Abstract
Amphiphilic diblock copolymers composed of methoxy poly ethylene glycol (MePEG) and poly epsilon caprolactone (PCL) were synthesized for the formation of micelles by ring opening mechanism using stannous octoate as a catalyst. The effects of the molecular weight of MePEG and the copolymer ratio on the properties of micelles were investigated by Nuclear Magnetic Resonance (1H-NMR), Fourier Transform Infrared Spectroscopy (FT-IR), and Gel Permeation Chromatography (GPC). The diblock copolymers were self-assembled to form micelles and their hydrophobic core was used for the encapsulation of the anti-cancer drug (etoposide) in aqueous solution. The sizes of micelles were less than 250 nm with a narrow size distribution with monodispersed unimodal pattern. Differential Scanning Calorimetric (DSC) thermogram was done for etoposide-loaded micelles to understand the crystalline nature of the drug after entrapment. A drug loading capacity up to 60% (w/w) with an entrapment efficiency of 68% was achieved as determined by reverse phase high performance liquid chromatography (RP-HPLC). In vitro release kinetics showed a biphasic release pattern of etoposide for 2 weeks. The cytotoxic efficacy of the etoposide-loaded micelles demonstrated greater anti-proliferative activity (IC50 = 1.1 µg/ml) as compared to native drug (IC50 = 6.3 µg/ml) in pancreatic cancer cell line MIA-PaCa-2. Thus, etoposide-loaded MePEG/PCL block copolymeric micelles can be used as an efficient drug delivery vehicle for pancreatic cancer therapy.
Introduction
Drug delivery systems for controlled delivery of the drugs at the desired site of action in the body have generated great interest in the scientific community. There have been tremendous efforts to develop an efficient system for site-specific delivery. One of the possible means of reaching such a goal may be delivery via particulate drug delivery systems (CitationScholz et al., 1995; CitationLa et al., 1996). Particulate drug delivery systems due to their small size may be useful as sustained-release injections or for the delivery of a drug to a specific organ or target site. Recent studies of polymeric micelles have gained considerable attention as a versatile nanomedicine platform with greatly improved drug pharmacokinetics and efficacious response in cancer treatment (CitationSutton et al., 2007; CitationAliabadi et al., 2008; CitationMikhail & Allen, 2009; CitationShin et al., 2009). Micelles formed from amphiphilic diblock copolymers have recently attracted significant attention in the diverse field of medicine and biology. The micelles are the spherical colloidal nanoparticles formed by self-assembly of amphiphilic polymeric molecules. The micellar cores are usually constructed with biodegradable polymers such as aliphatic polyester and polypeptides, and water-soluble poly ethylene glycol which is most frequently used to build micelle corona because it can efficiently stabilize the micelles in blood compartments and reduce the uptake at the reticuloendothelial sites (e.g. Liver and Spleen). In these micellar delivery systems, the hydrophobic core of the micelle is a carrier compartment that accommodates water-insoluble anti-cancer drugs and the shell consists of a brush-like protective corona that stabilizes the micellar nanoparticles in aqueous solution (CitationKataoka & Kwon, 1993; CitationKataoka et al., 2001; CitationAdams et al., 2003). By encapsulating drugs within the micelles, solubility limits for hydrophobic drugs can be increased substantially. Polymeric micelles have been shown to increase the aqueous solubility of chemotherapeutic drugs and prolong there in vivo half-lives with narrowed systemic toxicity. This is demonstrated in an early phase of clinical trials in Japan, Korea, and the US (CitationDanson et al., 2004; CitationMatsumura et al., 2004).
The major factors that influence the performance of polymeric micelles for drug delivery are loading capacity, release kinetics, circulation times, biodistribution, and stability. Recent studies have shown that the in vivo anti-tumor activity of a drug incorporated into the polymeric micelles is positively correlated with the stability of micelles in vitro. The formation of micelles is thermodynamically favorable only above a specific concentration of the amphiphilic molecules called critical micellar concentration (CMC). When the concentration of the copolymer is below the CMC the micelles tend to disassemble. Such, thermodynamic instability of micelles below the CMC is one of the concerns for their application in vivo. For the preparation of degradable polymeric micelles with the desired feature of long circulating drug carriers, amphiphilic block copolymeric micelles based on methoxy (poly ethylene glycol) MePEG and PCL were prepared, as shown in . As a hydrophilic segment, we choose MePEG because of its non-toxic nature and it is approved by Food and Drug Administration (FDA) for internal use in the human body (CitationGref et al., 1996; CitationShuai et al., 2004; CitationSutton et al., 2007). PCL is a hydrophobic block and was chosen as a promising candidate for the biodegradable and biocompatible polymers, and its biodegradability can be enhanced greatly by copolymerization (CitationHuatan et al., 1995).
Figure 1. MePEG/ϵ-CL diblock copolymer (a), structural formula of Etoposide (b).
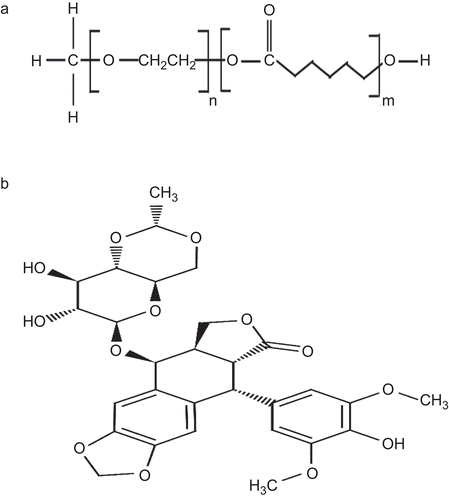
Etoposide, 4′-demethyl-epipodophyllotoxin9-[4,6-O-(R)-ethylidene-beta-D-glucopyranoside], 4′ -(dihydrogen phosphate), as shown in , is a semi-synthetic derivative of podophyllotoxin that exhibits anti-tumor activity. Etoposide is widely used in cancer chemotherapy to treat various types of cancer such as lung cancer, Hodgkin’s disease, non-Hodgkins lymphoma, AIDS related Kaposi’s sarcoma, gastric cancer, breast cancer, ovarian cancer, etc. (CitationSmit et al., 1989; CitationLampretch & Benoit, 2006). Etoposide inhibits Deoxyribonucleic acid (DNA) synthesis by forming a complex with topoisomerase II and DNA. This complex induces breaks in double stranded DNA and prevents repair by topoisomerase II binding. This accumulated break in DNA prevents the entry into the mitotic phase of cell division, and leads to cell death. Etoposide acts primarily in the G2 and S phases of the cell cycle (CitationGros et al., 2003; CitationSalido et al., 2007). Being hydrophobic in nature the water solubility of the drug has been a major problem in drug formulation and drug absorption. Etoposide has low water solubility (less than 0.1 mg/ml) and short biological half-life (3.6 h) (CitationReddy et al., 2006; CitationRaju et al., 2007). Apart from the solubility, the major drawbacks of the drug are the acute toxicity to normal tissue and inherent multi-drug resistance effects. To reduce the acute toxicity of the free drugs and to improve their therapeutic efficacy, various drug delivery systems like liposomal carrier and polymeric micellar systems are designed to act as delivery vehicles for etoposide, which increases the anti-tumor efficacy and reduces the adverse effects associated with etoposide. The pharmacokinetics property of etoposide and stability of the drug increases in the nanorange formulation, combating the acute toxicity and multidrug resistant activity (CitationSengupta et al., 2000; CitationLampretch & Benoit, 2006; CitationSnehalatha et al., 2008; CitationShin et al., 2009). CitationTang et al. (2010) loaded etoposide in poly(ethylene glycol)-co-poly(sebacic acid) (PEG–PSA), which demonstrated enhanced cytotoxic activity against a human lung tumor cell line in vitro and in a xenograft mouse model. Etoposide was encapsulated in a novel block ionomer complexes (BIC) composed of graft-comb copolymers of Pluronic and poly(acrylic acid) (Pluronic–PAA) with a cationic surfactant, hexadecyltrimethylammonium bromide (HTAB) can be used as promisive nanocarriers for pharmaceuticals (CitationOh et al., 2006). Earlier, the use of polymeric micelles as carriers of hydrophobic anti-cancer drugs has advanced greatly by the work of Kataoka groups (CitationKataoka & Kwon, 1993; CitationKwon & Karaoka, 1995; CitationKataoka et al., 2001; CitationKakizawa & Kataoka, 2002). Depending on the chemical structures of the drug either the chemical conjugation or physical entrapment method are employed for the incorporation of hydrophobic drugs into the micellar core (CitationWhitesides et al., 1991; CitationTorchilin, 2007).
In this paper, we have described the synthesis of block copolymeric micelles composed of MePEG and PCL and evaluated the possibility of these micelles as drug delivery vehicles. Therefore, we investigated the block copolymeric micellar system and developed the aqueous-based formulation of etoposide. The characteristics of these micelles were investigated through dynamic light scattering (DLS), NMR, GPC, FT-IR techniques. The drug loading efficiency of the incorporated-etoposide in MePEG/PCL block copolymeric micelles were investigated by RP-HPLC. Furthermore, we have evaluated the potential of these micelles as efficient carriers for the drug etoposide by examining the cytotoxicity against the pancreatic cell line MIA PaCa-2.
Materials and methods
Materials
Etoposide was obtained from Sigma (Sigma-Aldrich, Germany). Methoxy poly (ethylene glycol) (MePEG, Mn= 5000 by supplier, Mn= 5541 by our GPC measurements) was supplied by Fluka (Sigma-Aldrich, St. Louis, MO), and purified by azeotropic distillation with dry toluene (Ranbaxy Fine Chem., India), and dried to constant weight under vacuum at 30°C before use. ϵ-caprolactone (PCL) (Mw = 114.14) was purchased from Fluka and was dried over calcium hydride for 48 h at room temperature and then distilled under reduced pressure prior to polymerization. Stannous-II octoate (Sn (Oct)2), Pyrene from Sigma-Aldrich (St. Louis, MO) was used as received. Distilled-deionized water was prepared with Milli-Q plus System (Elix 10, Millipore Corp. India). All other chemicals used were of reagent grade and used as purchased without further purification.
Synthesis of MePEG/PCL diblock copolymers
The polymers were activated by removing the entrapped moisture. PEG (molecular weight, 5000 Da) was subjected to azeotropic distillation with toluene for 6 h, and excess toluene was removed by rota vapor. PCL was activated by keeping it in activated molecular sieves for 24 h. MePEG/PCL diblock copolymers were synthesized by a ring opening polymerization of PCL using MePEG homopolymer as a micro-initiator and Sn (Oct)2 as a catalyst; 5 mg of different ratios of MePEG and PCL were mixed in a 10 ml round bottom flask connected with a vacuum joint. The mixture was degassed with vacuum pump (0.28 mbar) and kept at 160°C. When the polymerization was completed, the reaction product was cooled at an ambient temperature, and then dissolved in dichloromethane. The obtained solution was precipitated in excess amount of cold methanol to remove MePEG homopolymers and any residual caprolactone monomers. The precipitate was collected by filtration and washed several times with diethyl ether. The resulting product was dried in a vacuum oven at 40°C for 3 days (CitationZhou et al., 2003). All other copolymers with different (MePEG: PCL) ratios (80:20, 60:40, 40:60, 20:80) were synthesized by adopting the above method by keeping the final weight of the mixture constant at 5 mg.
Characterization of the diblock copolymers
Fourier-transform infrared (FT-IR) spectral studies were carried out with a Perkin Elmer (Model Spectrum 1) spectrophotometer in the range between 4000–1000 cm−1, with a resolution of 2 cm−1. Powdery samples were compressed into potassium bromide (KBr) pellets for the FT-IR measurements. The molecular weight distribution of MePEG/ϵ-CL diblock copolymers were characterized by elution time relative to polystyrene monodisperse standards from GPC apparatus (Shimadzu Model LC-10AD HPLC pump, Japan) with Class Vp software programme. Ultrastyragel tetrahydrofuran (THF) column (7.8 × 300 mm I.D. H-4, Waters, Milford, MA) and SIL-10ADVP Shimadzu, differential refractometeric detector were used. The mobile phase was chloroform with a flow rate of 1 ml/min. The injection volume was usually 100 µl of stock solutions (0.1∼0.5 w/v%). The calibration curve was prepared before measurement by using standard polystyrene (molecular weight: 5.6 × 103, 1.25 × 104, 2.7 × 104, 5.1 × 104, 1.25 × 105, and 2.75 × 105 respectively, Polymer Standard Services, Warwick, RI). In addition, the composition and number average molecular weight of each copolymer in CDCl3 solution were determined by 300 MHz 1H NMR (Bruker Avane, DPX300). The transition temperatures and melting temperature point of polymers were measured by DSC (Mettler Toledo DSC 821, configured to Mettler Toledo Star with Solaris V4.1a operating systems). The scans of polymers were recorded at a heating rate of 10°C/min in the range of 40–180°C, under nitrogen atmosphere in a sealed crucible.
Preparation of etoposide-loaded micelles
MePEG/PCL block copolymeric micelles containing etoposide were prepared according to the dialysis method (CitationChasin & Langer, 1990). Briefly, 100 mg of MePEG/PCL block copolymer (PGL3) was dissolved in 10 ml of dimethylformamide (DMF) organic solvent and to that different ratios of drugs such as 10–60% were loaded and micellar solution were named EPG1–EPG 4, respectively (shown in ). To form etoposide-loaded micelles and remove free etoposide, solution were dialyzed at room temperature for 24 h against 3 l of ultra pure water using cellulose dialysis membranes (Molecular weight cut-off: 12 kDa, size: 21/35, Sigma-Aldrich, St. Louis, Missouri, USA). The micellar solution were collected and sonicated at 39 W for 30 s, using Sonics Vibra cell VC505 Sonicator (Sonics & Materials, Inc. Newtown, USA). The micellar solution obtained in this process was frozen at −80°C and then lyophilized using a lyophilizer (FEEZONE 6lt, Labconco Corp, Kansas City, MO) to obtain dry powder of etoposide-loaded micellar products, as listed in .
Table 1. Summary of etoposide loaded MePEG/PCL block copolymeric micelles using PGL3 diblock copolymers.
Table 2. Summary and physical characterization of MePEG/PCL block copolymers and blank micelles.
Particle size distribution and surface characterization of micelles
To determine the hydrodynamic diameter of MePEG/PCL diblock copolymers and etoposide-loaded MePEG/PCL block copolymeric micelles were carried out using a Zetasizer (Model: ZEN3600, NanoZS, Malvern Instruments Ltd, UK) at a wavelength of 633 nm at 25°C. The intensity of the scattered light was detected at 90°C to the incident beam. The micelles (~ 1 mg) were dispersed in deionized water and sonicated in probe sonicator (Model: VCX750, Sonics & Materials, Inc. Newtown, USA) for 20 s in an ice bath for both particle size and zeta potential measurement. Measurements for size (average of five measurements for one batch) and zeta potential (average of 20 measurements for a batch) were taken for analysis (Shin et al., 1998). An average size distribution of aqueous micellar solution was determined based on the dispersion technology software (DTS Nano) and the surface charge from zeta potential measurement (Model: ZEN3600, Nano ZS, Malvern Instruments Ltd, UK).
Determination of critical micellar concentration (CMC)
In order to determine the CMC of MePEG/PCL block copolymeric micelles, fluorescence measurements were carried out by the fluorescence spectrophotometer (Synergy HT, Bio Tek Instruments, Inc., Winooski, VT) using pyrene as a fluorescent probe. The fluorescence spectra of pyrene at various concentrations of MePEG/PCL diblock copolymers were taken. The pyrene concentration was kept at 6.0 × 10−7 M and the excitation wavelength was 390 nm (CitationKim et al., 1998).
Drug loading and encapsulation efficiency by RP-HPLC method
The encapsulated drug content was estimated by dissolving 10 mg of etoposide-loaded micelles in 10 ml of organic solvent in the ratio of DMF:methanol (1:1 v/v), it was sonicated in an ice bath for 20 s, at 39 W (Model: VCX750, Sonics and materials Inc, Newtown, USA). The solution was centrifuged at 17458 × g for 10 min at 10°C (Sigma1-15K, Germany). The supernatant was taken out for analysis by reverse phase isocratic mode of HPLC with slight modification (CitationShirazi et al., 2001). The Agilent 1100 (Agilent technologies, Waldbronn Analytical Division, Germany) which consists of column (Zorbax Eclipse XDB-C18,150 × 4.6 mm, i.d) were used for analysis. To this 20 µl of sample was injected manually in the injection port and analyzed with the mobile phase of methanol:water (45:55 v/v), delivered at a flow rate of 1 ml/min with a quaternary pump (Model No -G1311A) at 60°C with thermo start (Model No -G1316A). The etoposide levels were quantified by UV detection at 220 nm with diode array detector (DAD, Model -G 1315A). The amount of etoposide in the sample was determined from the peak area correlated with the standard curve. The standard curve of etoposide was prepared under identical conditions. Drug loading was defined as the amount of drug encapsulated in 100 mg of micelles and was represented as % w/w. The encapsulation efficiency (%) was determined as the percentage of drug entrapped inside the micelles with respect to the initial amount of drug added in the formulation.
In vitro drug release studies
To know the amount of drug released in in vitro conditions, 10 mg of etoposide-loaded micelles were dissolved in 3 ml of PBS (0.01 M, pH 7.4 containing 0.1 % w/v of Tween 80) (CitationBisht et al., 2007). Tween 80 was used to maintain the sink condition under in vitro conditions for the drug release (CitationSahoo et al., 2004). Then it was mixed properly by vortexing and kept in a shaker at 37°C, rotating at 150 rpm in an orbit shaking incubator (Wadegati Lab equip, India). The samples were collected at pre-determined time intervals and replaced with fresh PBS (0.01 M, pH 7.4 containing 0.1% w/v of Tween 80). The collected supernatants were lyophilized for 48 h, and then it was dissolved in DMF and methanol (1:1 v/v) for the dissolution of the drug and centrifuged at 17458 x g for 10 min at 10°C, (Sigma 1-15K, Germany). The supernatant was taken out and subjected for RP-HPLC analysis for the determination of the amount of drug released with respect to different time intervals.
Cell culture
MIA PaCa-2 pancreatic cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and were grown in DMEM supplemented with 10% fetal bovine serum and 1% L-glutamine, 1% sodium pyruvate, 1% non-essential amino acids and 1% penicillin G/streptomycin (Gibco BRL, Grand Island, NY) at 37°C in a humidified, 5% CO2 atmosphere.
Mitogenic assay
MIA PaCa-2 cells were seeded at 5000 per well density in 96-well plates, and cells were allowed to attach for 24 h. A stock solution of etoposide was prepared in dimethyl sulfoxide (DMSO) (1 mg/ml) and stored at −70°C. Different aliquots of the above stock etoposide solution following suitable dilution in medium were added to the wells. Concentration of DMSO in the medium was kept < 0.1% so that it has no effect on cell proliferation (CitationSahoo et al., 2004). Native drug dissolved in DMSO or encapsulated in micelles at different concentrations in culture medium (5–100 ng/ml) were added to separate wells. Culture medium and void micelles (without drug) served as the respective controls. In a dose–response study, the cell viability was determined at 48 h following the drug treatment. After the incubation period, the cells were treated with fresh MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] reagent (250 µl, 5 mg/ml) and further incubated for 4 h, then treated with fresh solvent consisting of 20% (w/v) sodium dodecyl sulfate (SDS) dissolved in water at 37°C mixed with an equal volume of DMF. The solvent pH was adjusted to 7.4 using 2.5% of 80% acetic acid and 1% of 1 N HCl. The absorbance was read on a microplate reader at 550 nm using microplate reader (BT 2000 Microkinetices Reader, BioTek Instrument, Inc., Winooski, VT) (CitationKotze et al., 1997). The anti-proliferative effects of different treatments were calculated as a percentage inhibition of cell growth with respect to the respective controls.
Results and discussion
Synthesis of MePEG/PCL diblock copolymers
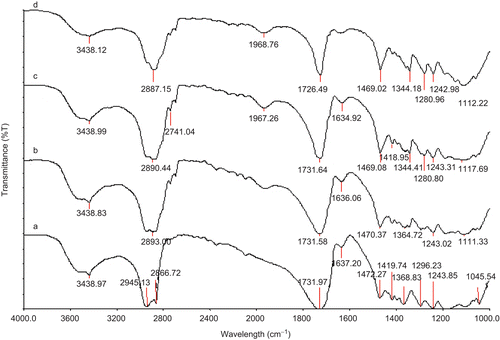
In this study, we have synthesized amphiphilic diblock copolymers (PGL series) composed of MePEG and polycaprolactone (PCL) by ring opening polymerization of PCL using MePEG prepolymers as a micro-initiator and stannous octoate as the catalyst . For the hydrophobic segment of amphiphilic diblock copolymer PCL was chosen, as its hydrophobicity facilitates the long in vivo degradation time of carrier vehicle (CitationDezhu et al., 1996) (). Moreover, it also controls the vital properties of micelles such as stability, drug loading capacity, and drug release profiles. In this regard, we have prepared four different ratio of MePEG/PCL diblock copolymeric micelle by varying the MePEG and PCL ratio, as depicted in . Further, the structure of four different ratio of MePEG/PCL diblock copolymer synthesized was identified by FT-IR spectroscopy. FT-IR results showed that the different ratio of MePEG/PCL copolymer exhibited peak characteristics of both PEG and PCL. The absorption band at 1726 cm−1 is due to the C=O (carbonyl bond) stretching vibrations of the ester carbonyl group, attributed to the formation of diblock copolymers of MePEG and PCL. Further, with increasing in molar ratio of MePEG/PCL diblock copolymers ([M]/[I], where [M] = PCL units, [I] =MePEG homopolymer) the aliphatic C-H stretching band of PCL at 2945 cm−1 was increased. Similarly, 1243 cm−1 were attributed to the characteristics -COO- bonds stretching vibrations of PEG. The absorption band at 3438 cm−1 is assigned to terminal hydroxyl groups in the copolymer from which PEG homopolymers has been removed. All the C-H stretching bands are centred at 2945 cm−1 and 2887 cm−1. Similar types of stretching vibrations for synthesized MePEG/PCL block copolymer micelles were also observed earlier by Shin et al. (1998). All these observed signals characterized the successful integrity of diblock copolymer in our formulated MePEG/PCL micelle (CitationZhu et al., 1997).
Figure 2. FTIR spectra of MePEG/ϵ-CL block copolymers synthesized with varying molar compositions (a) PGL1 (b) PGL2, (c) PGL3, and (d) PGL4 (see for the meaning of abbreviations).
Composition and molecular weight measurement
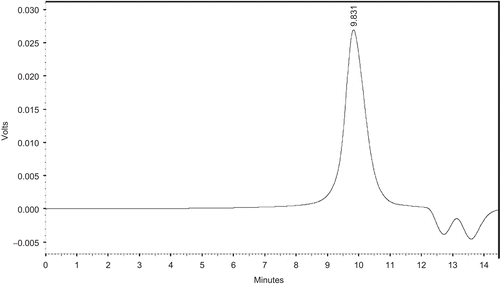
The composition and molecular weights of the diblock copolymers were determined by 1H NMR spectroscopy and GPC measurement. 1H NMR spectrum of MePEG/PCL block copolymer was shown in . We have estimated the unit ratio of MePEG (signals at 3.64 ppm) and PCL (signals at 4.06 ppm) by measuring the peak intensities of the methylene proton of the PEG chain and the ϵ-methylene proton in PCL units, respectively. The calculated results were summarized in . The molecular weight distributions of MePEG/PCL block copolymer measured by GPC analysis were depicted in . The composition was estimated from the number-average molecular weight (Mn) of MePEG/PCL block copolymer and MePEG homopolymer. The results from 1H NMR and GPC measurement exhibited almost identical molecular weight values and relatively similar values to the calculated molecular weight of copolymer. The copolymers formed have narrow Mw distributions with Mw/Mn ratios varying from 1.22–1.51, as mentioned in . From GPC curve analysis the emergence of a single peak suggested the monodispersion of Mw and absence of any homopolymer of PCL and PEG monomers, as depicted in . These observations indicated that no transesterification and/or backbiting reactions occurred during the copolymerization (CitationPeter et al., 1997).
Figure 3. 1H-NMR spectrum of MePEG/ϵ-CL block copolymers (PGL4) (see for the meaning of abbreviations).
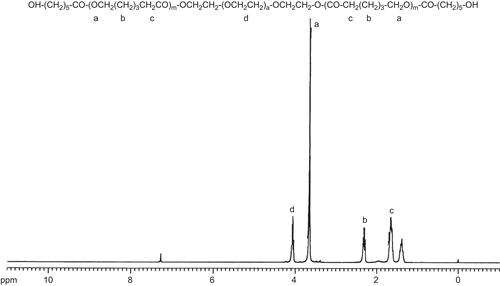
Figure 4. A typical GPC curve of PGL4 copolymers (see for the meaning of abbreviations).
Determination of critical micellar concentration
The CMC values from the fluorescence excitation of pyrene molecule at various concentrations of MePEG/PCL diblock copolymers were depicted in . Pyrene was chosen as a suitable effective probe because of its fluorescence photochemical properties. It has a significant long life time of pyrene monomers and an efficient formation of eximers (CitationKalanasundaram & Thomas, 1977; CitationYekta et al., 1993). Pyrene has a strong hydrophobic character and low solubility in water (CitationKalanasundaram & Thomas, 1977). When the micelles are formed, pyrene partitioned preferably towards the hydrophobic domain afforded by the micellar core, thus experiences a non-polar environment. As a result pyrene could be solubilized into the interior of the hydrophobic region of these MePEG/PCL diblock copolymers, so that it could be moved from the water environment to hydrophobic micellar cores. The fluorescence intensity of the copolymer changed with increased ratios of the hydrophobic PCL molecule. The excitation wavelength of pyrene shifted from 333 to 336 nm as the probe partitioned from the aqueous environment to the hydrophobic core of the micelle. The ratio of the fluorescence intensity at 336 nm to the fluorescence intensity 333 nm was calculated for each micellar diblock. The increase in ratio was attributed to the probe incorporating into the hydrophobic core of the micelles. From this the CMC values were calculated. For the measurement the emission wavelength of pyrene, it was kept at 390 nm and the concentration constant at 6.0 × 10−7 M. The CMC values were calculated to be 1.51 × 10−7 mol/l, 2.47 × 10−7 mol/l, 3.47 × 10−7 mol/l, 4.47 × 10−7 mol/l, for PGL 1, PGL 2, PGL 3, PGL 4, and depicted in . As the hydrophobic length of the micellar component in a copolymer increased, the CMC values decreased, as we have observed in our studies. The stability of the micelles both in vitro and in vivo depends on their CMC values. The hydrophobic length plays a critical role; for example, an increase in the length of the hydrophobic block at a given length of the hydrophilic block causes a significant decrease in the CMC values observed from our studies (PGL 1, PGL 2). When the length of the hydrophobic is significantly shorter than that of hydrophilic part, the block copolymers form aggregates (i.e PGL 4). Therefore the molecular design of the block copolymers were optimized to have a optimum ratio of hydrophilicity and hydrophobicity chain length, suitable for the micelle formation (i.e. PGL 3). This might be due to the force balance effect of the hydrophobic parts degree of stretching, interfacial energy of the micellar core with water, and interaction between hydrophilic chain as a shell (CitationZhu & Jiang, 2007). Similar related CMC values were also obtained with MePEG/PCL diblock copolymers where pyrene preferentially perturbates the inner core of MePEG/PCL diblock copolymers micelle in the proportion of hydrophobic chain length (CitationKim et al., 1998; Shin et al., 1998).
The obtained results were in good agreement with the results published by other groups (CitationAstafieva et al., 1993; CitationGao & Eisenberg, 1993). The micellization mainly depends on the nature and length of the hydrophobic chain length; whereas the nature of the soluble hydrophilic block has only trivial dependence on the onset of micellization. The micellization process of the diblock copolymers in appropriate solvent was thermodynamically favorable for one block, but unfavorable to another block. At the concentration below the CMC, all the diblock copolymers were in single chain form. At the CMC the collapsed block began to associate to form loose aggregates. At this stage the insoluble blocks remain in their individual collapsed states in order to maintain equilibrium with the single chain. As micelle formation required the presence of two opposing forces, one was attractive force between amphiphiles leading to aggregation, the other was the repulsive force that prevents the unlimited growth of the micelles (CitationAstafieva et al., 1993). When the concentration of the block copolymer molecule increased equilibrium, the state shifted to micellar forms. The insoluble blocks in the micellar core rearranged to find their low energy conformation and then solvent molecule gradually driven out of the micellar cores.
The hydrodynamic diameter and surface charge of the micelles
Particle size (hydrodynamic diameter) and zeta potential of MePEG/PCL blank copolymeric micelles (PGL 1–PGL 4) were analyzed using a dynamic laser light scattering method. The results of blank micellar size measurement revealed that with increase in molecular weight and hydrophobic chain length (PCL) of copolymer, the size of blank MePEG/PCL micelles were increased from137 nm to168 nm, depicted in . Similar studies conducted by Shin et al. and Shuai et al. (Shin et al.,1998; CitationShuai et al., 2004) also reported that with increase in molecular weight of copolymer the size of block copolymeric micelles were increased and copolymer with large PCL blocks formed larger micelle particles. This analysis demonstrated that the molecular weight of copolymer and the hydrophobic chain length of MePEG/PCL polymeric micelle were the major determinant for particle size. All the formulations of blank MePEG/PCL micelle showed a narrow range of zeta potential in between −5 to −8 mV, and the polydispersity index (PDI) value demonstrated relatively a low value in between 0.08–0.33, as determined by DLS. This result reinforced the opinion that the micellization takes place as a result of molecular association rather than aggregation of smaller micelles. It is well known that small size of the particle necessitated to enhance the circulation half-lives and reduced reticulo endothelial system (RES) clearance. Hence, considering the small size we have selected PGL 3 and PGL 4 among other formulations for etoposide loading. Besides offering comparable small size these selected formulations have shown hydrophilic-lipophilic balance (HLB value) above 6, which was the appropriate ratio for the micelle formation (CitationBecher & Schick, 1987). The anti-cancer drug etoposide was successfully loaded in both PGL 3 and PGL 4 formulations. However, it was observed that at higher drug loading (e.g. 60%) the PGL 4 formulation was precipitated compared to PGL 3 formulation. This could be due to its lower percentage of PCL block, which was responsible for binding and encapsulating the hydrophobic drug. As a result, we have selected PGL 3 polymer for drug-loaded micelle preparation with an aim to get small size and better stability. In this way, we have prepared four formulations of etoposide-loaded polymeric micelles with different weight ratios of etoposide-to-diblock polymer (PGL 3), as shown in . These etoposide-loaded MePEG/PCL micelles are now termed as EPG 1, EPG 2, EPG 3, and EPG 4 of variable drug loading such as 10%, 20%, 40%, and 60%, respectively. The size (hydrodynamic diameter) analysis of etoposide-loaded MePEG/PCL micelles demonstrated that the sizes of micelles were increased with an increased amount of drug loading and have a narrow size distribution, as depicted in . As the loading content of etoposide increased, the size of MePEG/PCL micelles increased from 170 nm to 264 nm. Similar trends were also obtained with taxol-loaded MePEG/PCL micelles, where reported micelle sizes were increased with increase in loading content of drug taxol (CitationKim & Lee, 2001). Beside size measurement the surface charge of different formulations of etoposide-loaded MePEG/PCL micelles were studied by DLS measurement. These analyses showed that the surface charge were in the range of −7.53 to −14.06 mV in PBS (0.01 M, pH 7.4). However, the zeta potential of the PCL particles was found to be ~ −45 mV at the same buffer. Another experiment conducted by CitationLee et al. (2009) reported that pure PCL particles were having the zeta potential ~ −50 mV, while the zeta potential values for MePEG-PCL block copolymer particles were ranging from −20 to 0 mV. The decrease in the absolute value of zeta potential is attributed to the presence of MePEG moiety at the corona of the micellar particle. The presence of PEG can shield surface ion charge of the micelles. With an increased amount of the PEG content in the copolymers, the absolute value of zeta potential was reduced. Moreover, the selected micellar system had a longer PEG chain, hence exhibited higher thickness of aqueous layer, and could offer better stabilization of the micellar system with enhanced circulation half-lives (CitationShi et al., 2005).
Drug loading, encapsulation efficiency, and state of the drug incorporated in the micelles
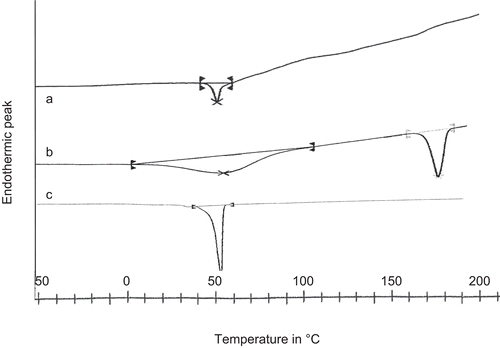
The amount of etoposide incorporated into MePEG/PCL micelles was calculated by the weight ratio of the amount of encapsulated etoposide in micelles to the amount of etoposide loaded for the micellar preparation (as depicted in ). The drug loading efficiency increased with the increased ratio of drug to polymer, as observed from . However, during the preparation of etoposide-loaded micelles, with increase of the drug ratio to the polymer, a large amount of aggregation was found. Therefore, an appropriate amount of etoposide was introduced into the micelle by controlling the weight ratio between polymer and drug, as shown in . So in drug loading experiments, the weight ratio of etoposide-to-polymer was kept in the range of 10–60%. A similar observation was found with the preparation of indomethacin-loaded MePEG/PCL micelles (CitationKim et al., 1998). The state of incorporated drug in the micelles is an important factor for determining the release profile of a drug delivery system. illustrated the DSC thermogram of etoposide-loaded micelle, native etoposide, and void micelle. The native etoposide showed an endothermic melting peak at 189°C. In drug-loaded micelles, no peak was observed at the temperature range of 100–250°C, showing the molecular dispersion of the etoposide in the copolymer, and the peak observed ∼ 55°C was the melting peak of PCL crystal. However, in the void micelles only PCL peak was observed ∼ 55°C. It could thus be said that etoposide entrapped inside the formulated micelles was in an amorphous or disordered crystalline phase form of a molecular dispersion in the polymer matrix.
Figure 5. DSC thermogram of (a) Etoposide-loaded micelle, (b) Etoposide native drug, and (c) Void micelle.
Releasing profiles of etoposide from polymeric micelles
The release profiles of etoposide from the MePEG/PCL micelles were studied in vitro. showed a release profile of etoposide-loaded micelles in PBS (0.01 M, pH, 7.4). A typical two phase release profile was observed in all the formulations of the drug-loaded micelles. There was a relatively rapid release in the first stage (~ 40% of the entrapped etoposide release in 24 h) in all the formulations, followed by a sustained and slow release over a prolonged time period; ~ 90% of the drug was released in 2 weeks. A regression analysis of the slow terminal rate of the drug release for the micelles observed was approximately (in the burst release phase) 3.38% per hour (r2 = 0.8967) with a maximum of 48% release in first 24 h; in the slow release phase, drug release was 0.113% per hour (r2 = 0.9631) with a maximum of 90% release in 2 weeks. The formulations (EPG 1–EPG 4) showed more or less the same behavior for the drug release pattern. The low drug loading encapsulation (10% and 20%) showed early release, when compared to the higher drug loading (40–60%). From these results we could consider that drug binding affinity and polymer degradation are two related phenomena. Generally it is assumed that the drug is released by several processes (CitationGref et al., 1996); (i) a diffusion through the polymeric matrix, (ii) release by polymer degradation (either surface or bulk), and (iii) a solubilization and diffusion through microchannels which exist in the polymer matrix or are formed by erosion. Being moderately lipophilic in nature, etoposide was physically entrapped in the hydrophobic core of the micelles (CitationSengupta et al., 1998; CitationTyagi et al., 1999). Accordingly the in vitro release behavior of the drug from these polymeric micellar systems was affected by its inner core with hydrophobic properties.
Figure 6. In vitro release profile of etoposide from micelles (EPG1, EPG2, EPG3, and EPG4). Data as mean ± sem (n = 3).
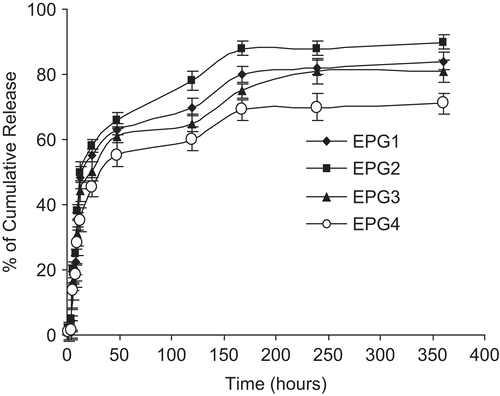
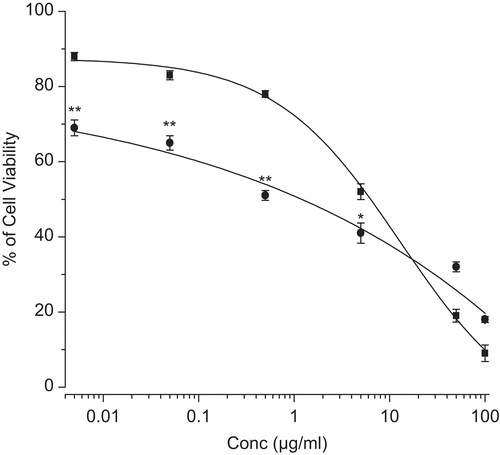
Figure 7. Dose-dependent cytotoxic effect of etoposide in solution and etoposide in micelles (EPG4). Different concentration of etoposide either as solution (filled square) or encapsulated in micellar (filled circle) was added to wells with micelles (without drug) or medium acting as respective controls. The percentage of survival was determined by standardizing the absorbance of controls to 100%. Data as mean ± sem (n = 3). Values of * p < 0.05 etoposide-loaded micelles vs native etoposide and ** p < 0.005 etoposide-loaded micelles vs native etoposide.
Proliferation assay
The pharmacokinetics properties and stability of the anti-cancer drug increases when encapsulated in a nanocarrier delivery system. This mode of delivery system combats the acute toxicity and multi-drug resistant activity (CitationSengupta et al., 2000; CitationLampretch & Benoit, 2006; CitationSnehalatha et al., 2008; CitationShin et al., 2009). In our study the growth inhibitory effect of etoposide was evaluated in vitro by using human pancreatic cancer cell line MIA PaCa-2. The selected etoposide loaded micelles (EPG 4) were taken for proliferation assay for their enhanced entrapment efficiency compared to other formulations. The MIA PaCa-2 cells were treated with a series of equivalent concentrations of either native etoposide or etoposide-loaded micelles (5–1000 ng/ml) for 48 h of incubation.
The etoposide-loaded micelles were dissolved in culture medium and native etoposide was dissolved in DMSO. The cultured medium-treated cells served as control for the cytotoxicity experiment (). The result showed IC50 values of etoposide entrapped in micellar system and native etoposide was 1.1 µg/ml and 6.3 µg/ml, respectively. Further, it was confirmed that the etoposide-loaded in micelles had better anti-proliferative activity compared to native etoposide at lower concentrations, but at higher concentration (100 µg/ml) native drug was more effective than the micelles.
It was well-known that etoposide inhibits DNA synthesis by forming a complex with topoisomerase II and DNA; by inducing breaks in double stranded DNA it prevents repair by topoisomerase II binding and ultimately influenced apoptosis in diverse cell types (CitationSalido et al., 2007). Experimental evidence showed that at 5 µM, concentration of etoposide was sufficient for inducing anti-proliferation activity in MIA PaCa-2 cells (CitationSato et al., 2000). Further, anti-proliferation activity of MIA PaCa-2 was confirmed by the [3H] thymidine incorporation test in which the DNA synthesis decreased after exposure of etoposide. In another study, etoposide encapsulated in star amphiphilic block copolymer showed a significant level of cytotoxicity activity in porcine kidney epithelial cells (LLC-PK) when compared with native etoposide (CitationWang et al., 2005). It was well-known that the major drawback in cancer treatment is the multi-drug resistance activity due to the over-expression of P-glycoprotein (P-gp) pumps in malignant cell, which effluxed out the delivered anti-cancer drugs from inside the tumor cells. Further, it was reported that anti-cancer drugs like etoposide were strongly affected by the multi-drug resistance phenomenon (Abe et al., 1994; Koike et al., 1996). A study conducted by CitationElamanchili et al. (2008) suggested that amphiphilic diblock copolymers composed of methoxypolyethylene glycol-block-polycaprolactone (MePEG-b-PCL) enhanced the cellular accumulation of Pgp substrates by modulating the function of this drug efflux transporter. The diblock copolymer demonstrated enhanced accumulation of various Pgp substrates in Pgp over-expressing MDR cells but did not influence substrate accumulation in non-Pgp expressing cells. These results highlighted the potential of MePEG-b-PCL diblock copolymer to reverse drug resistance in MDR cancer cells through inhibition of Pgp function, making it a promising candidate for overcoming MDR. The experiment conducted by CitationLampretch and Benoit (2006) conformed the etoposide-loaded lipid nanocapsules developed by their group with polyethylene glycol hydroxystearate (as the surfactant) reduced the cell growth of glioma cell lines C6, F98, and 9L by reversing the multi-drug resistance of P-gp and further the carriers ensured endocytosis by increasing the cytotoxicity of etoposide-loaded lipid nanocapsules in glioma cell lines. In another study CitationPark et al. (2005) incorporated the hydrophobic anti-cancer drug paclitaxel in folate-conjugated methoxy (poly ethylene glycol)/poly(epsilon-caprolactone) (MePEG/PCL) micelles which showed higher and selective cytotoxic activity for the cancer cell lines such as MCF-7 and HeLa. However, the paclitaxel-loaded MePEG/PCL micelles without folate groups showed similar cytotoxicity for both the normal (human fibroblasts) and the cancer cell lines MCF-7 and HeLa.
The amphiphilic block copolymers, for cancer therapy, are used looking at the physiological aspects of the tumor tissues. Most solid tumors possess unique physiological characteristics that are not observed in normal tissues or organs, such as extensive angiogenesis and hence hypervasculature, defective vascular architecture, impaired lymphatic drainage system, and greatly increased production of a number of permeability mediators. The above-mentioned phenomenon is known as the enhanced permeability and retention (EPR) effect. The EPR effect provides a great opportunity for more selective targeting of lipid- or polymer-conjugated anti-cancer drugs to the tumors. Through the EPR effect liposomes and micellar formulation tend to accumulate in tumor tissues much more than the normal tissues (CitationMaeda et al., 2000; CitationTorchilin 2007; CitationMikhail & Allen, 2009). Therefore, amphiphilic block copolymeric micelles have been shown to increase drug accumulation at solid tumor sites via passive targeting which relies on the EPR. In fact, passive targeting of drugs to tumors using colloidal systems has been reported to result in as much as a 10–50-fold increase in drug accumulation in tumors relative to normal tissues (Iyer et al., 2006). For example, CitationKwon and Kataoka (1995) reported that the doxorubicin conjugated polyethylene glycol-b poly aspartic acid) (PEG-b-PAsp-Dox) micelle system increased the tumor accumulation of the encapsulated doxorubicin than free doxorubicin following i.v. administration in tumor-bearing mice. The formulated etoposide-loaded methoxy (poly ethylene glycol)/poly (epsilon-caprolactone) (MePEG/PCL) might have overcome the multi-drug resistance related P-glycoprotein pump by acting as an anti-cancer drug reservoir maintaining higher anti-cancer drug concentration inside the cancer cells and releasing the drug in a sustained manner ensuring it as a promising candidate for drug delivery.
Conclusions
In the above studies, biodegradable MePEG/PCL diblock copolymers of different molecular weights and compositions were synthesized for the delivery of model anti-cancer drug etoposide. These amphiphilic diblock copolymers were characterized by using DLS, NMR, GPC, DSC, and FT-IR techniques. It was successfully self-assembled into micelle and etoposide was efficiently entrapped in the hydrophobic core of the resultant MePEG/PCL micelles. The ideal formulation selected for in vitro studies showed an average size of 250 nm, negative zeta potential (∼ −14 mV) with high drug loading (up to 68%). We hypothesize this small size of the resultant etoposide-loaded micelles may serve as a promising candidate for passive targeting to tumors through the EPR effect. Furthermore, it was observed that etoposide encapsulated micellar formulation was comparatively more anti-proliferative than native etoposide under in vitro conditions as evaluated in pancreatic cancer cell line. This is due to sustained release of encapsulated drug for a longer time, resulting in reduction of cell viability. Thus, the discussed comprehensible results justified this MePEG/PCL micellar drug delivery approach can be used as a future avenue to improve the potentiality of etoposide without affecting its anti-cancer properties for the treatment of cancer.
Declaration of interest
SKS would like to thank the Department of Biotechnology, Government of India for providing the grants no. BT/04(SBIRI)/48/2006-PID and BT/PR7968/MED/14/1206/2006.
References
- Abe, T., Hasegawa, S., Taniguchi, K., Yokomizo, A., Kuwano, T., Ono, M., Mori, T., Hori, S., Kohno, K., Kuwano, M. (1994). Possible involvement of multidrug-resistance-associated protein (MRP) gene expression in spontaneous drug resistance to vincristine, etoposide and adriamycin in human glioma cells. Int J Cancer. 58:860–864.
- Adams, M.L., Lavasanifar, A., Kwon, G.S. (2003). Amphiphilic block copolymers for drug delivery. J Pharm Sci. 92:1343–55.
- Aliabadi, H.M., Shahin, M., Brocks, D.R., Lavasanifar, A. (2008). Disposition of drugs in block copolymer micelle delivery systems: from discovery to recovery. Clin Pharmacokinet. 47:619–34.
- Astafieva, I., Zhong, X.F., Eisenberg, A. (1993). Critical micellization phenomena in block polyelectrolyte solution. Macromolecules. 26:7339–52.
- Becher, P., Schick, M. (1987). Nonionic surfactants physical chemistry. Marcel Dekker, New York..
- Bisht, S., Feldmann, G., Soni, S., Ravi, R., Karikar, C., Maitra, A., Maitra, A. (2007). Polymeric nanoparticle-encapsulated curcumin (‘nanocurcumin’): a novel strategy for human cancer therapy. J Nanobiotechnol. 5:1–18.
- Chasin, M., Langer, R. (1990). Biodegradable polymers as drug delivery systems. USA: Marcel Dekker.
- Danson, S., Ferry, D., Alakhov, V., Margison, J., Kerr, D., Jowle, D., Brampton, M., Halbert, G., Ranson, M. (2004). Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br J Cancer. 90:2085–91.
- Dezhu, M., Xiaolie, L., Ruiyun, Z., Nishi, T. (1996). Miscibility and spherulites in blends of poly(e-caprolactone) with ethylene terephthalate-caprolactone copolyester. Polymer. 37:1575–81.
- Elamanchili, P., McEachern, C., Burt, H. (2008). Reversal of multidrug resistance by methoxypolyethylene glycol-block-polycaprolactone diblock copolymers through the inhibition of P-glycoprotein function. J Pharm Sci. 98:945–58.
- Gao, Z., Eisenberg, A. (1993). A model micellization for block copolymers in solutions. Macromolecules. 26:7353–60.
- Gref, R., Mainamitake, Y., Peracchia, M.T., Langer, R. (1996). Microparticulate systems for the delivery of protein and vaccines. USA: Marcel Dekker, New York.
- Gros, L., Delaporte, C., Frey, S., Decesse, J., de Saint-Vincent, B. R., Cavarec, L., Dubart, A., Gudkov, A.V., Jacquemin-Sablon, A. (2003). Identification of new drug sensitivity genes using genetic suppressor elements: protein arginine N-methyltransferase mediates cell sensitivity to DNA-damaging agents. Cancer Res. 63:164–71.
- Huatan, H., Collett, J.H., Attwood, D. (1995). The microencapsulation of protein using a novel ternary blend based on poly(epsilon-caprolactone). J Microencapsul. 12:557–67.
- Iyer, A.K., Khaled, G., Fang, J., Maeda, H. (2005). Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006, 17-18:812–8.
- Kakizawa, Y., Kataoka, K. (2002). Block copolymer micelles for delivery of gene and related compounds. Adv Drug Deliv Rev. 54:203–22.
- Kalanasundaram, K., Thomas, J.K. (1977). Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J Am Chem Soc. 99:2039–44.
- Kataoka, K., Harada, A., Nagasaki, Y. (2001). Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 47:113–31.
- Kataoka, K., Kwon, G.S. (1993). Block copolymer micelles as vehicles for drug delivery. J Cont Rel. 24:119–32.
- Kim, S.Y., Shin, I.G., Lee, Y.M., Cho, C.S., Sung, Y.K. (1998). Methoxy poly(ethylene glycol) and epsilon-caprolactone amphiphilic block copolymeric micelle containing indomethacin. II. Micelle formation and drug release behaviours. J Contr Rel. 51:13–22.
- Kim, S.Y., Lee, Y.M. (2001). Taxol-loaded block copolymer nanospheres composed of methoxy poly(ethylene glycol) and poly(epsilon-caprolactone) as novel anticancer drug carriers. Biomaterials. 22:1697–704.
- Koike, K., Abe, T., Hisano, T., Kubo, T., Wada, M., Kohno, K., Kuwano, M. (1996). Overexpression of multidrug resistance protein gene in humane cancer cell lines selected for drug resistance to epipodophyllotoxin. Jpn J Cancer Res. 87:765–772.
- Kotze, A.F., de Leeuw, BJ., Lueben, HL., de Boer, AG., Verhoef, JC., Junginger, HE. (1997). Chitosans for enhanced delivery of therapeutics peptides across intestinal epithalia: in vitro evaluation in Caco-2 cell monolayers. Int J Pharm. 159:243–53.
- Kwon, G.S., Kataoka, K. (1995). Block copolymer micelles as long circulating drug vehicles. Adv Drug Deliv Rev. 16:295–309.
- La, S.B., Okano, T., Kataoka, K. (1996). Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly(ethylene oxide)-poly(beta-benzyl L-aspartate) block copolymer micelles. J Pharm Sci. 85:85–90.
- Lampretch, A., Benoit, J.-P. (2006). Etoposide nanocarriers suppress glioma cell growth by intracellular drug delivery and simultaneous P-glycoprotein inhibition. J Contr Rel. 112:208–13.
- Lee, J.S., Hwang, S.J., Lee, D.S., Kim, S.C., Kim, D.W. (2009). Formation of poly(ethylene glycol)-poly(ϵ-caprolactone) nanoparticles via nanoprecipitation. Macromol Res. 17:72–8.
- Maeda, H., Wu, J., Sawa, T., Matsumura, Y., Hori, K. (2000). Tumour vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Contr Rel. 65:271–84.
- Matsumura, Y., Hamaguchi, T., Ura, T., Muro, K., Yamada, Y., Shimada, Y., Shirao, K., Okusaka, T., Ueno, H., Ikeda, M., Watanabe, N. (2004). Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br J Cancer. 91:1775–81.
- Mikhail, S., Allen, C. (2009). Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels. J Contr Rel. 138:214–23.
- Oh, K.T., Bronich, T K., Bromberg, L., Alan, H,T., Kabanov, A V. (2006). Block ionomer complexes as prospective nanocontainers for drug delivery. J Contr Rel. 115:9–17.
- Park, E.K., Kim, S.Y., Lee, S.B., Lee, Y.M. (2005). Folate-conjugated methoxy poly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric micelles for tumour-targeted drug delivery. J Contr Rel. 109:158–68.
- Peter, J., Ester, M., Peter, V., Jennie, H., Peter, J., Jan, F. (1997). Melt block copolymerisation of e-caprolactone and lactide. J Polym Sci A. 35:219–26.
- Raju, Y.P., Garbhapu, A., Prasanna, S.A.L., Rao, B.S., Murthy, R. (2007). Studies on enhancement of dissolution rate of etoposide. Indian J Pharm Sci. 69:269–73.
- Reddy, L.H., Adhikari, J S., Dwarakanath, B.S.R., Sharma,, R K., Murthy, R.R. (2006). Tumouricidal effects of etoposide incorporated into solid lipid nanoparticles after intraperitoneal administration in dalton’s lymphoma bearing mice. AAPS J. 8:E254–62.
- Sahoo, S.K., Ma, W., Labhasetwar, V. (2004). Efficacy of a transferrin-conjugated paclitaxel loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 112:335–40.
- Salido, M., Gonzalez, J., Vilches, J. (2007). Loss of mitochondrial membrane potential is inhibited by bombesin in etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol Cancer Ther. 6:1292–9.
- Sato, N., Mizumoto, K., Kusumoto, M., Nishio, S., Maehara, N., Urashima, T., Ogawa, T., Tanaka, M. (2000). Up-regulation of telomerase activity in human pancreatic cancer cells after exposure to etoposide. Br J Cancer. 82:1819–26.
- Scholz, G., Lijima, M., Nagasaki, Y., Kataoka, K. (1995). A novel reactive polymeric micelle with aldehyde groups on its surface. Macromolecules. 28:7295–7.
- Sengupta, S., Velpandian, T., Sapra, P., Sanyal, M., Gupta, S.K. (1998). Etoposide encapsulated long circulating liposomes: improved antitumour efficacy with reduced adverse effects. Naunyn Schmeideberg’s Arch Pharmacol. 358:R531.
- Sengupta, S., Tyagi, P., Velpandian, T., Gupta, Y.K., Gupta, S.K. (2000). Etoposide encapsulated in positively charged liposomes: pharmacokinetic studies in mice and formulation stability studies. Pharmacol Res. 42:459–64.
- Shi, B., Fang, C., You, XM., Zhang, Y., Fu, S., Pei, YY. (2005). Stealth MePEG-PCL micelles: effects of polymer composition on micelle physicochemical characteristics, in vivo drug release, in vivo pharmacokinetics in rats and biodistribution in S180 tumour bearing mice. Colloid Polym Sci. 283:954–67.
- Shin, H.-C., Alani, A.W.G., Rao, D.A., Rockich, N.C., Kwon, G.S. (2009). Multi-drug loaded polymeric micelles for simultaneous delivery of poorly soluble anticancer drugs. J Contr Rel. 140:294–300.
- Shin, I.G., Kim, S.Y., Lee, Y.M., Cho, C.S., Sung, Y.K. (1998b). Methoxy poly(ethylene glycol)/epsilon-caprolactone amphiphilic block copolymeric micelle containing indomethacin. I. Preparation and characterization. J Contr Rel. 51:1–11.
- Shirazi, F.H., Bahrami, G., Stewart, D.J., Tomiak, E., Delorme, F., Noel, D., Goel, R. (2001). A rapid reversed phase high performance liquid chromatographic method for determination of etoposide (VP-16) in human plasma. J Pharm Biomed Anal. 25:353–6.
- Shuai, X., Ai, H., Nasongkla, N., Kim, S., Gao, J. (2004). Micellar carriers based on block copolymers of poly(epsilon-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J Contr Rel. 98:415–26.
- Smit, E.F., Carney, D.N., Hurford, P., Sleijfer, D.T., Postmos, P.E. (1989). Phase II study oral etoposide in elderly patients with small cell lung cancer. Thorax. 44:631–3.
- Snehalatha, M., Venugopal, K., Saha, R.N., Babbar, A.K., Sharma, R.K. (2008). Etoposide loaded PLGA and PCL nanoparticles II: biodistribution and pharmacokinetics after radiolabeling with Tc-99m. Drug Deliv. 15:277–87.
- Sutton, D., Nasongkla, N., Blanco, E., Gao, J. (2007). Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 24:1029–46.
- Tang, B.C., Fu, J., Watkins, D.N., Hanes, J. (2010). Enhanced efficacy of local etoposide delivery by poly(ether-anhydride) particles against small cell lung cancer in vivo. Biomaterials. 31:339–44.
- Torchilin, V.P. (2007). Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 24:1–16.
- Tyagi, P., Sengupta, S., Velpandian, T., Gupta, Y.K., Kochupillai, V., Gupta, S.K. (1999). Evaluations of the antitumour activity of liposomal formulations of etoposide against choriocarcinoma xenograft in Balb/c nu/nu mice. Pharm Pharmacol Commun. 5:595–8.
- Wang, F., Bronich, T.K., Kabanov, A.V., Rauh, R.D., Roovers, J. (2005). Synthesis and evaluation of a star amphiphilic block copolymer from poly(epsilon-caprolactone) and poly(ethylene glycol) as a potential drug delivery carrier. Bioconjug Chem. 16:397–405.
- Whitesides, G.M., Mathias, J.P., Seto, C.T. (1991). Molecular self-assembly and nanochemistry: a chemical strategy for the synthesis of nanostructures. Science. 254:1312–9.
- Yekta, A., Duhamel, J., Brochard, P., Adiwidjja, H., Winnik, M.A. (1993). A fluorescenct probe study of hydrophobically modified poly (ethylene oxide). Macromolecules. 26:1829–36.
- Zhou, S., Deng, X., Yang, H. (2003). Biodegradable poly(epsilon-caprolactone)-poly(ethylene glycol) block copolymers: characterization and their use as drug carriers for a controlled delivery system. Biomaterials. 24:3563–70.
- Zhu, J., Jiang, W. (2007). Self assembly of block copolymers in selective solvents. In: Chemical physics research trend. Vol. 252, Chapter 2. Sameul V Arnold (Ed) Nova Science publishers, Inc, New York:65–116.
- Zhu, Z.X., Xiong, C.D., Zhang, L.L., Deng, X. (1997). Synthesis and characterisation of poly(e-caprolactone)-poly(ethylene glycol) block copolymer. J Polym Sci A. 35:709–14.