Abstract
Sodium alendronate is an effective treatment for osteoporosis, but its oral administration is associated with adverse gastrointestinal effects. The aim of this work was to evaluate gastroresistant sodium alendronate-loaded microparticles prepared by spray-drying using Eudragit® S100 or a blend of Eudragit® S100/Methocel® E4M. Both formulations presented high encapsulation efficiencies, mean diameters below 17 μm, and similar collapsed shape. Dissolution experiments showed good gastro-resistance for the microparticles at pH 1.2. At pH 6.8, the blended microparticles retarded the drug release. In vivo studies showed that the formulations were able to protect the rat stomachs against ulcer formation by sodium alendronate. In conclusion, the microparticles seems to be promising oral carriers for sodium alendronate.
Introduction
Sodium alendronate is an important drug in the treatment of post-menopausal osteoporosis and Paget’s disease (CitationLin, 1996; CitationPerez-Lopez, 2004). Due to its ability to interact with the calcium hydroxyapatite crystals, sodium alendronate accumulates in bone, inhibiting the activity of osteoclasts, the bone resorbing cells (CitationYates & Rodan, 1998; CitationEbetino et al., 2005). Clinical studies with post-menopausal women have demonstrated that the reduction of bone resorption due to the sodium alendronate administration leads to an increase of bone mineral density with a decreased fracture incidence (CitationAdami et al., 1995; CitationPerez-Lopez, 2004).
Like other bisphosphonates, oral sodium alendronate treatment is associated with injuries of the upper gastrointestinal tract, most notably in the esophagus (Citationde Groen et al., 1996; CitationGraham & Malaty, 1999; CitationLanza et al., 2000). Adverse effects such as gastroesophageal inflammation and ulceration, esophageal erosions, and gastrointestinal bleeding have been associated with the ability of sodium alendronate to damage the epithelium by topical irritant effects (CitationLanza, 2003; CitationSener et al., 2005; CitationMarshall et al., 2006). Patients presenting gastrointestinal problems or esophageal abnormalities such as achalasia ought to avoid the sodium alendronate treatment (CitationLanza, 2003). Moreover, sodium alendronate should be used with caution in patients who simultaneously require treatment with non-steroidal anti-inflammatory drugs, because the combination of both can accentuate the mucosal injury (CitationGraham & Malaty, 1999).
The dose instructions of sodium alendronate are designed to prevent the gastro-esophageal irritation and to optimize its absorption (CitationLi & Kendler, 2004). To avoid the occurrence of side-effects the sodium alendronate tablets should be swallowed with a full glass of water, and the patients should not lie down for at least 30 min in order to facilitate esophageal passage and to decrease mucosal adherence (Citationde Groen et al., 1996). Like other bisphosphonates, sodium alendronate presents low bioavailability after oral dose (less than 1%). Besides, its absorption decreases with food and drinks (CitationLin, 1996). For this reason, the tablet must be taken at least 30 min before the first food, beverage, or medication of the day with water only. Other beverages (including mineral water), food, and some medications are likely to reduce sodium alendronate absorption (CitationLin, 1996).
The cautions that must be taken in the ingestion of a tablet of sodium alendronate are inconvenient and lead to a poor therapeutic adherence (CitationReginster, 2006). The development of sodium alendronate oral formulations safer for the patient could enhance the therapeutic efficacy. The oral route is the most common and convenient for patients, and a reduction in dosing frequency enhances compliance (CitationLavelle, 2001). Among the systems designed for oral drug delivery, the polymeric microparticles are an interesting and new alternative to overcome the problems associated with sodium alendronate oral administration.
Over the last decades, polymeric microparticles have been widely studied to control and/or target the release of drugs (CitationFreiberg & Zhu, 2004; CitationVarde & Pack, 2004). Regarding the oral administration, polymeric microparticles have been developed in order to modify the pharmacokinetic profiles of drugs and to increase their bioavailability, giving a more uniformly spread out in the gastrointestinal tract and more reproducible gastrointestinal transport (CitationPalmieri et al., 2000; CitationFreiberg & Zhu, 2004; CitationKurkuri & Aminabhavi, 2004). These systems have also been studied to reduce the drug toxicity, mainly in the gastrointestinal tract (CitationPalmieri et al., 2000; CitationLuppi et al., 2009).
The advantages of the drug release rate manipulation by using polymer blend microparticles have been previously reported (CitationBlanco-Prieto et al., 2004; CitationLionzo et al., 2007; CitationPoletto et al., 2007), and a successful approach developed in our group showed that gastroresistance and controlled release microparticles containing pantoprazole, an anti-ulcer agent, can be prepared using Eudragit® S100 and hydroxypropyl methylcellulose (HPMC) (CitationRaffin et al., 2006; Citation2007; Citation2008).
Up to now, few reports in the literature show the preparation of bisphosphonate-loaded microparticles. Some studies were carried out to develop formulations for implantation or injection of the drug for the treatment of bone diseases (CitationPatashnik et al., 1997; CitationPerugini et al., 2001; CitationWeidenauer et al., 2003; Citation2004; CitationCzuryszkiewicz et al., 2005; CitationSamdancioglu et al., 2006; CitationNafea et al., 2007; CitationHuang et al., 2009; CitationShi et al., 2009a; Citationb). To the best of our knowledge, there is no report in the literature concerning the microencapsulation of sodium alendronate aiming the improvement of its gastrointestinal tolerance following oral administration. Taking those considerations into account, the objective of this work was to evaluate the ability of sodium alendronate-loaded gastroresistant microparticles in reducing the gastrointestinal toxicity of this drug after oral administration in rats. We hypothesized that the adequate selection of polymers to prepare the blend could furnish sodium alendronate-loaded microparticles capable to reduce the drug mucosal injury. In this way, Eudragit® S100 was chosen to confer gastroresistance, and HPMC was chosen to control the sodium alendronate release. Viewing a comparison, microparticles prepared solely with Eudragit® S100 were also investigated.
Materials and methods
Monosodium alendronate trihydrate was purchased from Henrifarma (São Paulo, Brazil). Eudragit® S100 was obtained from Almapal (São Paulo, Brazil) and Methocel® E4M was supplied by Colorcon (Cotia, Brazil). o-Phthalaldehyde was obtained from Invitrogen (Carlsbad, California, USA) and 2-mercaptoethanol was acquired from Acros Organics (Geel, Belgium). All other chemicals and solvents were of pharmaceutical grade and used as received.
Preparation of sodium alendronate-loaded microparticles
Microparticles were prepared by the spray-drying technique. For the preparation of Eudragit® S100 microparticles (MP-EUD), the polymer (8.0 g) was dissolved in 0.05 M NaOH (500 mL) at 50°C. Sodium alendronate (2.0 g) was added into the solution before spray-drying (MSD 1.0, LabMaq, Brazil). The operational conditions were: feed rate of 0.40 L/h, air flow rate of 500 NL/h, atomizing air pressure of 3.7 kgf/cm2, inlet temperature of 150°C, and nozzle diameter of 1.2 mm.
For the preparation of microparticles using a blend of Eudragit® S100 and HPMC E4M (MPE-E4M), the Eudragit® S100 (5.0 g) was dissolved in 0.05 M NaOH (500 mL) under magnetic stirring at 50°C. After dissolution, the mixture was added of HPMC (2.5 g) and kept under mechanical stirring for 10 min. The resultant gel was kept at 4°C for 48 h until the complete dissolution of HPMC. At 25°C, the formed gel presented a viscosity of 4.66 cP (DV-II+ PRO Digital Viscometer, Brookfield, Massachusetts, USA). Sodium alendronate (2.0 g) was added into the solution before spray-drying. The samples were collected from the cyclone and the powder collector. The operational conditions were the same as described above for the preparation of MP-EUD. Eudragit® S100 microparticles and Eudragit® S100/HPMC blend microparticles prepared without the drug (placebos) were used for comparison.
Determination of yield and encapsulation efficiency
The yield of the process was calculated by the ratio of the experimental weight and the sum of the weights of all components, discounting the water content. Concerning the encapsulation efficiency, a spectrophotometric method based on the derivatization of sodium alendronate with o-phthalaldehyde was used (CitationAl Deeb et al., 2004; CitationCruz et al., 2009). A portion of microparticles equivalent to 10 mg of sodium alendronate was dissolved in 50 mL of 0.2 M NaOH and filtered using a filter paper (Whatman n° 40). Ten milliliters of the filtrate were transferred to another 50 mL volumetric flask. Then, 4 mL of derivatizing reagent were added and the volume was completed with 0.2 M NaOH. After 30 min, the absorbance was measured at 333 nm (Cary 50 UV-Vis, Vankel). The encapsulation efficiency of each formulation was calculated based on the relation of the theoretical and the experimental sodium alendronate concentrations, and expressed as percentage. Each sample was assayed in triplicate.
Particle size measurements
Microparticles were analyzed for their size and size distributions by laser diffractometry (Malvern Mastersizer 2000, Malvern Instruments, Worcestershire, UK) after dispersion of microparticles in iso-octane. The particle sizes were expressed as the mean diameter over the volume distribution d(4.3) and the size distributions (span) were calculated using equation (1).
where d(0.1), d(0.5), and d(0.9) are, respectively, the particle diameters at 10%, 50%, and 90% of the undersized particle distribution curve.
Morphology of the particles
Shape and surface of the microparticles were examined by means of a scanning electron microscopy (SEM) (Jeol Scanning Microscope, JSM-5800, Tokyo, Japan). The powders were carbon and gold sputtered (Jeol Jee 4B SVG-IN, Tokyo, Japan) before analyses (Centro de Microscopia Eletrônica-UFRGS, Porto Alegre, Brazil) (CitationLionzo et al., 2007).
In vitro drug release studies
Drug release experiments were studied in a dissolution apparatus (Vankel VK7010, VanKel Technology, Cary, California, USA) at 37°C, using the basket method at a rotation speed of 100 rpm. The experiments were carried out as described in our previous work (CitationCruz et al., 2009), in order to evaluate the gastroresistance at pH 1.2 and the sodium alendronate release profiles at pH 6.8.
Data analysis
The release kinetics was determined using the model-dependent monoexponential (equation 2) and biexponential (equation 3) approaches.
where Mt is the amount of the drug released at time t, M∞ is the initial concentration of the drug, k, α and β are the apparent release kinetic rate constants, and A and B are the fractions of the drug that contributed to the burst and sustained phases, respectively (CitationCruz et al., 2006a; Citationb).
To determine the release mechanism of sodium alendronate from microparticles, drug release profiles were analyzed by fitting experimental data to Korsmeyer-Peppas model (CitationKorsmeyer et al., 1983) (equation 4). In this equation, ft is the ratio of absolute cumulative amount of the drug released at time t and at infinite time, a is a constant incorporating structural and geometric characteristic of the carrier, and n is the release exponent, indicative of the mechanism of the drug release. For a drug delivery system presenting spherical geometry, n value of 0.43 corresponds to a Fickian diffusion of the drug, while n values ranging from 0.43–0.85 indicate that the mechanism is dependent on the drug diffusion and the swelling of the polymer (anomalous transport). Finally, n values equal to or higher than 0.85 correspond to a case II transport (relaxation controlled delivery) (CitationPeppas, 1985).
In all cases the fit was carried out using the Scientist 2.0 software (Micromath, Saint Louis, Missouri, USA). The selection of the model was based on the best coefficient of determination and the best model selection criteria (MSC), both provided by the software, and the best graphic adjustment.
Gastrointestinal tolerance
The protocol has been approved by the Ethical Comittee of the Universidade Federal do Rio Grande do Sul (protocol # 2006637, Brazil). Gastrointestinal tolerance was evaluated following oral administration of the formulations to rats by gastric gavage. Female Wistar rats weighing 200–250 g were used (Biotério Central, UFRGS, Brazil). After their arrival at the laboratory, the animals were kept in separate cages at a maximum of five animals per cage and maintained on a diet of tap water and standard laboratory rat food until the beginning of the experiments.
Gastrointestinal tolerance was evaluated by oral administration of the formulations to fasted rats by gastric gavage. On the day of the experiment, the animals were divided into six groups of six animals per group: three negative control groups received water (1) or placebo formulations (unloaded microparticles): MP-EUDp (2) and MPE-E4Mp (3); one positive control group received sodium alendronate aqueous solution (80 mg/kg) (4); and, finally two groups received microparticle formulations (80 mg/kg of drug): MP-EUD (5) and MPE-E4M (6). The formulations were dispersed in water and administered daily during 15 days. On the last day of the experiment, the rats were sacrificed by CO2 asphyxiation and the stomachs were removed, opened along the greater curvature, and cleaned with water to be photographed later. The images were analyzed by specific software ‘EARP’ to measure each lesion point. The ulcers were classified as level I, ulcer area < 1 mm2; level II, ulcer area 1–3 mm2; and level III, ulcer area > 3 mm2. The following parameters were determined: (i) Ulcerative Lesion Index (ULI) as 1x (number of ulcers level I) + 2x (number of ulcers level II) + 3x (number of ulcers level III); (ii) total area of lesion; and (iii) percentage of the lesion area calculated by dividing the lesion area by the total stomach area multiplied by 100 (CitationAndrade et al., 2006).
Results and discussion
In this study, Eudragit® S100 and HPMC microparticles were prepared by spray-drying for the delivery of sodium alendronate by oral route. All formulations were obtained with a similar yield of preparation of ∼ 52%. The microparticles containing the drug (MP-EUD and MPE-E4M) showed high values of encapsulation efficiencies and of drug loadings (). For the formulation based exclusively on Eudragit® S100, an increase of the encapsulation efficiency was observed, probably due to the higher solubility of the drug in the feed solution of this formulation. The mean diameters of microparticles ranged from 7.3–17.1 μm with a span close to 2, indicating narrow size distributions. Comparing the size of unloaded and loaded-sodium alendronate formulations, the presence of the drug did not influence the particle sizes.
Table 1. Physico-chemical characteristics of microparticles.
Shape and morphology of microparticles formulations were observed by SEM (). The microparticles presented a similar collapsed shape and smooth surface demonstrating that the presence of HPMC in the blended microparticles did not affect the morphology of the microparticles. No drug crystals were observed on the microparticles surface.
Figure 1. Photomicrographs of MP-EUD (A) and MPE-E4M (B) formulations (bar = 10 μm).

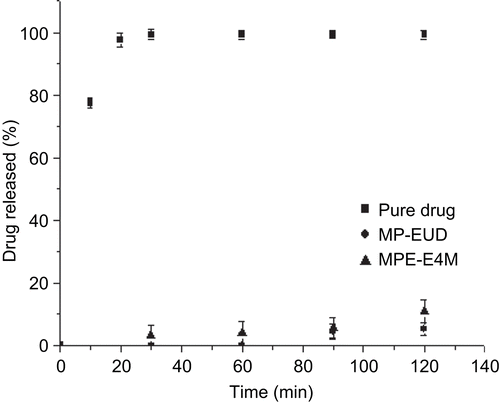
In order to verify the ability of microparticles to resist the acidic environment of the stomach without the release of the drug, a dissolution test was performed in 0.1 M HCl at pH 1.2. Within 2 h, 5.2% and 10.8% of the drug were released from the formulations MP-EUD and MPE-E4M, respectively (). The values could be attributed to the drug adsorbed on the surface of the microparticles.
Figure 2. Dissolution profiles of MP-EUD and MPE-E4M formulations in 0.1 M HCl pH 1.2.
shows the in vitro release profiles at pH 6.8. The dissolution of pure sodium alendronate was fast and 99.7% of the drug was dissolved within 10 min. On the other hand, the release of sodium alendronate from the microparticles MP-EUD and MPE-E4M were 95.6% in 150 min and 94.3% in 540 min, respectively. It is important to note that the release of the drug from MPE-E4M was remarkably slower than from MP-EUD. Regarding the literature, HPMC is widely used as a rate-controlling agent for drug delivery systems due to its high swellability, which affects the drug release kinetics (CitationSiepmann & Peppas, 2001).
Figure 3. Dissolution profiles of pure sodium alendronate, MP-EUD and MPE-E4M formulations in phosphate buffer solution pH 6.8.
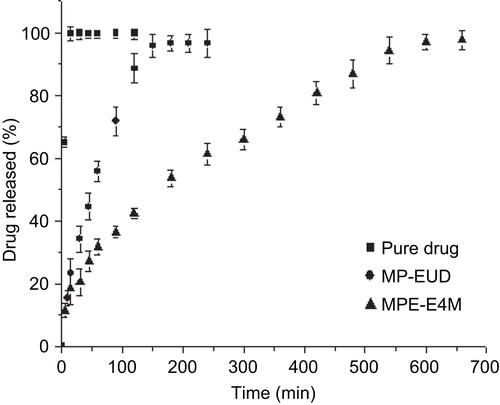
The drug release profiles were analyzed by fitting dissolution data to monoexponential and biexponential equations. Regarding the values of coefficient of determination and MSC, the release of sodium alendronate from MP-EUD was better described by the monoexponential equation (). The observed rate constant was 0.0161 min−1 ± 0.0012 min−1 and the half-life of the release of sodium alendronate was 43 min ± 3. For the formulation prepared with the blend, the best fitting was observed using the biexponential model (). In this case, the drug was released in a biphasic manner. The two phases are correlated to the burst effect (parameter A and kinetic constant α) and to the sustained release (parameter B and kinetic constant β) (CitationCruz et al., 2006a; CitationPoletto et al., 2007). Parameter A showed that the initial concentration of sodium alendronate for the burst phases was ∼ 11% (). Taking that into account, the immediate release of a small fraction of sodium alendronate from the microparticles can be related to the portion of the drug which is adsorbed on the surface of the microparticles. In this way, this result corroborates the hypothesis used to explain the fraction of sodium alendronate released in the gastroresistance evaluation.
Table 2. Parameters calculated by monoexponential, biexponential and power law models for MP-EUD and MPE-E4M.
Regarding the sustained phase, the mathematical modeling showed that 81% of sodium alendronate was released with a kinetic constant β of 0.0040 ± 0.0012 min−1 and the half-life of the drug release from MPE-E4M was 173 ± 7 min.
The release data were also analyzed by the Korsmeyer-Peppas model (equation 4). Regarding the data reported in , the microparticles showed a good fit with the Korsmeyer-Peppas model presenting coefficient of determination close to 0.99. The values of n for the formulations indicated that the mechanism of the drug release is non-Fickian (anomalous transport). In the case of the microparticles prepared only with Eudragit® S100, the release mechanism is dependent on the polymer swelling and dissolution besides the drug diffusion. For the blend microparticles (MPE-E4M), the anomalous transport is a consequence of the swelling of the particles and slow dissolution of the Eudragit® S100 due to the HPMC hydrogel, besides the drug diffusion.
To further evaluate the microparticles gastrointestinal tolerance in vivo, the formulations MP-EUD and MPE-E4M were administrated to the animals by oral gavage. In previous studies, the dose of 80 mg/kg/day during 15 days was determined as efficient to produce mucosal damage in Wistar rats (data not shown). displays the results obtained for the gastrointestinal toxicity. The negative control groups (water and placebo microparticles) did not present ulcerative lesions. The sodium alendronate solution group presented ULI of 6.5 ± 0.61, total area of lesion of 3.99 ± 0.90 mm2, and percentage of the lesion area of 0.53 ± 0.12%. In addition, for this group hemorrhagic points all over the stomach were verified and 1 day before the end of the study, one animal died due to a perforation in the stomach. Concerning the tested groups, it was observed that the treatment with 80 mg/kg of sodium alendronate-loaded microparticles significantly reduced the ULI in comparison with the sodium alendronate solution control group (p < 0.01). For the groups which received the formulations MP-E and MPE-E4M, no ulcer was observed, but few hemorrhagic points were visualized.
Table 3. Effects of the treatments on ulcerative lesion index, total area of lesion, and percentage of lesion area.
Although there are some reports that demonstrate the appearance of duodenal ulcers following sodium alendronate oral administration (CitationLanza, 2003), it is important to mention that in the present work no lesion in the duodenum, jejunum, and ileum was observed for all the groups. The occurrence of damage only in the level of the stomach can be explained as a function of pH conditions. In very low pH (pH < 2) the drug is in its acid form, which is more irritating than the sodium salt form (CitationPeter et al., 1998). After an overnight fasting, the pH of the empty stomach is between 1.0–2.0, and the drug is converted to the free acid form. When the drug arrives in the duodenum, the pH increases, and the drug is converted to the sodium salt form, which is less irritating. Additionally, one should take into account the lower susceptibility of the animal model in the formation of duodenal ulcers. The inverse situation is observed for non-steroidal anti-inflammatory drugs. In humans peptic ulcers are reported, while in rat model duodenal ulcers are observed (CitationGuterres et al., 2001; CitationBeck et al., 2005).
Finally, the gastroresistant polymer Eudragit® S100 played an important role in the reduction of the side-effects caused by sodium oral administration. Moreover, the multiparticulate nature of the formulations also contributes to minimize the side-effects since they can reduce the possibility of a high local concentration of the drug.
Conclusion
In conclusion, the results showed that the gastroresistant microparticles prepared using Eudragit® S100 or a blend of Eudragit® S100 and HPMC had a protective effect against mucosal injury induced by sodium alendronate oral administration. In addition, the release of this hydrophilic drug was delayed for all formulations and the presence of HPMC in the blended microparticles provided slower release in comparison with the formulation prepared exclusively with Eudragit® S100. In this way, those polymeric microparticles are a promising platform to deliver bisphosphonate drugs for the oral route.
Acknowledgement
The authors thank CNPq/MCT, CAPES/COFECUB, FAPERGS.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Adami, S., Passeri, M., Ortolani, S., Broggini, M., Carratelli, L., Caruso, I., Gandolini, G., Gnessi, L., Laurenzi, M., Lombardi, A., Norbiato, G., Pryortillotson, S., Reda, C., Romanini, L., Subrizi, D., Wei, L., Yates, A.J. (1995). Effects of oral alendronate and intranasal salmon-calcitonin on bone mass and biochemical markers of bone turnover in postmenopausal women with osteoporosis. Bone. 17:383–90.
- Al Deeb, S.K., Hamdan, I.I., Al Najjar, S.M. (2004). Spectroscopic and HPLC methods for the determination of alendronate in tablets and urine. Talanta. 64:695–702.
- Andrade, S.F., Antoniolli, D., Comunello, E., Cardoso, L.G.V., Carvalho, J.C.T., Bastos, J.K. (2006). Antiulcerogenic activity of crude extract, fractions and populnoic acid isolated from Austroplenckia populnea (Celastraceae). Z Naturforsch C. 61:329–33.
- Beck, R.C.R., Pohlmann, A.R., Benvenutti, E.V., Costa, T.D., Guterres, S.S. (2005). Nanostructure-coated diclofenac-loaded microparticles: preparation, morphological characterization, in vitro release and in vivo gastrointestinal tolerance. J Braz Chem Soc. 16:1233–40.
- Blanco-Prieto, M.J., Campanero, M.A., Besseghir, K., Heimgatner, F., Gander, B. (2004). Importance of single or blended polymer types for controlled in vitro release and plasma levels of a somatostatin analogue entrapped in PLA/PLGA microspheres. J Contr Rel. 96:437–48.
- Cruz, L., Assumpção, E., Guterres, S.S., Pohlmann, A.R. (2009). High encapsulation efficiency of sodium alendronate in Eudragit S100/HPMC blend microparticles. Quim Nova. 32:1170–4.
- Cruz, L., Schaffazick, S.R., Dalla Costa, T., Soares, L.U., Mezzalira, G., da Silveira, N.P., Schapoval, E.E.S., Pohlmann, A.R., Guterres, S.S. (2006a). Physico-chemical characterization and in vivo evaluation of indomethacin ethyl ester-loaded nanocapsules by PCS, TEM, SAXS, interfacial alkaline hydrolysis and antiedematogenic activity. J Nanosci Nanotechnol. 6:3154–62.
- Cruz, L., Soares, L.U., Dalla Costa, T., Mezzalira, G., da Silveira, N.P., Guterres, S.S., Pohlmann, A.R. (2006b). Diffusion and mathematical modeling of release profiles from nanocarriers. Int J Pharm. 313:198–205.
- Czuryszkiewicz, T., Areva, S., Honkanen, M., Linden, M. (2005). Synthesis of sol-gel silica materials providing a slow release of biphosphonate. Colloids Surf A Physicochem Eng Asp. 254:69–74.
- De Groen, P.C., Lubbe, D.F., Hirsch, L.J., Daifotis, A., Stephenson, W., Freedholm, D., PryorTillotson, S., Seleznick, M.J., Pinkas, H., Wang, K.K. (1996). Esophagitis associated with the use of alendronate. N Engl J Med. 335:1016–21.
- Ebetino, F.H., Roze, C.N., McKenna, C.E., Barnett, B.L., Dunford, J.E., Russell, R.G.G., Mieling, G.E., Rogers, M.J. (2005). Molecular interactions of nitrogen-containing bisphosphonates within farnesyl diphosphate synthase. J Organomet Chem. 690:2679–87.
- Freiberg, S., Zhu, X. (2004). Polymer microspheres for controlled drug release. Int J Pharm. 282:1–18.
- Graham, D.Y., Malaty, H.M. (1999). Alendronate gastric ulcers. Aliment Pharmacol Ther. 13:515–9.
- Guterres, S.S., Muller, C.R., Michalowski, C.B., Pohlmann, A.R., Dalla Costa, T. (2001). Gastro-intestinal tolerance following oral administration of spray-dried diclofenac-loaded nanocapsules and nanospheres. STP Pharma Sci. 11:229–33.
- Huang, W., Wang, Y., Ren, L., Du, C., Shi, X. (2009). A novel PHBV/HA microsphere releasing system loaded with alendronate. Mater Sci Eng C. 29:2221–5.
- Korsmeyer, R.W., Gurny, R., Doelker, E., Buri, P., Peppas, N.A. (1983). Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 15:25–35.
- Kurkuri, M.D., Aminabhavi, T.M. (2004). Poly(vinyl alcohol) and poly(acrylic acid) sequential interpenetrating network pH-sensitive microspheres for the delivery of diclofenac sodium to the intestine. J Contr Rel. 96:9–20.
- Lanza, F. (2003). Bisphosphonate mucosal injury—the end of the story? Dig Liver Dis. 35:67–70.
- Lanza, F., Schwartz, H., Sahba, B., Malaty, H.M., Musliner, T., Reyes, R., Quan, H., Graham, D.Y. (2000). An endoscopic comparison of the effects of alendronate and risedronate on upper gastrointestinal mucosae. Am J Gastroenterol. 95:3112–7.
- Lavelle, E.C. (2001). Targeted delivery of drugs to the gastrointestinal tract. Crit Rev Ther Drug Carrier Syst. 18:341–86.
- Li, W.W., Kendler, D.L. (2004). Pharmaceutical care and community pharmacists understanding of bisphosphonate dosing information. J Clin Pharm Ther. 29:531–6.
- Lin, J.H. (1996). Bisphosphonates: a review of their pharmacokinetic properties. Bone. 18:75–85.
- Lin, J.H., Chen, I.W., Deluna, F.A. (1994). Nonlinear kinetics of alendronate—plasma-protein binding and bone uptake. Drug Metab Dispos. 22:400–5.
- Lionzo, M.I.Z., Re, M.I., Guterres, S.S., Pohlmann, A.R. (2007). Microparticles prepared with poly(hydroxybutyrate-co-hydroxyvalerate) and poly(epsilon-caprolactone) blends to control the release of a drug model. J Microencapsul. 24:175–86.
- Luppi, B., Bigucci, F., Zecchi, V., Cerchiara, T. (2009). Gastroresistant microcapsules: new approaches for site-specific delivery of ketoprofen. Drug Deliv. 16:24–9.
- Marshall, J.K., Thabane, M., James, C. (2006). Randomized active and placebo-controlled endoscopy study of a novel protected formulation of oral alendronate. Dig Dis Sci. 51:864–8.
- Nafea, E.H., El-Massik, M.A., El-Khordagui, L.K., Marei, M.K., Khalafallah, M.N. (2007). Alendronate PLGA microspheres with high loading efficiency for dental applications. J Microencapsul. 24:525–38.
- Palmieri, G.F., Michelini, S., Di Martino, P., Martelli, S. (2000). Polymers with pH-dependent solubility: possibility of use in the formulation of gastroresistant and controlled-release matrix tablets. Drug Dev Ind Pharm. 26:837–45.
- Patashnik, S., Rabinovich, L., Golomb, G. (1997). Preparation and evaluation of chitosan microspheres containing bisphosphonates. J Drug Target. 4:371–80.
- Peppas, N.A. (1985). Analysis of fickian and non-fickian drug release from polymers. Pharm Acta Helv. 60:110–1.
- Perez-Lopez, F.R. (2004). Postmenopausal osteoporosis and alendronate. Maturitas. 48:179–92.
- Perugini, P., Genta, I., Conti, B., Modena, T., Pavanetto, F. (2001). Long-term release of clodronate from biodegradable microspheres. AAPS PharmSciTech. 2:1–10.
- Peter, C.P., Handt, L.K., Smith, S.M. (1998). Esophageal irritation due to alendronate sodium tablets—possible mechanisms. Dig Dis Sci. 43:1998–2002.
- Poletto, F.S., Jager, E., Re, M.I., Guterres, S.S., Pohlmann, A.R. (2007). Rate-modulating PHBHV/PCL microparticles containing weak acid model drugs. Int J Pharm. 345:70–80.
- Raffin, R.P., Colome, L.M., Haas, S.E., Jornada, D.S., Pohlmann, A.R., Guterres, S.S. (2007). Development of HPMC and Eudragit S100 (R) blended microparticles containing sodium pantoprazole. Pharmazie. 62:361–4.
- Raffin, R.P., Colome, L.M., Pohlmann, A.R., Guterres, S.S. (2006). Preparation, characterization, and in vivo anti-ulcer evaluation of pantoprazole-loaded microparticles. Eur J Pharm Biopharm. 63:198–204.
- Raffin, R.P., Colome, L.M., Schapoval, E.E.S., Pohlmann, A.R., Guterres, S.S. (2008). Increasing sodium pantoprazole photostability by microencapsulation: effect of the polymer and the preparation technique. Eur J Pharm Biopharm. 69:1014–8.
- Reginster, J.Y. (2006). Adherence and persistence: impact on outcomes and health care resources. Bone. 38:18–21.
- Samdancioglu, S., Calis, S., Sumnu, M., Hincal, A.A. (2006). Formulation and in vitro evaluation of bisphosphonate loaded microspheres for implantation in osteolysis. Drug Dev Ind Pharm. 32:473–81.
- Sener, G., Goren, F.O., Ulusoy, N.B., Ersoy, Y., Arbak, S., Dulger, G.A. (2005). Protective effect of melatonin and omeprazole against alendronat-induced gastric damage. Dig Dis Sci. 50:1506–12.
- Shi, X., Wang, Y., Ren, L., Gong, Y., Wang, D. (2009a). Enhancing alendronate release from a novel PLGA/Hydroxyapatite microspheric system for bone repairing applications. Pharm Res. 26:422–30.
- Shi, X., Wang, Y., Varshney, R.R., Ren, L., Zhang, F., Wang, D. (2009b). In-vitro osteogenesis of synovium stem cells induced by controlled release of bisphosphate additives from microspherical mesoporous silica composite. Biomaterials. 30:3996–4005.
- Siepmann, J., Peppas, N.A. (2001). Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv Drug Deliv Rev. 48:139–57.
- Varde, N.K., Pack, D.W. (2004). Microspheres for controlled release drug delivery. Expert Opin Biol Ther. 4:35–51.
- Weidenauer, U., Bodmer, D., Kissel, T. (2003). Microencapsulation of hydrophilic drug substances using biodegradable polyesters. Part I: Evaluation of different techniques for the encapsulation of pamidronate di-sodium salt. J Microencapsul. 20:509–24.
- Weidenauer, U., Bodmer, D., Kissel, T. (2004). Microencapsulation of hydrophilic drug substances using biodegradable polyesters. Part II: Implants allowing controlled drug release—a feasibility study using bisphosphonates. J Microencapsul. 21:137–49.
- Yates, A.J., Rodan, G.A. (1998). Alendronate and osteoporosis. Drug Discov Today. 3:69–78.