major drawback in the therapeutic application of this weakly acidic drug, as an oral dosage form, is its low aqueous solubility because of its hydrophobic structure (CitationBudavari, 1989; CitationKovacs et al., 2009). This results not only in slower dissolution and absorption rates on oral administration but it is also one of the causes for local irritation to the gastric mucosa, leading to gastrointestinal side-effects (CitationLelawongs et al., 1988; CitationGovindarajan & Nagarsenker, 2005).
Compounds with aqueous solubilities lower than 100 µg/ml often present dissolution limitations to absorption. The surface area of a drug available for dissolution depends on the particle size of the solid and its ability to be wetted by gastrointestinal fluids. Various physicochemical and physiological factors influence the saturation solubility of a drug in the gastrointestinal tract. One possibility for improving solubility by complexation is incorporation with cyclodextrins (CDs) (CitationHörter & Dressman, 1997; CitationChen et al., 2007).
CDs are a group of torus shaped oligosaccharides that are formed by the enzymatic cyclization of starch. They are cylindrical-shaped molecules with a hydrophilic outer surface and a rather hydrophobic cavity in the center. The CDs are capable of forming inclusion complexes with many drugs by taking up a whole drug molecule (guest molecule), or some part of it, into the central cavity. Such molecular encapsulation will affect many of the physicochemical properties of the drug, such as its chemical stability and solubility (CitationLoftsson et al., 2005; CitationBrewster & Loftsson, 2007).
Hydroxypropyl-β-cyclodextrin (HP-β-CD) has high solubility (> 50%) in water and is expected to be a useful drug carrier because of its excellent ability to improve the solubility dissolution rate and bioavailability of poorly water-soluble drugs (CitationPitha & Pitha, 1985; CitationSzente & Szejtli, 1999; CitationMiyake et al., 2000). HP-β-CD has been reported to form an inclusion complex with KET and improve its solubility (CitationEsclusa-Diaz et al., 1996a; Citationb; CitationTaneri et al., 2002). The reported studies have shown that HP-β-CD is able to increase the oral bioavailability of various drugs (CitationPuglisi et al., 1995; CitationBrewster et al., 1997).
The purpose of the present study was to evaluate the improvement of dissolution and oral bioavailability of KET after designed inclusion complexation with HP-β-CD in tablet formulations.
Materials and methods
Materials
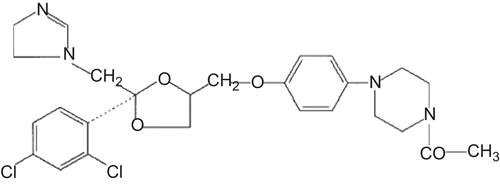
Ketoconazole (KET), cis-1-acetyl-4-{4-[2-(2,4-dichloro-phenyl)- 2-imidazol-1-ylmethyl-1,3-dioxalan-4-ylmethoxy]- phenyl} piperazine () was kindly donated by Ilsan Iltas Pharmaceuticals Co. (Istanbul, Turkey); hydroxypropyl-β-CD (HP-β-CD), DS: 5.5 was a generous gift from Cerestar, Inc. (Hammond, IN, USA); lactopress anhydrous and spray-dried were supplied from Borculo Domo Ingredients (Zwolle, Netherlands); clotrimazole (CLT) was kindly supplied by Orva Pharmaceuticals Ltd. (Izmir, Turkey). All other chemicals used were of analytical grade.
Figure 1. Chemical structure of KET.
Preparation of kneaded products (KPs)
Products of KET: HP-β-CD were prepared in 1:2 guest:host molar ratios by kneading method, as reported previously (CitationTaneri et al., 2003a; Citationb). Briefly, physical mixtures of KET and HP-β-CD were mixed in the same quantity of a methanol:water (1:1) mixture and kneaded until the bulk of the solvent mixture evaporated. Consequently, they were dried at room temperature and then in a drying oven at 105°C, for 24 h and 5 min, respectively. Next, they were pulverized and sieved to a fine particle size of 100 μm.
Preparation of oral tablets
Tablets containing 59.2 mg KET:HP-β-CD (1:2) KP product including magnesium stearate and lactopress anhydrous as excipients were prepared by using direct compression methods with a punch size of 4 mm and a hydraulic press at a constant pressure of 1.5 tons for 15 s. Tablets containing 10 mg KET alone were also prepared with the same excipients and methods for comparison. The compositions of the tablets are given in .
Table 1. Composition of tablet formulations containing different ingredients (mg).
Physical characterization of tablets
Formulated tablets were subjected to different physical characterization studies. The weight variation was determined on 20 tablets using an electronic balance (Sartorius, Germany). Tablet hardness was determined for a minimum of six tablets using a Monsanto (Standard type, Cambridge, UK) tablet hardness tester. Ten tablets were taken and their thickness was recorded by a digital caliper (Mitutoyo, Kawasaki, Japan). Friability was calculated as the percentage weight loss of 20 tablets after rotation using a Roche friabilator (Basel, Switzerland) for 4 min at 25 rpm.
Drug content of tablets
The drug content of the formulated tablets was estimated spectrophotometrically (Shimadzu UV-160A) after extraction in the methanol. Ten tablets were finely powdered; powder sample equivalent to 10 mg of KET was accurately weighed and transferred to a 100 mL volumetric flask. Initially ∼ 50 mL of methanol was added to the volumetric flask and allowed to stand for 6 h with intermittent sonication to ensure complete solubility of the drug. Then the volume was made up with methanol and the mixture was centrifuged. The supernatant liquid was filtered through a 0.2 µm membrane filter and KET was analysed spectrophotometrically at a wavelength of 245 nm.
In vitro dissolution studies
The dissolution studies were carried out on the tablets by using the USP dissolution apparatus (type DT, Offenbach/Main, Germany), with a modified paddle method at 100 rpm both in 100 mL simulated gastric fluid (SGF) (pH:1.2) and simulated intestinal fluid (SIF) without enzymes (pH:6.8) maintained at 37 ± 0.5°C during 2 h. Sampling was performed after 2.5, 5, 7.5, 10, 12.5, 15, 20, 25, 30, 40, 50, 60, 75, 90, 105, and 120 min and then fresh medium was added to maintain the original volume. The KET contents were determined spectrophotometrically at 272 and 292 nm for SGF and SIF, respectively. All the studies were carried out in triplicate.
In vivo studies
The followed protocol was approved by the Ege University, Faculty of Medicine, Ethical Committee. Tablets with 4 mm diameter were administered orally via a silicone rubber gastric intubation tube to male albino rabbits with avarage weight 2.5 kg which were housed individually in standard cages in a room with air, humidity, and temperature control. The rabbit’s mouth was opened, and a wooden rod was inserted between the jaws. A gastric tube was centrally placed to the hole. Tablets were set on the tip of a gastric intubation tube, and administered into the stomach of rabbits. After receiving the oral dose, 5 ml of water was administered to facilitate the accession of tablets (CitationMaeda et al., 1979; CitationIshikawa et al., 2000). Rabbits were fasted overnight but had free access to water; 2–3 mL blood samples were withdrawn from a heparinized branule (G22, G24), placed in the marginal vein, at each of the pre-determined times after administration: 10, 20, 30, 60, 90, 120, 180, 240, 300, 360, 420, 480, 600, and 720 min. Samples were transferred to 4 mL blood tubes and centrifuged at 3500 rpm for 10 min by IEC PR-6000 Centrifuge (Damon, USA). Separated supernatant was taken into Eppendorf tubes (Damon, Massachusetts, USA) and stored at −20°C until analyzed.
Analytical method
The KET and internal standard (CLT) were extracted from the serum by using solid-phase extraction (SPE) columns. Extraction columns (ODS C-18, Accubond II SPE, Agilent, Palo Alto, CA, USA) were conditioned with 6 mL of distilled water and methanol, respectively. After adsorption of 1 mL serum sample under 20 mm Hg pressure with 3 mL/min flow rate, alkalinized with 0.25 mL 0.1 M NaOH and 100 µL internal standard (3 mg/ mL CLT) is added to the columns under vacuum. Serum components are separated away by washing with 9 mL distilled water. KET is eluted from the column with 1 mL of methanol into a test tube. The methanolic extract is evaporated to dryness at 45°C under a gentle stream of nitrogen. The residue is reconstituted in 200 μL methanol after ultrasonication, filtrated from 0.45 μm pore sized membrane filter, and injected into the chromatographic column (CitationSwezey et al., 1982; CitationRiley, 1986; CitationPietra et al., 1992; CitationVelikinac et al., 2004).
The drug serum levels were determined by HPLC method. The HPLC system consisted of a LC-6A model pump, equipped with a UV detector operated at a 254 nm wavelength, an injector with a 100 µL loop, and a CR-4A recorder. The stationary phase was a Nucleosil C18 reversed phase column (250 mm × 46 mm, id., 5 µm mean particle size) maintained at 30°C. A mixture of methanol:acetonitrile:0.02 M KH2PO4 buffer (pH 6.8) in the ratio 35:30:35, delivered at a flow rate of 1.2 mL/min, was used as a mobile phase. The retention times of KET and CLT were determined as 22,318 min and 31,770 min, respectively. The overall analysis time was 90 min. The limit of detection was 7 µg/mL in serum. Standard curves were developed with rabbit blank serum in the range of 1–10 µg/µL. The method was linear between this range of concentrations (r2 = 1).
Pharmacokinetic and statistical analysis of the data
The KET concentration-time data were evaluated for the extent and rate of KET absorption. Pharmacokinetic parameters were evaluated by using the stripping method (Least Sum of Squares) and the results were examined according to the single compartment kinetic model. The area under the serum concentration vs time curve was determined by linear and log trapezoidal methods, from 0 to infinity (AUC). The maximum plasma concentration of drug in plasma (Cmax) was used as a measure of the extent of absorption, and the time to Cmax (tmax) was calculated.
All the data was statistically analyzed by analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. p-value of less than 0.05 was considered as evidence of a significant difference.
Results and discussion
The physicochemical, biopharmaceutical, and thermoanalytical properties of inclusion complexation of KET with CDs were investigated previously (particle size distribution, surface area, partition coefficient, heat of dissolution, and wettability studies). These results suggest that HP-β-CD form a relatively stable inclusion complex with KET (CitationTaneri et al., 2003a; Citationb).
Physical characterization of the tablets
Physical appearance, tablet hardness, friability, weight variation, and drug content uniformity of all formulations were found to be satisfactory, as can be observed from the data in . Average tablet weight of the formulations obtained of all the experimental runs had a range of 69.8–70.4 mg and was found to meet the pharmacopoeial requirements regarding the uniformity of weight. The drug contents of all the batches of tablets prepared were more than 99% of the theoretical content. Hardness values for all formulations varied from 2.5–3.5 kg. Friability of all tablets was found to be less than 0.8% (CitationVatsaraj et al., 2002). Although F1 and F2 had the same hardness, F2 showed a higher friability value when compared with F1. It could be explained with the amount of spray-dried lactopress (65.6 and 16.4 mg for F1 and F2, respectively) which consists mainly of spherical particles of lactose monohydrate, resulting in very good flow, excellent tableting properties, and also is characterized by superior binding properties. Tablet thickness had a range of 2.00–2.04 mm and was considered constant for all formulations. These results suggested that the method of preparation of formulation is an acceptable method for preparing good quality tablets of KET or KP.
Table 2. Physical properties of tablets.
In vitro dissolution evaluations
The dissolution profiles of the tablets in SGF and SIF are plotted in and . Nearly 100% of the drug dissolved within the first 15 min from all tablet formulations in SGF (). The reason for initial higher release in the rate of KET can be explained as the drug was highly soluble in the gastric pH. Since KET is a weak base with pKa values of 2.94 and 6.51, an acid medium is required to transform the drug into the soluble hydrochloride salt (CitationHoeschele et al., 1994; CitationCordoba-Diaz et al., 2001).
Figure 2. In vitro dissolution profiles of tablets in SGF (mean ± SD, n = 3).
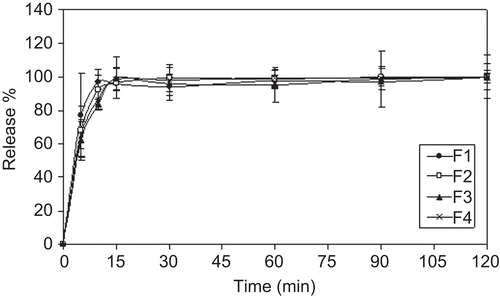
Figure 3. In vitro dissolution profiles of tablets in SIF (mean ± SD, n = 3).
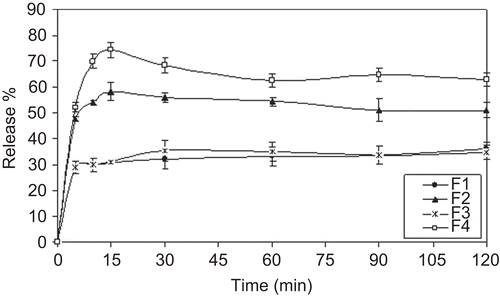
As pH increased, the rate and extent of dissolution slowed; for F1 and F3 coded tablets only 30% of KET was dissolved within the first 15 min in SIF (). KET precipitated in the dissolution medium depend on the high pH. CitationCarlson et al. (1983) reported the effect of pH on the in vitro dissolution and solubility of KET tablets. According to their results, KET precipitated rapidly from solution as the pH of the dissolution medium exceeded 5.5.
Depending on the in vitro release studies in SIF, F2, and F4 coded tablets showed better dissolution results compared to F1 and F3 coded tablets (). Moreover, the use of HP-β-CD increased the dissolution rate 2-fold. Within the first 10 min ∼ 70% of the drug dissolved from F4 tablets. This can be explained with the increase in solubility and wettability by inclusion complexation. The solubilizing effect of the HP-β-CD and complete amorphization of the drug resulted in improved dissolution of the drug from CD (CitationSavolainen et al., 1998).
In vivo studies
The increasing effects of HP-β-CD on the solubility and dissolution rate of KET are expected to enhance bioavailability of KET after oral administration. shows the plasma concentration-time profiles of KET after oral administration of the tablets containing KET and its HP-β-CD complexes to rabbits. The mean values of pharmacokinetic parameters for all formulations are summarized in .
Figure 4. KET serum concentration time profiles after oral administration of tablets in rabbits (mean ± SD, n = 6).
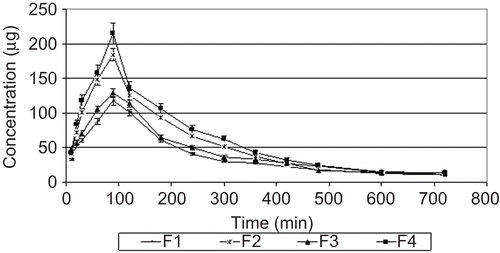
Table 3. Pharmacokinetic parameters for KET obtained following oral administration of tablet batch (n = 6).
Oral pharmacokinetics (Cmax, tmax, AUC0→720) were comparable after administration of tablets containing KET and tablets containing KP. Compared to administration of plain KET tablets, administration of inclusion complex tablets increased Cmax and AUC0→720 values of KET. F4 coded tablets showed 1.44- and 1.66-fold increase in AUC0→720 and Cmax values, respectively, when compared to F3 coded tablets. The ka value of F4 tablets showed a 1.73-fold increase compared with F3 tablets. These results support the observation of faster absorption of drug from CD, as suggested by a higher Cmax. A significant (p < 0.05) increase in the bioavailability is obtained with the tablets containing HP-β-CD inclusion complexes. These results indicate an improvement in the rate of absorption of the drug when it is administered as an inclusion complex with HP-β-CD. This proves the fact that the improvement in the dissolution rate of the drug improves its bioavailability.
In the literature, the bioavailability of cinnarizine was significantly enhanced, when administered in a capsule, by forming an inclusion complex with HP-β-CD, an effect attributed to the higher dissolution rate (CitationJarvinen et al., 1995). In another similar study, it has been observed that a greater therapeutic effect was obtained against the luminal infections in mouse with albendazole and HP-β-CD inclusion complexes (CitationEvrard et al., 2002).
Conclusions
In conclusion, this study indicates that tablets containing inclusion complexes could be useful in oral administration of KET to achieve increased solubility and improved bioavailability during the development of the formulations. Thus, HP-β-CD has a significant advantage for providing high aqueous solubility while maintaining a lack of toxicity and side-effects in oral pharmaceutical tablets of KET.
Acknowledgements
The authors wish to thank Professor M. Kata for providing scientific support.
Declaration of interest
This study is supported by Pharmaceutical Technology Research Support of Novartis Pharmaceuticals Co., Istanbul, Turkey. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bennett, J.E. (2001). Antifungal agents. In: Molinoff, P., ed. Goodman and Gilman’s The pharmacological basis of therapeutics, ed 10. New York: McGraw-Hill, 1175–90.
- Brewster, M.E., Anderson, W.R., Meinsma, D., Moreno, D., Webb, A.I., Pablo, L., Estes, K.S., Derendorf, H., Bodor, N., Sawchuk, R., Cheung, B., Pop, E. (1997). Intravenous and oral pharmacokinetic evaluation of a 2-hydroxypropyl-β-cyclodextrin-based formulation of carbamazepine in the dog: comparison with commercially available tablets and suspensions. J Pharm Sci. 86:335–9.
- Brewster, M.E., Loftsson, T. (2007). Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 59:645–66.
- Budavari, S. (1989). The Merck index. 11th ed. Budavari, S., ed. Rahway, NJ: Merck and Co., Inc., 835.
- Carlson, J.A., Mann, H.J., Canafax, D.M. (1983). Effect of pH on disintegration and dissolution of ketoconazole tablets. Am J Hosp Pharm. 40:1334–6.
- Chen, H., Gao, J., Wang, F., Liang, W. (2007). Preparation, characterization and pharmacokinetics of liposomes-encapsulated cyclodextrins inclusion complexes for hydrophobic drugs. Drug Deliv. 14:201–8.
- Cordoba-Diaz, D., Cordoba-Diaz, M., Awad, S., Cordoba-Borrego, M. (2001). Effect of pharmacotechnical design on the in vitro interaction of ketoconazole tablets with non-systemic antacids. Int J Pharm. 226:1–8.
- Esclusa-Diaz, M.T., Gayo-Otero, M., Pérez-Marcos M.B., Vila-Jato, J.L., Torres-Labandeira, J.J. (1996a). Preparation and evaluation of ketoconazole-β-cyclodextrin multicomponent complexes. Int J Pharm. 142:183–7.
- Esclusa-Diaz, M.T., Guimaraens-Mendez, M., Perez-Marcos, M.B., Vila-Jato, J.L., Torres-Labandeira, J.J. (1996b). Characterization and in vitro dissolution behaviour of ketoconazole: β- and 2-hydroxypropyl-β-cyclodextrin inclusion compounds. Int J Pharm. 143:203–10.
- Evrard, B., Chiap, P., DeTullio, P., Ghalmi, F., Piel, G., Van Hees T., Crommen, J., Losson, B., Delattre, L. (2002). Oral bioavailability in sheep of albendazole from suspension and from a solution containing hydroxypropyl-β-cyclodextrin. J Contr Rel. 85:45–50.
- Govindarajan, R., Nagarsenker, M.S. (2005). Formulation studies and in vivo evaluation of a flurbiprofen-hydroxypropyl β-cyclodextrin system. Pharm Dev Tech. 1:105–14.
- Hoeschele, J.D., Roy, A.K., Pecoraro, V.L., Carver, P.L. (1994). In vitro analysis of the interaction between sucralfate and ketoconazole. Antimicrob Agents Chemother. 38:319–25.
- Hörter, D., Dressman, J.B. (1997). Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 25:3–14.
- Ishikawa, T., Watanabe, Y., Takayama, K., Endo, H., Matsumoto M. (2000). Effect of hydroxypropylmethylcellulose (HPMC) on the release profiles and bioavailability of a poorly water-soluble drug from tablets prepared using macrogol and HPMC. Int J Pharm. 202:173–8.
- Jarvinen, T., Jarvinen, K., Schwarting, N., Stella, V.J. (1995). β-cyclodextrin derivates, SBE- β-CD and HP- β-CD, increase the oral bioavailability of cinnarizine in beagle dogs. J Pharm Sci. 84:295–9.
- Karasulu, H.Y., Taneri, F., Sanal, E., Guneri, T., Ertan, G. (2002). Sustained release bioadhesive effervescent ketoconazole microcapsules tabletted for vaginal delivery. J Microencapsul. 19:357–62.
- Kovacs, K., Stampf, G., Klebovich, I., Antal, I., Ludányi, K. (2009). Aqueous solvent system for the solubilization of azole compounds. Eur J Pharm Sci. 36:352–8.
- Lelawongs, P., Barone, J.A., Colaizzi, J.L., Hsuan, A.T.M., Mechlinski, W., Legendre, R., Guarnieri, J. (1988). Effect of food and gastric acidity on absorption of orally administered ketoconazole. Clin Pharm. 7:228–35.
- Loftsson, T., Hreinsdottir, D., Mason, M. (2005). Evaluation of cyclodextrin solubilization of drugs. Int J Pharm. 302:18–28.
- Maeda, T., Takenaka, H., Yamahira, H., Noguchi, T. (1979). Use of rabbits for GI drug absorption studies: physiological study of stomach-emptying controlled rabbits. Chem Pharm Bull. 27:3066–72.
- Miyake, K., Arima, H., Hirayama, F., Yamamoto, M., Horikawa, T., Sumiyoshi, H., Noda, S., Uekama, K. (2000). Improvement of solubility and oral bioavailability of rutin by complexation with 2-hydroxypropyl-cyclodextrin. Pharm Dev Tech. 5:399–407.
- Pietra, A.M., Cavrini, V., Andrisano, V., Gatti, R. (1992). HPLC analysis of imidazole antimycotic drugs in pharmaceutical formulations. J Pharm Biomed Anal. 10:873–9.
- Pitha, J., Pitha, J. (1985). Hydroxypropyl-β-cyclodextrin: preparation and characterization; effects on solubility of drugs. Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J Pharm Sci. 74:987–90.
- Puglisi, G., Ventura, C.A., Spadaro, A., Campana, G., Spampinato, S. (1995). Differential effects of modified β-cyclodextrins on pharmacological activity and bioavailability of 4-biphenylacetic acid in rats after oral administration. J Pharm Pharmacol. 47:120–3.
- Riley, C.M. (1986). Determination of ketoconazole in the plasma, liver, lung and adrenal of the rat high-performance liquid chromatography. J Chrom Biomed Appl. 377:287–94.
- Savolainen, J., Jarvinen, K., Matilainen, L., Jarvinen, T. (1998). Improved dissolution and bioavailability of phenytoin by sulfobutylether-β-cyclodextrin ((SBE)7m-β-CD) and hydroxypropyl-β-cyclodextrin (HP-β-CD) complexation. Int J Pharm. 165:69–78.
- Swezey, S.F., Giacomini, K.M., Abang, A., Brass, C., Stevens, D.A., Blaschke, T.F. (1982). Measurement of ketoconazole, a new antifungal agent, by high-performance liquid chromatography. J Chromatogr Biomed Appl. 227:510–5.
- Szente, L., Szejtli, J. (1999). Highly soluble cyclodextrin derivatives: chemistry, properties, and trens in development. Adv Drug Del Rev. 36:17–28.
- Taneri, F., Guneri, T., Aigner, Z., Berkesi, O., Kata, M. (2003a). Thermoanalytical studies on complexes of ketoconazole with cyclodextrin derivatives. J Therm Anal Calor. 74:769–77.
- Taneri, F., Guneri, T., Aigner, Z., Kata, M. (2002). Improvement in the physicochemical properties of ketoconazole through complexation with cyclodextrin derivatives. J Incl Phenom Macrocycl Chem. 44:257–60.
- Taneri, F., Guneri, T., Aigner, Z., Kata, M. (2003b). Influence of cyclodextrin complexation on the physicochemical and biopharmaceutical properties of ketoconazole. J Incl Phenom Macrocycl Chem. 47:15–23.
- Vatsaraj, N., Zia, H., Needham, T. (2002). Formulation and optimization of a sustained-release tablet of ketorolac tromethamine. Drug Deliv. 9:153–9.
- Velikinac, I., Cudina, O., Jankovic, I., Agbaba, D., Vladimirov, S. (2004). Comparison of capillary zone electrophoresis and high performance liquid chromatography methods for quantitative determination of ketoconazole in drug formulations. Il Farmaco. 59:419–24.