Abstract
The present investigation deals with the development of a pH and time-dependent press-coated pulsatile drug delivery system for delivering drugs into the colon. The system consists of a drug containing core, coated by a combination of natural polymer Delonix regia gum (DRG) and hydroxypropyl methylcellulose (HPMC K4M) in various proportions, which controls the onset of release. The whole system was coated with methacrylic acid copolymers, which not only prevents the drug release in the stomach, but also prolongs the lag time. Tramadol HCl was used as a model drug and varying combinations of DRG and HPMC K4M were used to achieve the desired lag time before rapid and complete release of the drug in the colon. It was observed that the lag time depends on the coating ratio of DRG to HPMC and also on press coating weight. Drug release was found to be increased by 15–30% in the presence of colonic microbial flora. The results showed the capability of the system in achieving pulsatile release for a programmable period of time and pH-dependent release to attain colon-targeted delivery.
Introduction
Oral controlled release dosage forms are known to provide a zero order or first order release in which the drug is released at a substantially steady rate per unit time. These dosage forms are satisfactory for the administration of pharmaceuticals. However, there are many cases where maintaining a constant concentration of a drug is not desirable. In such cases, a time-controlled pulsatile drug delivery system becomes more advantageous. Additionally, it is thought that time-release formulations could be an effective tool in chronopharmacotherapy, because their unique drug release properties could take advantage of circadian rhythms in physiologic and pathologic functions (CitationWhite et al., 1998). Oral time-controlled pulsatile release formulations possess the interesting characteristic that is the capability of giving one or more instant and transit release pulses at a pre-determined time period after a desired lag time or at specific sites. A timed-release formulation could allow drug release and a greater plasma drug concentration specifically when clinical signs are developed or aggravated. Disease conditions such as early morning symptoms of asthma, arthritis, and for ischemic heart disease, a time-controlled pulsatile drug delivery system, offer more benefits.
There are also cases in which a position-controlled drug delivery system (CitationKopecek et al., 1992; CitationNykanen et al., 2001; CitationKrishnaiah et al., 2002) uses the colon as an absorption site to increase the systemic absorption, to optimize the drug desirable effects and to reduce undesirable effects. Finally, a controlled drug delivery system (CDDS) would be advantageous when a delay in absorption is desirable from a therapeutic point of view, particularly for the treatment of diseases that have peak symptoms in the early morning and that exhibit circadian rhythms, such as nocturnal asthma, angina, and rheumatoid arthritis (CitationKinget et al., 1998; CitationHalsas et al., 1999; Citation2001). In rheumatoid arthritis, symptoms are more exaggerated in the early morning (CitationScott, 1960; CitationKowanko et al., 1981) and night time medication is more beneficial (CitationSchorn & Seymour, 1981). A pulsatile drug delivery system is helpful to avoid pharmacokinetic drug–drug interactions between concomitantly administered medications, to reduce dose size, to reduce dosing frequency so as to maximize patient compliance, to maximize desired effects, to minimize undesired effects, to localize drug at the right site in right quantity at right time, and to minimize systemic drug metabolism in the upper part of the gastrointestinal (GI) tract (CitationBussemer et al., 2001).
Tramadol hydrochloride is a centrally acting analgesic which has relatively low affinity for opioid receptors (i.e. µ type) and also appears to modify the transmission of pain impulses by inhibition of nor-epinephrine and serotonin uptake. In comparison with other opoids, it has limited side-effects in the treatment of acute and chronic pains. A slow drug release system is particularly suitable for analgesic agents, which may be advantageous due to reduction in frequency of administration while maintaining the analgesic effect for a longer duration, for example overnight (CitationObaidat & Obaidat, 2001; CitationTiwari et al., 2003; CitationVarshosaz et al., 2006).
A number of oral systems are available through which a pulsatile release pattern can be achieved. Most systems contain a drug reservoir, surrounded by a barrier which either erodes, dissolves (CitationGazzaniga et al., 1994a; Citationb; CitationOtsuka & Matsuda 1994; CitationWilding et al., 1994; CitationMaggi et al., 1999), or ruptures (Ueda et al., 1989; CitationAmsden & Cheng, 1995; CitationMorita et al., 2000).
A dry-coated tablet was recently renewed as a novel system to deliver a drug in a pulsatile way at pre-determined times following oral administration (CitationTakeuchi et al., 2000; CitationGonzalez-Rodriguez et al., 2003).
Many authors have reported that compression coating with hydrogel around core tablet has been applied for time-controlled release or colon-specific drug delivery (CitationConte et al., 1993; CitationGazzaniga et al., 1994a). Press-coating technique is advantageous because the tablets can be prepared with various characteristics suitable for a number of uses (Maggi et al., 1993; CitationOtsuka & Matsuda, 1994; CitationPozzi et al., 1994).
Gastrointestinal transit of dosage forms is highly variable and affected by many factors, but small intestinal transit time is almost constant at 3 ± 0.5h, independent of the types of dosage forms and size as well as the fasted or fed state (CitationDavis et al., 1986) which forms the platform for the time-dependent systems for colon-specific drug delivery. Time controlled release systems have limitations for colonic delivery due to large variation of the gastric emptying time of dosage forms,a which varies from few minutes to more than 3 h. Thus, time prediction and site specificity is difficult. By a simple pH-dependent approach, drug could be located to the colon, but with lack of site specificity, because of varying pH of the GI tract and the pH drop of the colon (6.5) compared to that of the small intestine (7–7.8) (CitationEvans et al., 1998; CitationBajpai et al., 2003). Enteric-coated time-controlled press-coated drug delivery systems are helpful for targeting drug to colon (CitationFukui et al., 2000). Thus, combining time- and pH-dependent approaches may be a useful strategy for colon-specific drug delivery (CitationCheng et al., 2004).
Natural polymers that have been used previously to prepare press-coated colon-specific tablet dosage forms include guar gum (CitationKrishnaiah et al., 1998; Citation1999),pectin/chitosan (CitationFernandez-Hervas & Fell, 1998),pectin/ethyl cellulose (CitationAhrabi et al., 2000), and pectin/HPMC (CitationTurkoglu et al., 1999; CitationTurkoglu & Ugurlu 2002; CitationUgurlu et al., 2007).
In the present study, we have developed a pH- and time-dependent press-coated–enteric-coated pulsatile drug delivery system using natural polymer DRG isolated in our laboratory from the seeds of higher plant Delonix regia (Hook) Raf. Family-Fabaceae, for delivering drugs to the colon. The developed system, using tramadol HCl as the model drug, is made up of three compartments, i.e. a drug containing core tablet and two layers of coating (the outer enteric film-coating layer and the inner press-coating layer), which could have minimum influence of gastric emptying time on drug release and be trusted to reach the colon and release the drug therein.
Materials and methods
Tramadol hydrochloride (Cadila Healthcare Ltd., Ahmedabad, India), Eudragit L100 and S100 (Evonic Degussa India Pvt. Ltd., Mumbai, India), and Hydroxypropyl methylcellulose (HPMC K4M) (Colorcon Pvt. Ltd., Goa, India) were kindly supplied as gift samples. Delonix regia gum (DRG) was isolated and characterized in our laboratory (CitationTekade & Gattani, 2009). Spray-dried lactose and magnesium stearate (S. D. Fine Chemicals, Mumbai, India) were purchased. All other chemicals and solvents used were of analytical grade.
Methods
Preparation of enteric coated timed-release press-coated tablets
The experimental list showing coating weight and proportions of DRG and HPMC K4M is given in .
Table 1. Experimental list for press-coated tramadol HCl tablets.
Preparation of core tablets
The core tablets were prepared by direct compression method followed by press-coating and enteric-coating (). Tramadol hydrochloride, spray-dried lactose, magnesium stearate, and talc were mixed with each other according to the geometric dilution method. Rapid release core tablets were prepared by compressing 100 mg powder blend using 7 mm flat faced punches and die cavity on a rotary tablet press (Rimek Minipress I, Ahmedabad, India). These prepared core tablets were evaluated for weight variation, crushing strength, friability, thickness, content uniformity, and dissolution study. Here, we used amaranth red as a coloring agent to differentiate the inner core from both the outer coats in the photo image study.
Table 2. Compositions of Core, PCC, and PCEC tablets.*
In vitro release studies of tramadol HCl core tablets
Prepared tramadol HCl core tablets were placed in vessels of dissolution test apparatus containing 900 ml pH 6.8 or pH 7.4 phosphate buffer solutions as dissolution medium. Dissolution studies were performed using USP XXIII type 2 apparatus (Paddle method) at 50 rpm and 37 ± 0.5°C. At pre-determined time intervals, 5 ml samples were withdrawn and replaced with equal volumes of fresh buffer medium. Withdrawn test samples were filtered through a 0.22 µm filter and analyzed using a UV spectrophotometer (1700, Schimadzu, Japan).
Press-coating of core (PCC) tablets
Press-coated tablets were prepared according to the previously reported method (CitationSawada et al., 2004). DRG and HPMC were passed through a 120 μm sieve and used for release retarding outer shells. Half of the total quantity of coating powder blend was filled in die cavity to make a powder bed at the bottom. The previously compressed tablet using 8 mm flat faced punches and die cavities were then placed in the centre on the above powder blend. The remaining equivalent powder was filled in the die, and the content was compressed using a flat punch, 10 mm in diameter, as summarized in . Formulated press-coated tablets were evaluated for their physical properties ().
Table 3. Physical properties of tramadol HCl core and DRG-HPMC press-coated tablets.
Enteric coating of press-coated tablets
The core tablets were coated in a conventional coating pan. The enteric coating solution was prepared by dissolving 5% w/v Eudragit L100:S100 (1:2 ratio) containing 5% v/v di-butyl phthalate as a plasticizer in an aqueous isopropyl alcohol solution. Coating of the press-coated tablets was performed using a tablet coating machine (Spacelab, Nashik, India) under the following conditions, the coating pan set at an angle of 45°, rotating speed of the pan 15 rpm, 0.5 mL/min spraying rate, and drying temperature 55°C. The coated tablets were dried for 24 h at 45°C. The coating process continued until the amount of the coating was 52.5 mg (15%) per tablet. Here, we used methylene blue as a coloring agent to differentiate the outer coating layer from the inner coat.
Test method for DRG-HPMC coat erosion
After compressing the tablets, an erosion study was conducted (CitationTurkoglu & Ugurlu, 2002). The USP XXIII dissolution apparatus 2 (paddle method) was used at stirring rate of 50 rpm or 100 rpm at 37 ± 0.5°C. Initially the dissolution test was conducted using pH 1.2 buffer solution for 2 h. At the end of 2 h the medium was replaced with pH 7.4 phosphate buffer solution and the test was continued for an additional 3 h. Finally, medium was replaced with pH 6.8 phosphate buffer and the study was continued for the remaining time period. After 5 h, depending on the study design, 3 ml 4% w/v diluted rat caecal content was added to the dissolution vessels and the study was continued until 12 h. In order to induce the enzymes that are specific for the biodegradation of the DRG during its passage through the colon, the albino rats were intubated with teflon tubing and 1 ml of 1% w/v solution of DRG in water was administered directly into the stomach. This treatment was continued for 6 days in six different animals. The studies were carried out with 4% w/v caecal content obtained after 6 days of the enzyme induction (CitationChourasia & Jain, 2004). All the experiments were performed by continuously supplying nitrogen into the dissolution media. Tablets were dried overnight at 45°C in an oven and the remaining tablet mass was determined gravimetrically.
In vitro dissolution studies for press-coated–enteric-coated core tablets
An in vitro dissolution study was conducted at three stages to evaluate release profiles. First, on drug containing core tablets, press-coated tablets, followed by enteric-coated tablets. The dissolution study was performed according to method described in the United States Pharmacopoeia XXIII type 2 apparatus (Paddle method) with 900 ml of dissolution medium and stirring rate of 100 rpm at 37 ± 0.5°C. In vitro release test was performed for 2 h in pH 1.2 buffer (average gastric emptying time), 3 h in pH 7.4 phosphate buffer (average small intestinal transit time) (CitationSangalli et al., 2001), and the remaining 7 h in pH 6.8 phosphate buffer as the dissolution media (CitationZahirul Khan et al., 1999; CitationGang et al., 2004; CitationMastiholimath et al., 2007). At the end of 2 h, pH 1.2 buffer was replaced with fresh pH 7.4 phosphate buffer saline (PBS), then after 3 h, medium was removed and fresh pH 6.8 phosphate buffer was added for the remaining hours of study. To evaluate the effect of enzyme on the drug release rate, at the end of 5 h, freshly prepared 3 ml 4%w/v rat caecal content (CitationKrishnaiah et al., 1998) was added to the dissolution fluid that was previously bubbled with nitrogen to maintain an anaerobic condition, and the study was continued to find the effect of colonic content on release profile. At pre-determined time intervals test samples were withdrawn and assayed for the amount of tramadol hydrochloride released using a UV spectrophotometer (1700, Shimadzu, Japan) at λmax = 271 nm.
Results
As mentioned in the introduction, the objective of the present investigation was to develop a new press-coated pulsatile drug delivery system using a combination of natural polymer DRG and HPMC K4M as lag time controlling layer, with methacrylic acid copolymers (Eudragit L100 & S100) as enteric coating polymer. The whole system was designed into three steps: (i) preparation of drug containing core tablet; (ii) press-coating of core tablets using combination of polymers for controlling desired lag time; and (iii) enteric-coating of press-coated tablet using methacrylate polymers to prevent drug release in the stomach.
In vitro characterization of core tablets and press-coated tablets
The core tablets and press-coated tablets were prepared by direct compression method and were characterized for the quality control tests such as weight variation, crushing strength, thickness, friability, content uniformity, and in vitro release study. All the formulated core tablets comply with the pharmacopoeial limits. The image of press-coated–enteric-coated tablet was captured using a digital camera and is shown in . Other physical properties of core tablets and press-coated tablets are shown in .
Figure 1. Photo image of press-coated–enteric-coated core tablet.

In vitro dissolution profiles of core tablets
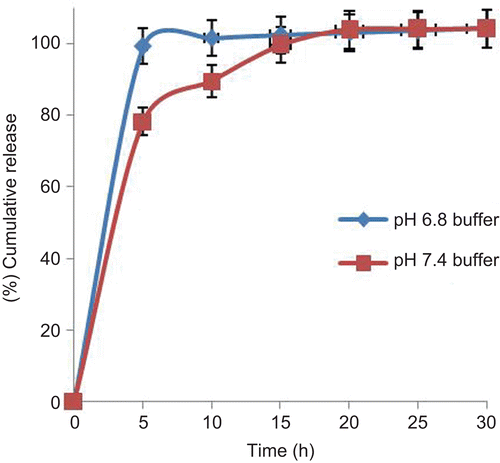
The core tablets were subjected to dissolution studies at pH 7.4 and pH 6.8 phosphate buffers to determine the release profile of core tablets in both media. There was observed more than 75% drug release within 5 min and 100% drug release within 15 min ().
Figure 2. In vitro release profile of tramadol HCl core tablets at pH 6.8 and pH 7.4 buffer media.
In vitro release studies of press-coated–enteric-coated tablets
The core tablets were subjected to press-coating and enteric-coating processes. The press-coated–enteric-coated tablets were studied for their dissolution profiles at pH 1.2 acid buffer solution for 2 h, at pH 7.4 phosphate buffer for 3 h, and at pH 6.8 phosphate buffer for remaining the hours of study (7 h). These studies were performed with or without rat caecal content to determine the effect of colonic bacteria on the release of the drug from the pulsatile system. Core tablets were press-coated with the same weight at various DRG-to-HPMC polymer ratios and with different weights at the same ratio. The experimental list showing coating weight and different proportions of DRG and HPMC K4M is shown in and release profiles are depicted in –. shows the effect of the presence of rat caecal content on the drug release profiles from the press-coated–enteric-coated pulsatile tablets. The photographs of tablets at different interval showing its intactness even after 6 h are shown in .
Figure 3. In vitro drug release profiles of press-coated tablets (200) mg at pH 1.2, pH 7.4, and pH 6.8 buffer solutions (n = 3, mean ± SD).
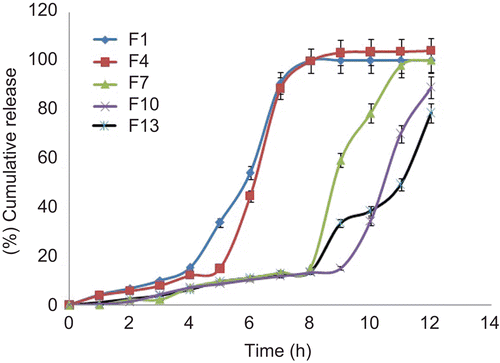
Figure 4. In vitro drug release profiles of press-coated tablets (250 mg) at pH 1.2, pH 7.4, and pH 6.8 buffer solutions (n = 3, mean ± SD).
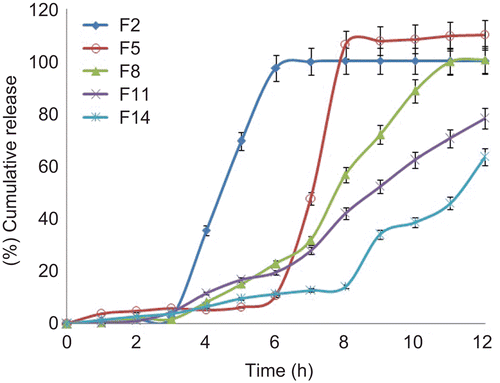
Figure 5. In vitro drug release profiles of press-coated tablets (300 mg) at pH 1.2, pH 7.4, and pH 6.8 buffer solutions (n = 3, mean ± SD).
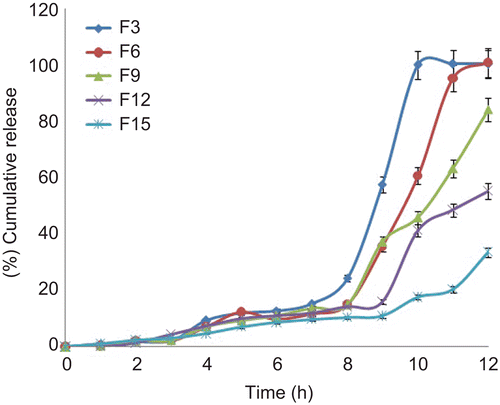
Figure 6. In vitro drug release profiles of press-coated–enteric-coated tablets (250 mg) at pH 1.2, pH 7.4, and pH 6.8 buffer solutions with or without rat caecal content (n = 3, mean ± SD).
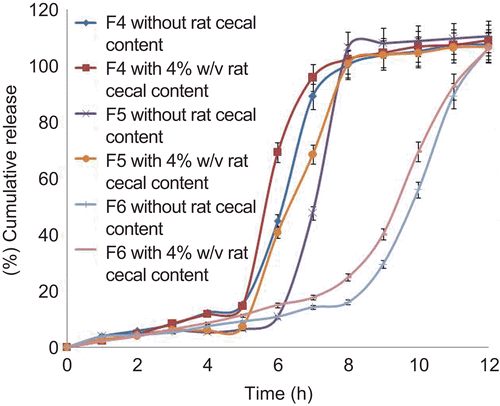
Figure 7. Photo images of tablets during in vitro dissolution study using digital camera at different time intervals.

Effect of coating weight and coating ratio of DRG to HPMC on drug release
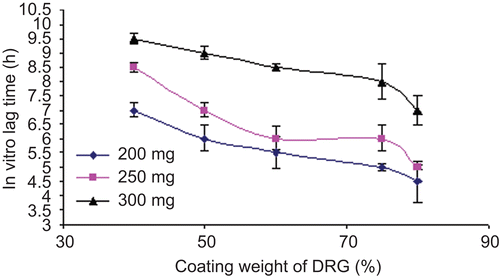
For the pulsatile drug delivery system, in vitro lag time is an important parameter to be optimized during the dissolution study. This lag time may get affected by coating weight, type of coating material, and ratio of coating material if they were used in combination. Here, we tried to find the effect of coating weight and coating ratio of DRG-to-HPMC to the core tablets during in vitro drug release studies. The results of effect of various coating weights at the same ratio and different coating ratio at the same coating weight on in vitro lag time are presented in and .
Figure 8. Effect of coating weight (mg) on in vitro lag time.
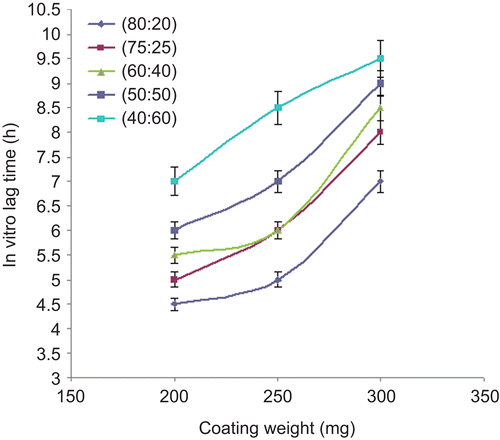
Figure 9. Effect of coating weight of DRG (%) on the in vitro lag time.
Coat erosion study
Natural polymer used as a compression coating material for colonic drug delivery system shows the coat erosion property (CitationUgurlu et al., 2007). This study was performed in the same manner as the dissolution study at pH 7.4 and pH 6.8 phosphate buffer solutions. This study was also conducted to find the effect of bacteria on the coat erosion. The results of coat erosion with or without rat caecal content are shown in and .
Figure 10. Coat erosion in the absence of rat caecal content.
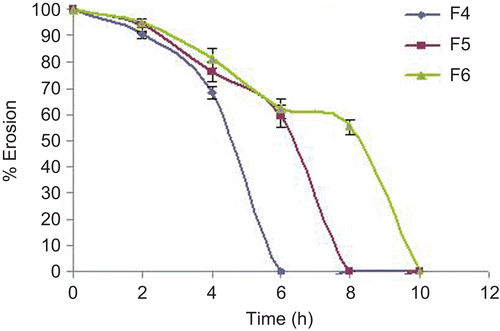
Figure 11. Coat erosion in the presence of rat caecal content (4%w/v).
Discussion
On the basis of desired lag time achieved (6 h) and thereafter pulsatile release (), formulation batch code F5 was selected as the optimized formulation for developing a time- and pH-dependent press-coated pulsatile drug delivery system. The in vitro release profiles of the optimized pulsatile drug delivery system were found to have good in vitro lag time controlling capability.
Core tablets show almost ther same release profiles in both pH 7.4 buffer and pH 6.8 buffer media. In both media, core tablets have shown complete drug release within 15 min, which shows the rapid and transient release profiles, satisfying the requirement of the pulsatile drug release system (i.e. instant and complete release) after the no release period. Coating percentage of DRG-to-HPMC of 80:20 in formulation batches F1–F3 shows more erosion as compared to other batches, which may be attributed to insufficient compressibility of DRG. We tried to compare the studies of various coating weight ratios at same coating weight and the results are given in –. The release of drug from all the batches mentioned in – shows that coat erosion decreases and drug release retards as the coating weight increases. This may be due to the decreased proportion of DRG polymer and increased proportion of HPMC. However, as the proportion of the HPMC increases more than 40% w/w in the formulation, the drug release from the system was found to be incomplete and the system becomes a sustained release delivery system, which may be due to the formation of a gel layer from which the drug may be released by the diffusion mechanism and, after 12 h, tablets were found to be less eroded. Lag time was found to be increased with increased HPMC proportion; however, above 40% w/w the system behaves more like a sustained release delivery system. On the other hand, as the proportion of DRG increases above 75% w/w, lag time was found to be decreased due to greater erosion of the system. From all the formulated batches studied, optimized F5 batch showed lag time controlling and pulse release properties. DRG is a natural polymer that show the biodegradation property by colonic microbial flora, so we tried to find the effect of colonic microbial flora on the drug release from the system in the presence of rat caecal content that was added at the end of the average gastric emptying time and small intestinal transit time (i.e. total 5 h). Photo images of optimized formulation batch F5 during dissolution study are shown in , which indicates erosion of coat occurs during in vitro release, and at the end of 6 h tablets were found to be fully exposed to dissolution fluid, thereafter complete release. In the presence of rat caecal content, drug release from the system (F4–F6) was found to be increased by 15–30% (). This clearly shows the effect of colonic microbial flora on the drug release rate due to degradation of natural polysaccharide (DRG), increased erosion, and therefore decreased lag time in the presence of rat caecal content. After complete wetting of systems and up to 50% erosion of coating material, the system shows complete drug release. These results were observed with formulation batches which contain more than 50% w/w of DRG in the total coating weight of the system. Formulation batch F5 shows the most appropriate release pattern of the pulsatile drug delivery system ().
Because the DRG polymer shows swelling properties in all the three media (pH 1.2 buffer, pH 7.4 buffer, and pH 6.8 buffer) (CitationTekade & Gattani, 2009), enteric-coating to the press-coat layer of the system could be useful to increase the lag time. Eudragit® L100 and S100 methacrylic acid pH sensitive copolymers in 1:2 w/w ratios dissolve at pH 6.8 (CitationMastiholimath et al., 2007). Thus, we have prepared the formulation in such a way that when it is administered at bedtime (10 pm) it may release the drug during the morning hours to give relief from the early morning pain of rheumatoid arthritis.
Conclusion
The present study demonstrates that tramadol HCl from the formulated system could be delivered when needed most by design of the time- and pH-dependent modified press-coated–enteric-coated tablet using a combination of natural polymer DRG and HPMC K4M. The lag time may be controlled by controlling press-coating weight as well as coating polymer ratio of DRG-to-HPMC. The optimized formulation gives a lag time of 6 h before rapid and transient release of the drug. Thus, if this formulation is administered at bedtime (10 pm), it may release the drug between 4–6 am, when the symptoms of rheumatoid arthritis are at its peak. However, in vivo study needs to be performed in order to confirm the findings of the in vitro study.
Declaration of interest
The authors are grateful to Cadila Healthcare Ltd., Ahmedabad, India, Evonic Degussa India Pvt. Ltd., Mumbai, India, and Colorcon Asia Pvt. Ltd., Goa, India for providing gift samples of Tramadol hydrochloride, Eudragit L100 and S100, and Hydroxypropyl methylcellulose (HPMC K4M), respectively. The management and principal (R. C. Patel Institute of Pharmaceutical Education and Research, Shirpur) are gratefully acknowledged for providing facilities to carry out this work.
References
- Ahrabi, S.F., Madsen, G., Dyrstad, K., Sande, S.A., Graffner, C. (2000). Development of pectin matrix tablets for colonic delivery of model drug ropivacaine. Eur J Pharm Sci. 10:43–52.
- Amsden, T., Cheng, L. (1995). A generic protein delivery system based on osmotically rupturable monoliths. J Contr Rel. 33:99–105.
- Bajpai, S.K., Bajpai, M., Dengree, R. (2003). Chemically treated hard gelatin capsules for colon-targeted drug delivery: a novel approach. J Appl Polym Sci. 89:2277–82.
- Bussemer, T., Otto, I., Bodmeier, R. (2001). Pulsatile drug-delivery systems. Crit Rev The Drug Carrier Syst. 18:433–58.
- Cheng, G., An, F., Zou, M., Sun, J., Hao, X., He, Y. (2004). Time and pH-dependent colon-specific drug delivery for orally administered diclofenac sodium and 5-aminosalicylic acid. World J Gastroenterol. 10:1769–74.
- Chourasia, M.K., Jain, S.K. (2004). Potential of Guar Gum microspheres for target specific drug release to colon. J Drug Target. 12:435–42.
- Conte, U., Magg, L., Torre, M.L., Giunchedi, P., La Manna, A. (1993). Press-coated tablets for time-programmed release of drugs. Biomaterials. 14:1017–23.
- Davis, S.S., Hardy, J.G., Fara, J.W. (1986). Transit of pharmaceutical dosage forms through the small intestine. Gut. 27:886–92.
- Evans, D.F., Pye, G., Bramley, R., Clark, A.G., Dyson, T.J., Hardcastle, J.D. (1998). Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut. 29:1035–41.
- Fernandez-Hervas, M.J., Fell, J.T. (1998). Pectin:chitosan mixtures as coatings for colon-specific drug delivery: an in vitro evaluation. Int J Pharm. 169:115–9.
- Fukui, E., Miyamura, N., Uemura, K., Kobayash, M. (2000). Preparation of enteric coated timed-release press-coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int J Pharm. 204:7–15.
- Gang, C., Feng, A., Mei-Juan, Z., Jin, S., Xiu-Hua, H., Yun-Xia, H. (2004). Time and pH-dependent colon- specific drug delivery for orally administred diclofenac sodium and 5- amino salicylic acid. Worid J Gastroenterol. 10:1769–74.
- Gazzaniga, A., Iamartino, P., Maffione, G., Sangalli, M.E. (1994a). Oral delayed-release system for colonic specific delivery. Int J Pharm. 108:77–83.
- Gazzaniga, A., Sangalli, M.E., Giordano, F. (1994b). Oral chronotopic drug delivery systems: achievement of time and/or site specificity. Eur J Pharm Biopharm. 40:246–50.
- Gonzalez-Rodriguez, M.L., Maestrelli, F., Mura, P., Rabasco, A.M. (2003). In vitro release of sodium diclofenac from a central core matrix tablet aimed for colonic drug delivery. Eur J Pharm Sci. 20:125–31.
- Halsas, M., Hietala, J., Veski, P., Jurjenson, H., Marvola, M. (1999). Morning versus evening dosing of ibuprofen using conventional and time controlled release formulations. Int J Pharm. 189:179–85.
- Halsas, M., Penttinen, T., Veski, P., Jurjenson, H., Marvola, M. (2001). Time controlled release pseudoephedrine tablets: bioavailability and in vitro/in vivo correlations. Pharmazie. 56:718–23.
- Kinget, R., Kalala, W., Vervoort, L., Van den Mooter, G. (1998). Colonic drug targeting. J Drug Target. 6:129–49.
- Kopecek, J.P., Kopeckova, H., Brondsted, R., Rathi, B., Rihova Yeh, P.-Y., Ikesue, K. (1992). Polymers for colon specific drug delivery. J Contr Rel. 19:121–30.
- Kowanko, I.C., Pownall, R., Knapp, M.S., Swannel, E.J., Mahoney, P.G. (1981). Circadian variations in the signs and symptoms of rheumatoid arthritis and in the therapeutic effectiveness of flurbiprofen at different times of the day. Br J Clin Pharmacol. 11:477–84.
- Krishnaiah, Y.S.R., Satyanarayana, S., Rama Prasad, Y.V. (1999). Studies of Guar Gum compression-coated 5-aminosalicylic acid tablets for colon-specific drug delivery. Drug Dev Ind Pharm. 25:651–7.
- Krishnaiah, Y.S.R., Satyanarayana, S., Rama Prasad, Y.V., Narasimha Rao, S. (1998). Evaluation of guar gum as a compression coat for drug targeting to colon, Int J Pharm. 71:137–46.
- Krishnaiah, Y.S.R., Satyanarayana, V., Kumar, B.D., Karthikeyan, R.S. (2002). In vitro drug release studies on guar gum-based colon targeted oral drug delivery systems of 5-fluorouracil. Eur J Pharm Sci. 16:185–92.
- Maggi, L., Conte, U., Gurni, R. (1999). Delivery device for the release of active ingredient in subsequent time. Proceedings of the International Controlled Release Bioactive Materials, Boston, MA, Vol. 26.
- Mastiholimath, V.S., Dandagi, P.M., Jain, S.S., Gadad, A.P., Kulkarni, A.R. (2007). Time and pH dependent colon specific, pulsatile delivery of theophylline for noctural asthma. Int J Pharm. 328:49–56.
- Morita, R., Honda, R., Takahashi, Y. (2000). Development of oral controlled release preparations, a PVP swelling controlled release system (SCRS): I. Design of SCRS and its release controlling factor. J Contr Rel. 63:297–304.
- Nykanen, P., Lempaa, S., Aaltonen, M.L., Jurjenson, H., Veski, P., Marvola, M. (2001). Citric acid as excipient in multiple-unit enteric-coated tablets for targeting drugs on the colon. Int J Pharm. 229:155–62.
- Obaidat, A.A., Obaidat, R.M. (2001). Controlled release tramadol hydrochloride from matrices prepared using glyceryl behenate. Eur J Pharm Biopharm. 52:231–5.
- Otsuka, M., Matsuda, Y. (1994). Controlled drug release of highly water-soluble pentoxifylline from time-imit disintegration type wax matrix tablets. Pharm Res. 11:351–4.
- Pozzi, F., Furlani, P., Gazzaniga, A. (1994). The TIME CLOCK system: a new oral dosage form for fast and complete release of drug after a predetermined lag time. J Contr Rel. 31:99–108.
- Sangalli, M.E., Maroni, A., Zema, L., Busetti, C., Giordano, F., Gazzaniga, A. (2001). In vitro and in vivo evaluation of an oral system for time and/or site-specific drug delivery. J Contr Rel. 73:103–10.
- Sawada, T., Kondo, H., Nakashima, H., Sako, K., Hayashi, M. (2004). Time-release compression-coated core tablet containing nifedipine for chronopharmacotherapy. Int J Pharm. 280:103–11.
- Schorn, D., Seymour, M.A. (1981). Indomethacin or sulindac in rheumatoid arthritis at night. S Afr Med J. 59:913–4.
- Scott, J.T. (1960). Morning stiffness in rheumatoid arthritis. Ann Rheum Dis. 19:361–8.
- Takeuchi, H., Yasuji, T., Yamamoto, H., Kawashima, Y. (2000). Spray-dried lactose composite particles containing an ion complex of alginate-chitosan for designing a dry-coated tablet having a time-controlled releasing function. Pharm Res. 17:94–9.
- Tekade, A.R., Gattani, S.G. (2009). Development and evaluation of pulsatile drug delivery system using novel polymer. Pharm Dev Tech. 14:380–7.
- Tiwari, S.B., Murthy, T.K., Pai, M.R., Mehta, P.R., Chowdaty, P.B. (2003). Controlled release formulation of Tramadol hydrochloride using hydrophilic and hydrophobic matrix system. AAPS Pharm Sci Tech. 4:1–6.
- Turkoglu, M., Takka, S., Baran, H., Sakr, A. (1999). Pectin hydroxyl propyl methyl cellulose drug delivery system for colon targeting. Pharm Pharmacol. 51:154.
- Turkoglu, M., Ugurlu, T. (2002). In vitro evaluation of pectin–HPMC compression coated 5-aminosalicylic acid tablets for colonic delivery. Eur J Pharm Biopharm. 53:65–73.
- Ugurlu, T., Turkoglu, M., Gurer, U.S., Akarsu, B.G. (2007). Colonic delivery of compression coated nisin tablets using pectin/ HPMC polymer mixture. Eur J Pharm Biopharm. 67:202–10.
- Varshosaz, J., Tavakoli, N., Kheirolahi1, F. (2006). Use of hydrophilic natural gums in formulation of sustained-release matrix tablets of tramadol hydrochloride. AAPS Pharm Sci Tech. 7:E1–7.
- White, W.B., Black, H.R., Weber, M.A., Elliott, W.J., Bryzinski, B., Fakouhi, T.D. (1998). Comparison of effects of controlled onset extended release verapamil at bedtime and nifedipine gastrointestinal therapeutic system on arising on early morning blood pressure, heart rate, and the heart rate-blood pressure product. Am J Cardiol. 81:424–31.
- Wilding, I.R., Davis, S.S., Pozzi, F. (1994). Enteric coated timed release system for colonic targeting. Int J Pharm. 111:99–102.
- Zahirul Khan, M.I., Zeljko, P., Nevenka, K. (1999). A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers I. Manipulation of drug releases using Eudragit L100-55 and Eudragit S100 combinations. J Contr Rel. 58:215–22.