Abstract
Efficient and specific delivery of antisenses (ASs) and protection of the sequences from degradation are critical factors for effective therapy. Sustained release nanoparticles (NP) offer increased resistance to nuclease degradation, increased amounts of AS uptake, and the possibility of control in dosing and sustained duration of AS administration. The biodegradable and biocompatible poly(D,L-lactic-co-glycolic acid) copolymer (PLGA) was utilized to encapsulate AS directed against osteopontin (OPN), which is a promising therapeutic target in mammary carcinoma. Whole body biodistribution of OPN AS NP was evaluated in comparison to naked AS, in intact and mammary carcinoma metastasis model bearing rats. Naked and NP encapsulated AS exhibited different biodistribution profiles. AS NP, in contrast to naked AS, tended to accumulate mostly in the spleen, liver, and at the tumor inoculation site. Drug levels in intact organs were negligible. The elimination of naked AS was faster, due to rapid degradation of the unprotected sequence. It is concluded that AS NP protect the AS from degradation, provide efficient AS delivery to the tumor tissue, and minimize AS accumulation in intact organs due to the AS sustained release profile as well as the favorable NP physicochemical properties.
Introduction
Nanocarrier systems, such as nanoparticles (NP), have been increasingly employed as drug delivery devices of proteins, macromolecules, small molecules, and nucleic acids (CitationFarokhzad & Langer, 2006; CitationZhang et al., 2008). The nanocarriers are utilized to protect molecules from biodegradation as well as to modify their pharmacokinetics and biodistribution (CitationLi & Huang, 2008). Antisense (AS) therapy provides a specific approach in down-regulating particular genes, related to tumor tissue and metastastic processes (CitationWang et al., 2001; CitationStahel & Zangemeister-Wittke, 2003; CitationTamm, 2006; CitationTamm & Wagner, 2006). Nanoparticulate technology is of particular use in developing a new generation of more effective cancer therapies capable of overcoming the many biological, biophysical, and biomedical barriers that the body stages against a standard intervention (CitationBrigger et al., 2002). Osteopontin (OPN) has been characterized as a promising target for therapy by antisense oligonucleotides directed against the RNA of this protein, thus preventing or reducing lytic skeletal metastasis (CitationRosol, 2000; CitationTuck & Chambers, 2001; CitationAgrawal et al., 2002; CitationRudland et al., 2002; CitationKang et al., 2003; CitationAdwan et al., 2004). Due to their large molecular size and high charge density the low cellular permeability of AS is a major problem encountered with their therapeutic use (CitationSmith, 1995; CitationKurreck, 2003). Preferable accumulation of the AS at the target tissue, and at the same time protecting the sequence from degradation, is essential conditions for AS therapeutic efficiency and minimal side-effects (CitationJuliano et al., 2009; CitationWhite et al., 2009).
Poly(lactic-co-glycolic acid) copolymers (PLGA) are among the few synthetic biocompatible biodegradable polymers approved for human gene therapy use (CitationShive & Anderson, 1997; CitationBala et al., 2004). Due to their nano-size range and surface charge, polymeric nanospheres containing AS can overcome the absorption barrier of the cell membrane by penetrating inside the cell via endocytosis (CitationCohen et al., 2000; CitationCohen-Sacks et al., 2002; CitationFattal & Bochot, 2008; Citationde Martimprey et al., 2009). Passive drug targeting to solid tumors can be achieved using NP due to the enhanced permeability and retention effect (EPR effect), based on abnormalities of tumor vasculature, such as hypervascularization, aberrant vascular architecture, extensive extravasation within tumor tissues, and impeded lymphatic drainage (CitationMaeda, 2001). Systemic AS therapy by non-targeted NP can be assisted by regional administration of injecting the NP near the diseased tissue, such as bathing the isolated arterial segment (CitationCohen-Sacks et al., 2002) or instilling the NP suspension in the supplying blood vessel. This approach, was utilized successfully in the treatment of experimental mammary carcinoma rat model treated with OPN NP (CitationElazar et al., 2010). However, in order to evaluate the potential therapeutic approach as well possible toxicity of the NP, uptake of the NP in the target tissue and body biodistribution should be examined.
In this work we examined the biodistribution of a controlled release NP delivery system for OPN AS sequences, following systemic administration. The biodistribution of AS NP was evaluated in healthy animals and in the mammary carcinoma bone metastasis model in comparison with naked AS sequences.
Materials and methods
PLGA, of a lactide-glycolide ratio of 50:50, inherent viscosity of 0.79 dl/g, and Mw of 75,200, was purchased from Birmingham Polymers (Birmingham, AL). OPN AS sequence, labeled with fluorescein (FITC), was purchased from Alpha DNA (Montreal, Quebec, Canada). Rhodamine-labeled PLGA was synthesized in our lab. Polyvinyl alcohol (PVA) with an average Mw range of 15,000–30,000 D, urea, polyacrylamide, TEMED, ammonium persulfate, Tris-Cl, TBE and TE buffers, and sodium acetate were obtained from Sigma (St Louis, MO). All the chemicals and solutions for internal labeling of oligonucleotides were obtained at the molecular biology grade. All other chemicals were obtained commercially at the highest analytical grade available. All buffers and solutions were filter sterilized.
AS NP preparation and characterization
A double emulsion system and the solvent evaporation technique were used to incorporate OPN AS sequences in PLGA (CitationElazar et al., 2010). Briefly, 300 µl of TE buffer (10 mM Tris and 1 mM EDTA, pH adjusted to 7.5) containing 1200 µg of AS (initial loading of 1.7 w/w%) were emulsified in 3% PLGA/chloroform solution (2.3 ml) and sonicated over an ice-bath using a microtip probe sonicator (Microson XL; Misonix, Farmingdale, NY) at 11 W for 30 s. The resulting primary emulsion was added dropwise to a 2% poly-vinyl-alcohol (PVA) solution (in 25 ml TE) containing 7.5 mM calcium chloride, and after sonicating this mixture for 1 min a double emulsion was formed. Following overnight chloroform evaporation at 4°C, the resulting particles were collected by ultracentrifugation at 80,000 g (TST28 rotor, Beckman’s polyallomer centrifuge tubes, 25 × 89 mm), washed three times with sterile double distilled water, resuspended in sterile 2% mannitol water solution, and lyophilized. Dry NP were stored in a vacuum desiccator at 4°C.
The amount of AS entrapped in the NP was analyzed by dissolving the NP (10 mg) in chloroform (2 ml), and extracting the AS from the polymer solution by repetitive addition of TE buffer (four times with 0.5 ml). The AS content was determined using the fluorescence Oligreen assay kit (Molecular Probes, Eugene, OR) with excitation at 480 nm and emission at 520 nm, and by UV spectroscopy at λ = 260 nm.
NP size and morphology were evaluated by dynamic light scattering, ALV (NIBS/HPPS GmbH, Langen, Germany), and a transmission electron microscope (TEM CN12, Philips, Eindhoven, The Netherlands). Average size and size distribution of empty NP and NP loaded with AS sequences were measured before and after lyophilization. For ALV size measurement, after lyophilization, 1 mg of dry NP was suspended in 1 ml of sterile double distilled water. Before the lyophilization, 30 µl of NP suspension were added to 1 ml of sterile double distilled water. Zeta-sizer (Malvern Instruments Ltd, UK) was used for evaluation of NP charge. The lyophilized empty and AS loaded particles were suspended in sterile double distilled sterile water or saline solution to reach the attenuation index 7–9, and the mean NP charge (mV ± SD) was measured.
OPN AS sequence was labeled with FITC at 5′end. PLGA was covalently conjugated with rhodamine. A double emulsion system and the solvent evaporation technique were used to incorporate FITC-labeled OPN sequences in NP, as described above; 10% of rhodamine labeled PLGA was used in the NP formulation.
Animals
Nude rats (athymic, RNU strain) were obtained from Harlan (Harlan comp, Borchen, Germany) at an age of 6–8 weeks. They were housed under specific pathogen-free conditions in a mini-barrier system of the central animal facility, conforming to the standards for care and use of laboratory animals of the DKFZ and NIH. Autoclaved feed and water was given ad libitum to the animals that were maintained under controlled conditions (21 ± 2°C room temperature, 60% humidity, and 12 h light–dark rhythm).
In-vivo metastasis model
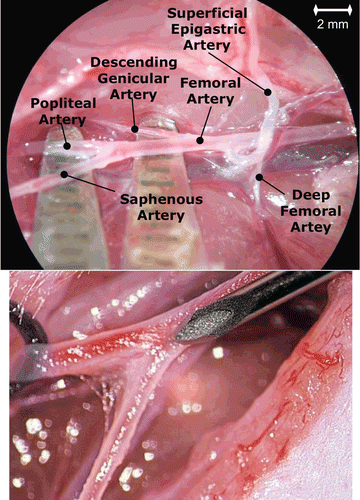
Sub-confluent MDA-MB-231 tumor cells were harvested using 2 mM EDTA in PBS− (phosphate-buffered saline without Ca++ and Mg++) and 0.25% trypsin (Sigma, Taufkirchen, Germany). Cells were counted in a Neubauer’s chamber and suspended in RPMI-1640 (5 × 105 cells in 1 ml). For tumor cell implantation, rats were anesthetized with a mixture of oxygen (0.5 l/min) and isoflurane (1–1.5 vol.%). For tumor cell inoculation the arterial branches of the right hind leg were dissected. The flow of the femoral artery (FA; ) was temporarily occluded by silk surgical sutures that were placed proximal and distal of the superficial epigastric artery’s (SEA; ) origin. The deep femoral artery (), which normally branches off the FA and supplies the medial and caudal muscles of the thigh, was also ligated. In addition, the SEA was ligated distally, which allowed the opening of this vessel without bleeding. A needle was inserted into the saphenous artery (SA), which is branching off the superficial epigastric artery and 105 MDA-MB-231 cells suspended in 0.2 ml RPMI-1640 were injected (), directed to the descending genicular and popliteal arteries, which supply the knee joint and the muscles of the leg. Resulting bone metastases were observed exclusively in the femur, tibia, and fibula of the respective hind leg. OPN AS was 5′-labeled with FITC and encapsulated in NP based on rhodamine labeled PLGA. Four weeks after tumor inoculation, the naked FITC-labeled AS and AS NP were administered into the superficial epigastric artery (n = 2, ). The AS NP were suspended with PBS to a concentration of 300 mg/ ml and injected 30 mg (in 100 µl PBS) per rat. Saline treated animals were used as control groups (n = 2). Egiht hours, 24 h, 72 h, and 7 days post-injection the rats were sacrificed and tissue (normal and the tumor inoculation site samples) were collected. Naked AS and AS NP biodistribution was evaluated by confocal microscopy of the tissue samples, utilizing appropriate filters for detecting FITC and/or rhodamine.
Figure 1. Anatomy of the right hind leg in the rat depicting tumor inoculation and NP injection. A needle was inserted into the saphenous artery, which is branching off the superficial epigastric artery and MDA-MB-231 cells were injected (upper). By using suturing material (surgical thread) the tumor cells were directed to the descending genicular and popliteal arteries, which supply the knee joint and the muscles of the leg. Four weeks after tumor inoculation, NP were administered into the superficial epigastric artery (bottom).
Quantitative biodistribution of AS NP
Preparation of 33P ATP labeled AS sequence and NP formulation 20 bp long OPN AS sequence (bp. 1343–1362: 5′-CTAACTTAAAAAACAAAAGA -3′) was internally labeled with 33P ATP (CitationBishop et al., 1996). Briefly, for OPN AS radio-labeling two short (10 bp long each) oligonucleotides corresponding to 5′-half (sequence A) and 3′-half (sequence B) of the final product, and third oligonucleotide complementary to oligos A and B (sequence T, 20 bp long) were synthesized (Alpha DNA, Montreal, Quebec, Canada). Oligo B was labeled with 33P ATP (Amersham Corp., UK) and T4 polynucleotide kinase (New England Biolabs, Beverly, MA) using standard methods (CitationSambrook et al., 1989). The labeled sequence was purified using Sep-Pak C18 cartridge (Waters Corp., Milford, MA) according to published procedures (CitationAlefelder et al., 1998). The 33P-labeled oligo B was then mixed with equimolar amounts of oligos A and T in 10mM Tris-Cl, pH 8.0, in eppendorf microcentrifuge tube, heated to 95°C, and cooled slowly to room temperature to allow oligos A and B to anneal to oligo C. Oligos A and B, which hybridized to oligo C, were then ligated by T4 DNA ligase (Promega Corp., Madison, WI) according to the manufacturer instructions using standard buffer conditions (CitationSambrook et al., 1989). The ligated material, corresponding to the desired radiolabeled product, was separated from oligo T and other oligonucleotides electrophoretically using 19% polyacrilamide, 7 M urea gels in TBE buffer (Sigma, St Louis, MO) using standard methods (CitationSambrook et al., 1989). Appropriate size standards were run in adjacent lanes to allow the positive identification of 20-base 33P-labeled final olinucleotide. The labeled material was eluted from the gel using water, desalted using Sep-Pak C18 cartridge, concentrated by lyophilization, resuspended in water, and stored at −20°C until needed. The double emulsion system and the solvent evaporation technique were used to incorporate 33P-labeled OPN sequences in PLGA as described above.
Biodistribution evaluation
Healthy male Sabra rats (Harlan Laboratories), weighing 350-400 g, were used in accordance with the guidelines for animal care of the Hebrew University of Jerusalem and National Institutes of Health (USA). Animals were fed standard laboratory chow and tap water ad libitum. The rats were randomly assigned to treatment or control groups. Naked AS (n = 4) or AS NP (n = 5) were injected into a rat tail vein at average 13.2*106 DPM/rat. Rats injected with saline were used as a control group (n = 2). The rats were kept in metabolic cages and their excrements were collected every day. The animals were euthanized 24 h, 72 h, and 7 days post-injection. One milliliter of blood, 0.5 ml of urine, whole organs, samples of skeletal muscles, and feces were taken from each rat for analysis. The samples were dissolved in 1 ml Solvable per 0.1 g of tissue (Perkin Elmer, The Netherlands), and incubated overnight at 60°C until complete solubilization. The obtained solution was decolorized with 0.2 ml of 35% hydrogen peroxide per 1 ml of solvable until the solution turned to pale yellow. The excess of hydrogen peroxide was inactivated by heating the samples at 60°C overnight. After mixing with 10 ml of Ultima Gold scintillation cocktail (Perkin Elmer, The Netherlands), the radioactivity (DPM) was counted by means of a liquid scintillation analyzer (Packard, Tri-carb 2900TR, USA) against a calibration curve of R2 = 0.986. The radioactivity was normalized to the animal weight and an organ accumulation was expressed as a percentage of total injected dose (mean ± SD).
Results
Formulation properties
AS sequence, targeting OPN was encapsulated in polymeric PLGA-based NP. The resulting formulation was homogenous in size (∼300 nm) and morphology (nanospheres), exhibiting a near neutral surface charge (−2.98 ± 0.52 mV), high AS encapsulation efficiency (51.7 ± 12%), and sustained release profile over 30 days (data not shown). It should be highlighted that the NP confer stability to the encapsulated AS in vivo, and structural integrity and activity of the AS NP is maintained for over 3 years when stored at 4°C in a vacuum desiccator (CitationElazar et al., 2010).
Qualitative AS biodistribution
In this study fluorescent (FITC) OPN AS, naked or encapsulated in fluorescent (rhodamine) NP, was administered to nude rats, previously injected with mammary carcinoma cells. The tissue distribution was monitored using confocal microscopy of tissue samples, collected at different time points. and show representative pictures of naked AS and AS NP accumulation in normal tissues 8 h (), 24 h (), 72 h () and 7 days () post-treatment, and in the metastasis inoculation site at the same time points (). At the beginning of the experiment most of the naked AS accumulated in the spleen and kidneys. Naked sequences also reached the liver, pancreas, and lungs, but to a lower extent. Minor accumulation of naked AS was found in the heart, gastrointestinal tract (GI), and at the inoculation site ( and ). No naked AS was detected in the brain 8 h post-treatment (). At the same time point, levels of AS NP in most tissues were negligible, except for the kidneys and at the metastasis inoculation site (). Levels of naked AS increased considerably in most tissues after 24 h, reaching highest values in the liver and kidneys (). The naked AS signal increased at 24 h also in the pancreas and the GI, but AS accumulation in the brain and the heart was still negligible (). By the time point of 24 h, AS NP accumulated in most tissues (), in the liver, spleen, pancreas, lung, and the heart. Although AS NP were detected in the GI, kidneys, and brain 24 h after the treatment, total accumulation in these organs was negligible (). In contrast, the AS NP level increased markedly at the inoculation site ().
Figure 2. The qualitative biodistribution of OPN AS NP in mammary carcinoma bone metastasis model bearing rats (intact organs). OPN AS was 5′-fluorescent labeled with FITC and encapsulated in fluorescent (rhodamine labeled) NP. Nude rats (350–400 g), implanted with mammary carcinoma cells, were treated with naked sequence or AS NP by IV injection to the rat femoral artery (n = 2). Saline treated animals were used as control groups (n = 2). Naked AS and AS NP biodistribution was evaluated by confocal microscopy of tissue samples collected after 8 h (a), 24 h (b), 72 h (c), and 7 days (d).
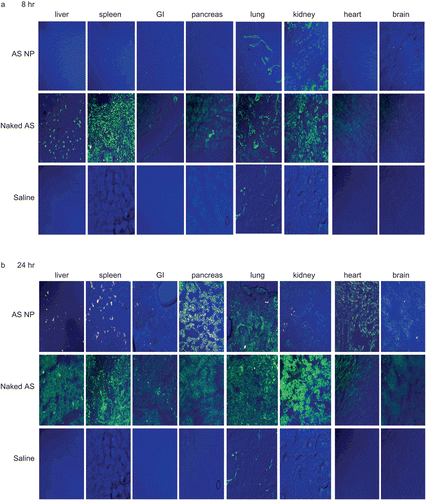
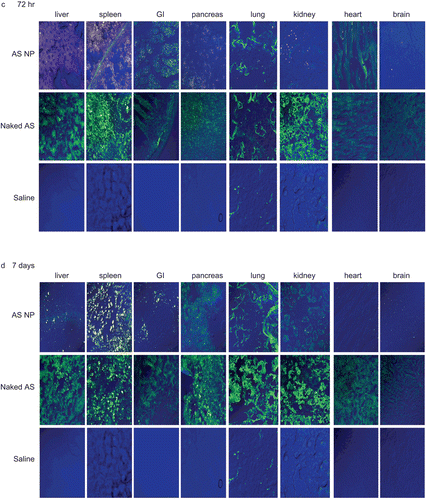
Figure 3. The qualitative biodistribution of OPN AS NP in mammary carcinoma bone metastasis model bearing rats at the metastasis inoculation site. OPN AS was labeled with FITC and encapsulated in fluorescent (rhodamine) NP. Nude rats (350–400 g), implanted with mammary carcinoma cells, were treated with naked sequence or AS NP by IV injection to the rat femoral artery (n = 2). Saline and empty rhodamine labeled NP were used as control groups (n = 2). Naked AS and AS NP distribution in the metastasis inoculation site was evaluated by confocal microscopy of tissue samples collected 8, 24, 72 h, and 7 days after the treatment.
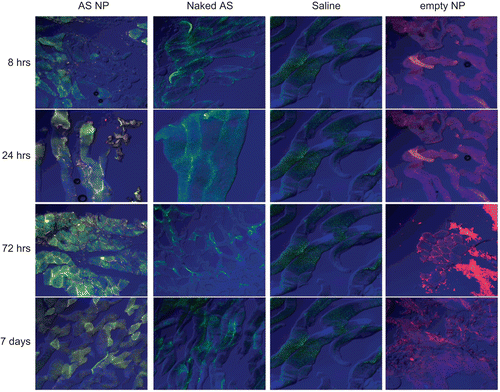
After 72 h, level of naked AS slightly increased in the spleen, with no change in the liver and kidneys, but in other tissues it was decreased considerably (). AS NP accumulated mostly in the spleen and the liver at 72 h, and also increased in the GI (). Marked accumulation of AS NP was found at the inoculation site (). Seven days after the beginning of the experiment naked AS tended to accumulate in excretion organs (). The levels remained almost unchanged in the liver, GI, and spleen, but were increased in the pancreas, lungs, and kidneys. Naked AS uptake in the brain and at the inoculation site remained insignificant during all the study period ( and ). In contrast, most of the AS NP were accumulated in the spleen and at the inoculation site after 7 days, whereas AS NP levels in other tissues were relatively low ( and ). AS NP uptake in the brain and the heart remained negligible at all time points (). AS NP were accumulated gradually at the inoculation site from the beginning of the experiment, reached a maximum at 72 h, and remained almost unchanged after 7 days ().
Quantitative AS biodistribution
Following the above experiment in certain tissues of animals with mammary carcinoma, a quantitative examination of the whole body biodistribution was performed comparing naked and NP encapsulated AS. shows the tissue distribution of naked AS and AS NP 24 h (), 72 h (), and 7 days () post-injection. presents the tissue distribution of naked or encapsulated AS, as a percentage of total body recovery at 24 and 72 h and 7 days.
Figure 4. The quantitative biodistribution of AS NP in whole body organs. Naked (n = 4) or NP (n = 6) encapsulated sequences were internally labeled with 33P ATP and were injected IV into a healthy male Sabra rats (350–400 g) tail vein (average 13.2 × 106 DPM/rat). Rats injected with saline were used as the control group (n = 2). Biodistribution was evaluated 24 h (a), 72 h (b), and 7 days (c) after treatment by beta-counter analysis of blood, organs, and excrement samples. The radioactivity was normalized to the animal weight and organ accumulation was expressed as a percentage of total injected dose (mean ± SD). Bars: blue – AS NPs, red – naked AS.
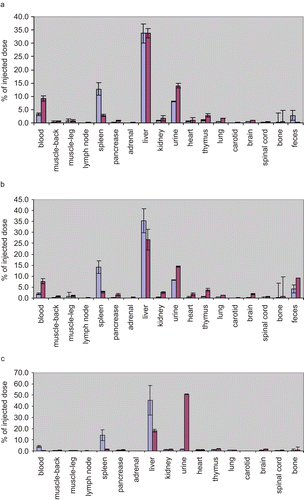
Figure 5. The quantitative biodistribution of AS NP in whole body organs presented as recovered amount (%). Naked (n = 4) or NP (n = 6) encapsulated sequences were internally labeled with 33P ATP and injected IV into a healthy male Sabra rats (350–400 g) tail vein (average 13.2 × 106 DPM/rat). Rats injected with saline were used as control group (n = 2). Biodistribution was evaluated at 24 h, 72 h, and 7 days by beta-counter analysis of blood, organs, and excrement samples. The radioactivity was normalized to the animal weight and organ accumulation was expressed as a percentage of total body recovery (mean ± SD).
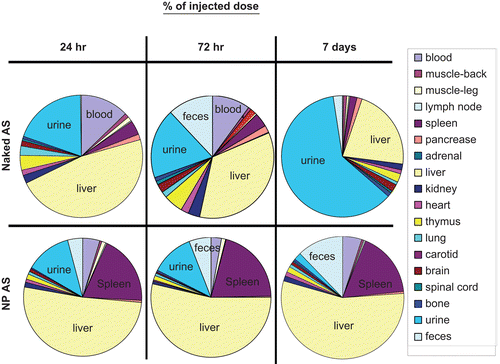
At the beginning of the experiment naked AS was found at higher levels in blood (, and ), in comparison to NP encapsulated sequences (9.3 ± 1.0% and 7.7 ± 1.2% naked AS vs 3.2 ± 0.5% and 1.9 ± 0.4% AS NP after 24 h and 72 h, respectively), but naked AS amount was insignificant (0.3 ± 0.0%) after 7 days, when AS NP level in blood started to increase (4.0 ± 0.9%). Throughout the time-course of the study, AS NP were distributed mainly in the liver and spleen ( and ). The total recovery from these two organs was as much as 46.4 ± 3.0% and 74.2 ± 7.4%, 24 h and 72 h after the treatment, respectively, and slightly decreased to 59.3 ± 8.9% after 7 days. In contrast, naked AS sequences accumulated in the spleen to a much lower extent (2.8% ± 0.4% after both 24 and 72 h, and 1.6% ± 0.1% after 7 days). Although relatively high amounts of naked AS reached the liver during the first half of the study (33.8% ± 1.7% and 26.7% ± 4.8%, after 24 and 72 h, respectively), only 18.1% ± 1.4% were found in this organ after 7 days ( and ). Throughout the study naked AS accumulated to a small extent in the thymus (2.8% ± 0.9%, 3.8% ± 4.8%, and 1.8% ± 0.3% after 24 h, 72 h, and 7 days, respectively). Relatively low levels of AS, both naked and NP encapsulated, were found in the kidneys at all time points, but always the amount of naked AS in the kidneys was higher than that of NP encapsulated sequences (1.7% ± 1.0%, 2.5% ± 0.3%, and 1.4% ± 0.3% naked AS vs 0.8% ± 0.1%, 0.5% ± 0.1%, and 1.2% ± 0.2%, AS NP after 24 h, 72 h, and 7 days, respectively). At all time points of the experiment, the accumulation of NP encapsulated AS was negligible in other harvested organs. Mostly ∼1% or less of the injected dose was found in the skeletal muscles, lymph node, pancreas, adrenal, brain, spinal cord, heart, and lungs. Naked AS was taken up by those tissues to a higher extent than AS NP ( and ).
In an effort to achieve a full mass balance, to evaluate clearance and calculate total recovery, the rats were kept in metabolic cages, so that urine and feces could be collected and analyzed. During the first 72 h both naked and encapsulated AS were cleared mostly in the urine (), but unprotected sequences were cleared faster (14.0% ± 4.2% and 14.5% ± 9.0%, naked AS vs 8.1% ± 3.6% and 8.6% ± 6.5% AS NP, after 24 and 72 h, respectively). After 7 days, most of the naked AS was found in the urine (50.8% ± 2.6%), whereas AS delivered from NP accumulated in the urine to a much lower extent (1.5% ± 1.3%). The amount of naked AS in feces was very low at 24 h (0.2 ± 0.1%), was increased 72 h after the treatment (9.1 ± 6.3%), and decreased after 7 days (2.3 ± 2.2%). In contrast, the levels of AS NP in the feces increased gradually throughout the study, in parallel to a decrease in its urine clearance (2.8 ± 1.9%, 4.3 ± 1.8%, and 9.3 ± 7.7% 24 h, 72 h and, 7 days after treatment, respectively, and ). Total body recovery of both naked and NP encapsulated AS was found to increase with time, beginning with 72.0 ± 6.2% and 65.6 ± 7.4% at 24 h, 77.4 ± 6.9% and 67.9 ± 12.4% at 72 h post-injection, and ending with 83.4 ± 6.0% and 80.7 ± 15.8% recovery after 7 days, for naked AS and AS NP, respectively.
Discussion
One of the major challenges in gene therapy is efficient cellular and tissue delivery, which could be achieved by NP delivery systems (CitationLedley, 1996; CitationMahato et al., 1997; CitationWeyermann et al., 2004; CitationFattal & Barratt, 2009). A polymeric NP formulation was developed (CitationCohen-Sacks et al., 2002) to improve cellular uptake of AS sequences and to provide their protection from degradation at the cellular and organism levels. This formulation was used to research tissues uptake and distribution of AS encapsulated in NP in comparison to naked AS. Body distribution, tissue affinity, cellular uptake, and activity of a formulation are influenced by surface charge (CitationJaaskelainen & Urtti, 2002; CitationBrannon-Peppas & Blanchette, 2004; CitationJuliano et al., 2008). It was shown that negative charge of nucleic acids, including AS oligonucleotides, can significantly decrease cellular and tissue uptake of these molecules, consequently affecting their therapeutic efficiency (CitationJuliano et al., 2009). In our formulation, the strong negative surface charge of AS was diminished by encapsulation in NP, tending to be near neutral, in spite of relatively high AS loading. Our NP formulation released the AS in a sustained manner, thus providing protection of the AS sequences from degradation, to increase their uptake, and to maintain higher AS amounts in the cell cytoplasm during longer periods of time (CitationCohen-Sacks et al., 2002).
The AS NP distribution in tissue and organs was studied in the mammary carcinoma bone metastasis model in addition to a quantitative whole body distribution study in healthy animals. The experiments in healthy rats showed different profiles of naked AS and AS NP distribution in blood and other tissues. Both naked AS and AS NP were injected directly to the blood (IV), but, due to their properties, AS NP were taken up by tissues, reflected by the reduced blood levels already 24 h after treatment, when the amount of naked AS was maximal ( and ). AS NP accumulation in tissues, mostly in the spleen and liver, continued throughout the study, and their amount in blood remained low ( and ). In addition, in mammary carcinoma bone metastasis study high AS NP accumulation was detected in the spleen and gradual accumulation of AS NP was found in the liver and the GI, and at the metastasis inoculation site ( and ).
NP size is a significant determinant of the formulation safety and efficacy (CitationMaeda, 2001; CitationAlexis et al., 2008). NP larger than 100 nm can escape renal clearance following systemic administration. Often NP do not penetrate the tight endothelial junctions of normal blood vessels, but they can extravasate in tumor vasculature and become trapped in the tumor tissue. With time the tumor concentration will build up reaching several folds higher than that of the plasma due to lack of efficient lymphatic drainage in solid tumor (CitationMaeda, 2001; CitationGreish, 2007; CitationAlexis et al., 2008). Moreover, the controlled release delivery mode of PLGA NP offers increased resistance to nuclease degradation, sustained duration of AS administration, and, consequently, prolonged antisense action. The examined formulation was characterized with homogenous morphology (spherical shape) and size distribution at ∼300 nm.
Naked AS, which was found mostly in the blood and the liver after 24 h, decreased gradually in these tissues due to enzymatic degradation of unprotected sequences, and it was almost undetectable in the blood after 7 days, when AS NP levels started to rise due to sustained release of the AS from the NP ( and ). Both in healthy and metastatic animals, AS was found in other tissues and organs, and usually the uptake of naked AS was higher than of AS NP. It is important to note that the extent of AS accumulation reported in these organs (naked AS, as well as AS NP) may slightly over-represent the actual accumulation since the organs were not thoroughly rinsed to remove residual blood. In addition, some levels of FITC-like auto-fluorescence were detected in the lungs, pancreas, smooth and heart muscles, and kidneys, which might give an additional fluorescent signal, higher than that of AS sequences accumulated in these organs (). The higher amount of naked AS in the kidneys, at all time points, might be due to more active urine clearance of the naked AS. Throughout the study naked AS accumulated to a small extent in the thymus and its accumulation profile was similar to that in the spleen, which might reflect the body immune response to AS sequences ( and ). In addition, ASs have been shown to be taken up to some extent by cells via an energy-dependent endocytosis (CitationCohen et al., 2000; CitationCohen-Sacks et al., 2002). Another reason for higher naked AS appearance in intact tissues could be due to degradation of the AS and uptake of its products labeled with radioactive or fluorescent markers (33P-ATP or FITC linked to AS fragments). Internal radioactive AS labeling was shown to reduce cleavage of radiolabeled mononucleotides from the sequence by unspecific enzymatic dephosphorylating activities, and, as a consequence, their amount in tissues, in comparison to terminal AS labeling (CitationSambrook et al., 1989; CitationZendegui et al., 1992; CitationBishop et al., 1996; CitationFrauendorf et al., 2003).
Naked AS and AS NP had different clearance profiles. Throughout the study course naked AS was excreted mainly and rapidly in urine, due to the highly hydrophilic nature of the AS and its degradation products ( and ). Already after 7 days more than half of the naked AS dose injected was cleared in the urine ( and ). In contrast, AS NP were excreted in the urine to a lower extent, and a significant amount was also found in the feces at all time points ( and ). After 7 days, the clearance of AS NP was mostly in feces, whereas only a small AS amount was found in the urine ( and ). The divergent clearance suggests different degradation rate and extent of naked and encapsulated AS. Following the treatment with naked AS, all the sequences can be rapidly degraded and cleared in urine. In AS NP formulation only the released AS is exposed to degradation, but encapsulated AS remains intact and generally can be found in tissues or excreted in the feces. Moreover, most of the encapsulated AS was released inside the cells, because of the rapid cellular uptake of NP, hence it is less exposed to degradation by nucleases existing in the extracellular matrix.
AS amount and location at the mammary carcinoma metastasis inoculation site were evaluated in order to examine the ability of the NP to deliver the AS to the tumor tissue. A relatively low signal of naked AS was detected at the inoculation site, and only between cells (), at all time points. A small increase in naked AS level was detected only after 24 h (), when (according to the whole body distribution data) its levels in blood should be relatively high. Therefore, it is apparent that most of the naked AS amount found at the metastasis inoculation site was inside blood vessels supplying the area, but not inside the tissue. In contrast, AS NP were detected in the inoculation site already 8 h after treatment, and their amount increased gradually throughout the experiment period (). Moreover, the fluorescent signal of rhodamine from the NP was found around and inside the cells starting from the 24 h time point, and FITC-AS delivered by NP was observed inside the cells at all time points ().
It can be concluded that AS NP protect the AS from degradation and provide efficient AS delivery to the tumor tissue. Moreover, administration by the NP delivery system minimizes AS accumulation in intact organs due to the AS sustained release profile, and the favorable NP physicochemical properties.
Declaration of interest
This study was supported in part by the BMBF BioDisc Cooperation (DKFZ-MOS), Grant No. 0315014/G401. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Adwan, H., Bauerle, T.J., Berger, M.R. (2004). Downregulation of osteopontin and bone sialoprotein II is related to reduced colony formation and metastasis formation of MDA-MB-231 human breast cancer cells. Cancer Gene Ther. 11:109–20.
- Agrawal, D., Chen, T., Irby, R., Quackenbush, J., Chambers, A.F., Szabo, M., Cantor, A., Coppola, D., Yeatman, T.J. (2002). Oste-opontin identified as lead marker of colon cancer progression, using pooled sample expression profiling. J Natl Cancer Inst. 94:513–21.
- Alefelder, S., Patel, B.K., Eckstein, F. (1998). Incorporation of terminal phosphorothioates into oligonucleotides. Nucleic Acids Res. 26:4983–8.
- Alexis, F., Pridgen, E., Molnar, L.K., Farokhzad, O.C. (2008). Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 5:505–15.
- Bala, I., Hariharan, S., Kumar, M.N. (2004). PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 21:387–422.
- Bishop, J.S., Guy-Caffey, J.K., Ojwang, J.O., Smith, S.R., Hogan, M.E., Cossum, P.A., Rando, R.F., Chaudhary, N. (1996). Intramolecular G-quartet motifs confer nuclease resistance to a potent anti-HIV oligonucleotide. J Biol Chem. 271:5698–703.
- Brannon-Peppas, L., Blanchette, J.O. (2004). Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 56:1649–59.
- Brigger, I., Dubernet, C., Couvreur, P. (2002). Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 54:631–51.
- Cohen, H., Levy, R.J., Gao, J., Fishbein, I., Kousaev, V., Sosnowski, S., Slomkowski, S., Golomb, G. (2000). Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Ther. 7:1896–905.
- Cohen-Sacks, H., Najajreh, Y., Tchaikovski, V., Gao, G., Elazer, V., Dahan, R., Gati, I., Kanaan, M., Waltenberger, J., Golomb, G. (2002). Novel PDGFbetaR antisense encapsulated in polymeric nanospheres for the treatment of restenosis. Gene Ther. 9:1607–16.
- De Martimprey, H., Vauthier, C., Malvy, C., Couvreur, P. (2009). Polymer nanocarriers for the delivery of small fragments of nucleic acids: oligonucleotides and siRNA. Eur J Pharm Biopharm. 71:490–504.
- Elazar, V., Adwan, H., Baeuerle, T., Rohekar, K., Golomb, G., Berger, M.R. (2010). Sustained delivery and efficacy of polymeric nanoparticles containing osteopontin and bone sialoprotein antisenses in rats with breast cancer bone metastasis. Int J Cancer. 126:1749–60.
- Farokhzad, O.C., Langer, R. (2006). Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 58:1456–9.
- Fattal, E., Barratt, G. (2009). Nanotechnologies and controlled release systems for the delivery of antisense oligonucleotides and small interfering RNA. Br J Pharmacol. 157:179–94.
- Fattal, E., Bochot, A. (2008). State of the art and perspectives for the delivery of antisense oligonucleotides and siRNA by polymeric nanocarriers. Int J Pharm. 364:237–48.
- Frauendorf, C., Hausch, F., Rohl, I., Lichte, A., Vonhoff, S., Klussmann, S. (2003). Internal 32P-labeling of L-deoxyoligonucleotides. Nucleic Acids Res. 31:e34.
- Greish, K. (2007). Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 15:457–64.
- Jaaskelainen, I., Urtti, A. (2002). Cell membranes as barriers for the use of antisense therapeutic agents. Mini Rev Med Chem. 2:307–18.
- Juliano, R., Alam, M.R., Dixit, V., Kang, H. (2008). Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 36:4158–71.
- Juliano, R., Bauman, J., Kang, H., Ming, X. (2009). Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol Pharm. 6:686–95.
- Kang, Y., Siegel, P.M., Shu, W., Drobnjak, M., Kakonen, S.M., Cordon-Cardo, C., Guise, T.A., Massague, J. (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 3:537–49.
- Kurreck, J. (2003). Antisense technologies. Improvement through novel chemical modifications. Eur J Biochem. 270:1628–44.
- Ledley, F.D. (1996). Pharmaceutical approach to somatic gene therapy. Pharm Res. 13:1595–614.
- Li, S.D., Huang, L. (2008). Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 5:496–504.
- Maeda, H. (2001). The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 41:189–207.
- Mahato, R.I., Takakura, Y., Hashida, M. (1997). Development of targeted delivery systems for nucleic acid drugs. J Drug Target. 4:337–57.
- Rosol, T.J. (2000). Pathogenesis of bone metastases: role of tumor-related proteins. J Bone Miner Res. 15:844–50.
- Rudland, P.S., Platt-Higgins, A., El-Tanani, M., De Silva Rudland, S., Barraclough, R., Winstanley, J.H., Howitt, R., West, C.R. (2002). Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 62:3417–27.
- Sambrook, J.E., Fritsch, E.F., Maniatis, T. (1989). Molecular cloning: A laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory.
- Shive, M.S., Anderson, J.M. (1997). Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 28:5–24.
- Smith, A.E. (1995). Viral vectors in gene therapy. Annu Rev Microbiol. 49:807–38.
- Stahel, R.A., Zangemeister-Wittke, U. (2003) Antisense oligonucleotides for cancer therapy-an overview. Lung Cancer. 41(Suppl 1):S81–8.
- Tamm, I. (2006). Antisense therapy in malignant diseases: status quo and quo vadis? Clin Sci. 110:427–42.
- Tamm, I., Wagner, M. (2006). Antisense therapy in clinical oncology: preclinical and clinical experiences. Mol Biotechnol. 33:221–38.
- Tuck, A.B., Chambers, A.F. (2001). The role of osteopontin in breast cancer: clinical and experimental studies. J Mammary Gland Biol Neoplasia. 6:419–29.
- Wang, H., Prasad, G., Buolamwini, J.K., Zhang, R. (2001). Antisense anticancer oligonucleotide therapeutics. Curr Cancer Drug Targets. 1:177–96.
- Weyermann, J., Lochmann, D., Zimmer, A. (2004). Comparison of antisense oligonucleotide drug delivery systems. J Contr Rel. 100:411–23.
- White, P.J., Anastasopoulos, F., Pouton, C.W., Boyd, B.J. (2009). Overcoming biological barriers to in vivo efficacy of antisense oligonucleotides. Expert Rev Mol Med. 11:e10.
- Zendegui, J.G., Vasquez, K.M., Tinsley, J.H., Kessler, D.J., Hogan, M.E. (1992). In vivo stability and kinetics of absorption and disposition of 3′ phosphopropyl amine oligonucleotides. Nucleic Acids Res. 20:307–14.
- Zhang, L., Gu, F.X., Chan, J.M., Wang, A.Z., Langer, R.S., Farokhzad, O.C. (2008). Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 83:761–9.