Abstract
Current research in the field of drug delivery devices, by which pulsatile release is achieved, has been intensified. In this article, an attempt has been made to discuss several types of drug delivery systems that show pulsatile drug delivery characteristics. As found frequently in the living body, many vital functions are regulated by pulsed or transient release of bioactive substances at a specific site and time. Thus it is important to develop new drug delivery devices to achieve pulsed delivery of a certain amount of drugs in order to mimic the function of the living systems, while minimizing undesired side-effects. Pulsatile delivery, which is meant as the liberation of drugs following programmed lag phases, has drawn increasing interest, especially in view of emerging chronotherapeutic approaches. This review article is an attempt to discuss various design strategies, chiefly including reservoir, capsular, and osmotic formulations, and drug delivery systems which cause the pulsed or triggered release of bioactive compounds induced due to certain stimuli like thermal, electrical, and magnetic.
| Abbreviations | ||
| PDDS, | = | Pulsatile drug delivery system; |
| Con A, | = | Concanavalin A; |
| DMAAm, N, | = | N-dimethylacrylamide; |
| EC, | = | Ethyl cellulose; |
| FLR, | = | Florite; |
| GI, | = | Gastro-intestinal; |
| GOD, | = | Glucose oxidase; |
| HA, | = | Hyaluronic acid; |
| HPMC, | = | Hydroxypropyl methylcellulose; |
| ISMN, | = | Isosorbide-5-mononitrate; |
| LCST, | = | Lower critical solution temperature; |
| L-HPC, | = | Low-substituted hydroxypropyl cellulose; |
| MAA, | = | Methacrylic acid; |
| MBAM, | = | Methylene bisacrylamide; |
| MCC, | = | Microcrystalline cellulose; |
| MEMS, | = | Micro-electrochemical system; |
| NPMA, | = | 4-Nitrophenyl methacrylate; |
| PAA, | = | Poly (acrylic acid); |
| PBMA, | = | Poly (butyl methacrylate); |
| PEG, | = | Polyethylene glycol; |
| PEO, | = | Polyethylene oxide; |
| PHEMA, | = | Polyhydroxyl ethyl methacrylate; |
| PIPAAm, | = | Poly N-isopropylacrylamide; |
| PLA, | = | Poly (lactic acid); |
| PLGA, | = | Poly (lactide-co-glycolide; |
| PMMA, | = | Poly (methacrylic acid); |
| PST, | = | Polystyrene; |
| PVA, | = | Polyvinyl alcohol; |
| PVP, | = | Polyvinyl pyrrolidone; |
| RES, | = | Reticuloendothelial system; |
| SIF, | = | Simulated intestinal fluid; |
| TES, | = | Time controlled explosion system. |
Introduction
‘Chronopharmaceutics’ consist of two words; chronobiology and pharmaceutics. Chronobiology is the study of biological rhythms and their mechanism. There are three types of mechanical rhythms in our body; namely, Circadian rhythm word originates from Latin word ‘circa’ means about and ‘dies’ means day, Ultradian rhythm means oscillation of shorter duration (more than one cycle per 24 h) and Infradian rhythm having oscillations longer than 24 h (less than one cycle per day). Circadian rhythm regulates many body functions in humans like metabolism, physiological behavior, sleep pattern, hormone production, etc.
PDDS is defined as the rapid and transient release of certain amount of drug molecules within a short time period immediately after a pre-determined off-release period, i.e. lag time (CitationKikuchi & Okano, 2002) ().
Figure 1. Drug release profiles from PDDS.
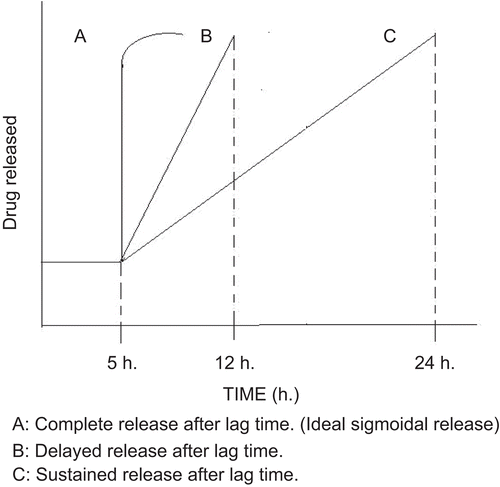
This novel drug delivery system has been attempted for the following reasons:
| i. | Chronopharmacotherapy of diseases which shows circadian rhythms in their pathophysiology. | ||||
| ii. | Avoiding degradation in upper gastrointestinal tract, e.g. proteins and peptides (CitationRubinstein et al., 1995). | ||||
| iii. | For time-programmed administration of hormones and many drugs such as isosorbide dinitrate, respectively, to avoid suppression of hormones in the body that can be hampered by constant release of hormone from administered dosage form and development of resistance. | ||||
| iv. | For drugs which develop biological tolerance, for the drug with extensive first pass metabolism, for drug targeted to specific site in the intestinal tract, e.g. colon. | ||||
PDDS is needed and found to be very useful in the treatment of diseases which shows circadian rhythms in their pathophysiology and the following table enlists all those diseases.
Advantages and disadvantage of PDDS
PDDS has unique advantages over other drug delivery systems as it does not release the drug before the desired lag time, resulting in less inter- and intra-subject variability, and also avoiding the risk of dose dumping, it releases drug to the right site at the right time and hence improves the bioavailability and stability, reduced adverse effects, and thus improve patient comfort and compliance. Although advantages of PDDS are many, its only disadvantage is that the rupture time cannot be always adequately manipulated as it is strongly correlated with the physicochemical properties of the polymer.
Classification of PDDS
This review article aimed at collating and understanding novelty and feasibility of different types of PDDS and upcoming technologies which are being exploited on an industrial scale. In this article, several approaches for the delivery of drugs in a pulsatile manner, mainly using time-controlled, stimuli-induced, externally-regulated systems () is reviewed. The following section deals in detail with all the above-mentioned approaches to develop PDDS.
Figure 2. Classification of PDDS.
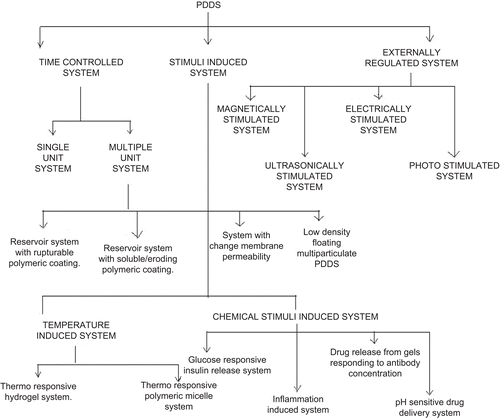
Time controlled system
As is often seen in human beings, hormone release is regulated with a certain rhythm, depending on the hormone type, in the order of several hours, days, or months. Similarly, the action of a certain drug should coincide with the proper site and time for optimal effect. Therefore, the development of a time controlled release system is desired for the treatment of patients. They are further classified into:
Single unit system
A single unit system mainly includes capsule-shaped and advanced osmotic devices. The capsule has the capability of delivering therapeutic agents into the body in a time- or position-controlled pulsatile release fashion. A swellable hydrogel plug was used to seal the drug contents into the capsule body. When this capsule comes in contact with the dissolution fluid, it swelled and, after a lag time, the plug pushed itself outside the capsule and rapidly releases the drug (CitationNeill et al., 1993). The lag time is controlled by a plug which is pushed away by swelling/erosion and the drug is released as a ‘pulse’ from the insoluble capsule body. Polymers used for designing this hydrogel plug are various viscosity grades of HPMC, polyvinyl alcohol, PVA, and PEO.
CitationBussemer et al. (2003a) developed a PDDS based on hard gelatin capsules coated with the rupturable polymer. The objective of their study was to develop and evaluate a PDDS based on drug contained hard gelatin capsules coated first with a swelling layer and then with an outer insoluble, water-permeable polymeric coating. An inner pressure developed by the swelling layer results in the rupture of the outer coating. Preliminary studies with a simulated rupture test demonstrated that the lag time prior to rupture was observed to depend on the properties of the coating such as its water permeability and mechanical strength. The result of their study revealed that the lag time increased with a higher coating level, but decreased with the addition of the hydrophilic pore former, HPMC. The coated capsules took up release medium at a nearly constant rate until a critical maximum was reached, where the swelling pressure was sufficient to rupture the outer coating. The rate of medium uptake decreased with increasing coating level while the extent of medium uptake was almost the same for the different coating levels. CitationBussemer et al. (2003a) found that the test conditions, such as surfactant addition to the release medium or floating vs complete immersion of capsules in the medium, affected the lag time.
CitationLi et al. (2008) developed a novel system for three-pulse drug release based on ‘tablets in capsule’ device. The objective of their study was to obtain programmed drug delivery from a novel system, which contained a water-soluble cap, impermeable capsule body, and two multilayered tablets, and it was seen that the types of materials for the modulating barrier and its weight can significantly affect the lag time. They selected sodium alginate and HPMC E5 as the modulating barrier material candidate. Through adjusting ratio of sodium alginate/lactose, lag time was controllable between the first two pulsatile releases, and the linear relationship was observed between the ratio and the lag time, whereas lag time between the second and the third pulse can be successfully modulated by adjusting the ratio of HPMC E5/lactose. In further studies, CitationLi et al. (2008) improved the drug release rate of the second pulsatile dose by adding a separating layer between the second and third layer and the modulating barrier layer in the three layered tablet. To evaluate the contribution of bulking agent to drug release rate they tried lactose, sodium chloride, and effervescent blend, and observed no superiority using sodium chloride and effervescent blend. However, lactose favored it. CitationLi et al. (2008) concluded from the outcome of their research that programmed delivery of the drug can be obtained from the tablets in the capsule system by systemic formulation approach to achieve pulsatile release three times daily.
CitationNiwa et al. (1995) prepared a novel capsule made from EC for the time-controlled release of drugs in the colon. The EC capsules were prepared by coating the gelatin capsule with EC. The thickness of the capsule body was varied by varying the coating of EC and the effect of wall thickness on the release of drugs was investigated. Mechanically large numbers of micropores (400 µm) were made at the bottom EC capsules. A swellable layer, consisting of L-HPC, was located in the bottom of the capsule body. Above the swellable layer there was the drug reservoir, composed of a mixture of the model drug, fluorescein, and a bulking agent, such as lactose or starch. The capsule was capped and sealed with a concentrated EC solution. After administration of drug containing capsule, water molecules penetrated the capsule through the micropores at the bottom of the capsule body. Hydration and swelling of the HPMC induce an increase in the internal osmotic pressure resulting in the ‘explosion’ of the capsule, and a burst-like drug release was then observed.
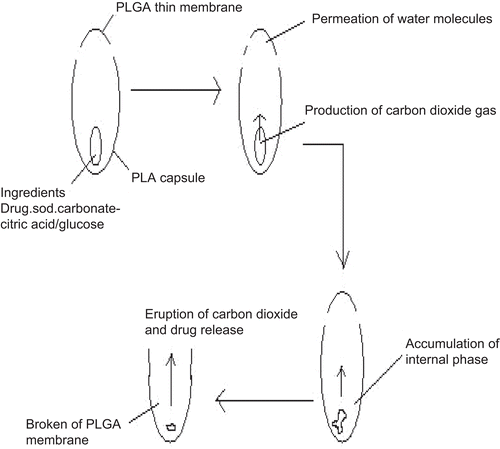
A similar approach for the pulsatile release of drugs was reported by CitationJimoh et al. (1995). In this study they utilized hollow biodegradable capsules with a thinner membrane at one end to control the lag time of PDDS. A schematic representation of the capsule formulation is shown in . In PLA capsule, the effervescent agents like citric acid/sodium bicarbonate were entrapped. As water penetrated into the capsule through the thin PLGA membrane side, an effervescent reaction is generated. Generated carbon dioxide gas accumulates in the capsule which ultimately ruptures the thin membrane. CitationJimoh et al. (1995) reported that the burst time, i.e. lag time of drug release, can be modulated by varying the dimensions (thickness of the membrane and size) of the capsule and the amount of the effervescent agents.
Figure 3. Scheme of the pulsed release biodegradable capsules.
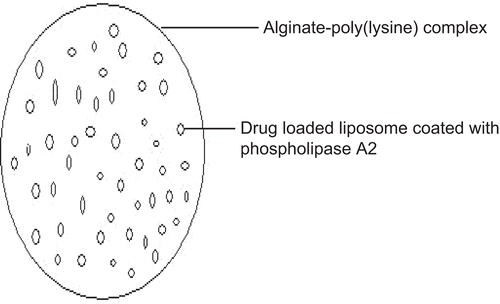
Liposomes though have been used as drug delivery carriers due to their structural similarity to cell membranes; still their uptake by the RES or a destabilization due to the adsorption of plasma proteins on the lipid bilayer have been major drawbacks that have limited the application of these formulations. To overcome these drawbacks of liposomes, CitationLanger et al. (1990) incorporated drug loaded liposomes into microcapsules of alginate hydrogels. The hydrogel matrix was designed to protect the liposomes from degradation and/or dispersion in vivo as well as possibly to regulate the release rate of the incorporated drug molecules. To achieve a pulsatile release of drug molecules the liposomes inside the microcapsules were coated with phospholipase A2 (). Phospholipase A2 was shown to gather at the water/liposome interfaces and remove an acyl group from the phospholipid in the liposome, resulting in the destabilization of the liposome. Destabilized liposomes thus release the drug molecules from the interior allowing release to be regulated by the rate determining microcapsule membrane.
Figure 4. Schematic representation of liposome-loaded alginate–poly (L-lysine). microcapsules.
CitationRahemba et al. (2009)developed an osmotic controlled PDDS for targeted drug delivery. The tablet core contained a high drug load in addition to several osmotic agents, swellable polymers, and the surrounding osmotic coating consists of a semi-permeable membrane that has been scored with a razor blade in several key locations. The components in the tablet core attract water into the core leading to its swelling, and propagate the scores in the coating along the length of the tablet. The coating bursts open after the scores have been fully propagated and release the tablet core contents into the surrounding media. In this study they investigated variables like configuration of the scores in the coating, the length of the scores, and the distance between the scores. They proved that the delivery system developed in this work was able to generate a reproducible dissolution profile consisting of a specific targeted lag time between 5 min and 2 h followed by immediate release of the drug from the core. The performance of the system was validated in vitro using salicylic acid as a drug. The outcome of the study revealed that the developed system was better than several other pulsatile systems as it was able to accommodate higher drug loading levels, has demonstrated more reproducible burst time, and was easier to manufacture.
In general, we can say that there are various factors that affect the degree and manner in which the pulsatile effect can be controlled like the swelling/erosion mechanism, water-permeable or enzyme-protective coating, the choice of elastic material, the thickness of the wall section, the configuration, location of the orifice, the viscosity, and the surface tension of the beneficial formulation agents like osmogens, bulking agents, effervescent agents, and surfactant.
Multiple unit/multi-particulate system
The purpose of designing a multi-particulate dosage form is to develop a reliable formulation that has all the advantages of a single unit formulation and yet is devoid of the danger of alteration in drug release profile and formulation behavior due to unit-to-unit variation. However, the multi-particulate PDDS has some drawbacks like low drug loading, proportionally higher need for excipients, lack of manufacturing reproducibility and efficacy, a large number of process variables, multiple formulation steps, higher cost of production, and need of advanced technology. The expected drug release mechanism and corresponding target bimodal plasma concentration profile of the above designed multi-particulate pulsatile system is depicted in . Details about the recent innovations made in multi-particulate PDDS have been given in the following sections.
Figure 5. Hypothetical design and plasma drug profile of a multi-particulate PDDS. (a) Design of a pellet with multiple coatings, (b) Predicted bi-modal plasma concentration profile.
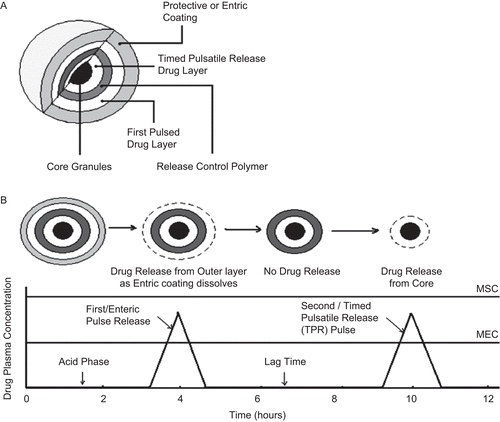
Reservoir system with rupturable polymeric coating.
Most multi-particulate PDDS are reservoir type devices coated with a rupturable polymeric layer. Upon water ingress, drug is released from the core after rupturing of the surrounding polymer layer due to pressure build-up within the system. The pressure necessary to rupture the coating can be achieved with swelling agents, gas-producing effervescent excipients, or osmotic agent. Water permeation rate and mechanical resistance of the outer membrane are the key factors that decide the lag time.
CitationUeda et al. (1989; Citation1994a; Citationb; Citationc) reported a TES, where drug was released neither by diffusion control nor by dissolution control, but by a quite novel mechanism, i.e. explosion of the outer membrane. This mechanism is especially useful with water insoluble drugs in which those prior art delay mechanisms related to diffusion of the drug through a permeable coating would not be effective. TES were developed for both single and multiple unit dosage forms. In both cases, a core contains drug plus an inert osmotic agent and suitable disintegrants. Individual units can be coated by a protective layer and then by a semi-permeable layer which is the rate controlling membrane for the influx of water into the osmotic core. The core ultimately explodes due to build up of the osmotic pressure by water ingress leading to immediate release of the drug. The explosion of the formulation can also be achieved through use of swelling agents and different drug release patterns can be achieved with the change of the form of TES. It was observed that, in tablets, drug was released quickly after the explosion of the outer membrane while in the case of TES in the form of beads or granules; drug was released with zero order pattern after a definite lag time because of the time variance of the explosion of the outer membrane in each bead or granule. Drug release was time-controlled due to the rupturing of the external water-insoluble membrane caused by an explosive swelling effect of swelling agents. It can be concluded from the outcome of the research carried out by Ueda et al., that the lag time increases with increasing coating level and with the higher amounts of talc or lipophilic plasticizer in the coating.
CitationChen (1993a; Citationb; Citation1995) proposed two approaches to overcome the drawbacks associated with TES and to have better control over the drug release pattern. First, the incorporation of water-soluble polymer in insoluble polymeric membrane of TES. This water-soluble polymer is of the enteric coating polymer type in which the polymer becomes soluble only at pH values above certain specific values which prevents the polymer dissolution in the stomach. As soon as the pellet reaches the elevated pH of the intestine, the polymer begins to dissolve and weaken the membrane coating. The explosion of the weakened membrane can be assured after a pre-determined time of exposure to the intestinal environment by varying the proportion of soluble and insoluble material in the coating as well as the coating thickness, the time delay before explosion can be prolonged with better control and reliability, with eventual disintegration of the coating ensuring release of the drug. However, the disadvantage of a water-soluble polymer to regulate time release is that once the portions of water-soluble polymers are dissolved, an active agent may start leaching out, leading to premature drug release and inconsistent drug efficacy. Second, an increase in the lag time of the TES systems by incorporating water impermeable materials in the coating. By reducing the rate of influx of water into the interior of the pellet containing the swelling agent, the rate of swelling can be reduced and the time to explosion can be prolonged and controlled.
CitationHartman et al. (2000) developed a multi-particulate PDDS with release properties dependent on the swelling of a UV cross-linked coating and described drug release properties of the developed systems based on a mathematical model. The core consists of MCC and milled sodium chloride coated with a copolymer of methacrylic acid and ethyl acrylate containing trifunctional acrylic monomer pentaerythritol triacrylate as a cross-linking agent and a photo-initiator, 2,2-dimethoxy-2-phenylacetophenone. Upon water ingress, the coating swells in such a way that the diffusion coefficient of water increases, making it permeable to the dissolved components in the core. However, the mixed nature of the polymers used in this work resulted in a decrease in the diffusion coefficient initially with an increase in water activity (below 0.4). Thus, CitationHartman et al. (2000), from the outcome of this study, reported that the lag time can be controlled by amount of cross-linking, coating thickness, and duration of the UV cross-linking time. The mathematical model developed in this investigation gives good estimation of the lag-time.
CitationGuo et al. (2003) developed diclofenac sodium pulsatile release pellets by extrusion-spheronization technology. The drug-loaded pellets were coated with a swelling material as the inner coating swelling layer and EC aqueous dispersion as the outer coating controlled layer in a mini-fluidized bed spray-coater. The delayed release time and release rate were influenced by the swelling material, the coating level of the inner swelling layer, and the outer controlled layer. Pharmacokinetic and bioavailability studies in eight human subjects revealed that the lag time for pulsed delivery of diclofenac in vitro and in vivo is in good agreement.
CitationBussemer et al. (2003b) studied the swelling characteristics of various swellable polymers in swelling layers that induce rupturing of an outer polymer coating in PDDS. An apparatus was designed to measure the swelling energy/force and water uptake of discs, made of polymers simultaneously. The swelling energy of several excipients was found to decrease in the following order: croscarmellose sodium (AcDiSol®) > L-HPC > sodium starch glycolate (Explotab) > crosspovidone (Kollidon CL) > HPMC (Methocel K100 M).
CitationDashevsky and Mohamad (2006) developed a rupturable multi-particulate PDDS consisting of a drug core; a swelling layer, comprising of a super-disintegrant and a binder; and an insoluble, water-permeable polymeric coating Aquacoat® ECD. Lag time was shorter for theophylline layered on sugar cores, compared to cores of 100% theophylline. A fast and complete release after lag time was achieved with cross-linked carboxymethyl cellulose (AcDiSol®), in comparison to a sustained release pattern with L-HPC and sodium starch glycolate (Explotab®) as swelling agents. Lag time was controlled by the coating level of the outer membrane which was brittle and ruptured sufficiently to ensure fast drug release. The brittleness of membrane was increased with the addition of talc. CitationDashevsky and Mohamad (2006) concluded from the outcome of their study that upon water ingress the swellable layer expands which results in the rupturing of the outer membrane with subsequent rapid drug release.
Reservoir systems with soluble or eroding polymer coatings.
Another class of reservoir type multi-particulate PDDS is based on soluble/erodible layers in place of rupturable coatings. The barrier dissolves or erodes after a specific lag time followed by burst release of drug from the reservoir core. In general, for this kind of system the lag time prior to drug release can be controlled by the thickness of the coating layer.
A pH sensitive multi-particulate system comprised of Eudragit S100 coated pellets was designed for chronotherapeutic delivery of diltiazem hydrochloride for treating angina pectoris. The drug-loaded pellets were produced by an aqueous extrusion spheronization technique using MCC as a spheronizing aid and PVP K-30 as a binder. Different coat weights of Eudragit S100 were applied to the drug-loaded pellets to produce the pH sensitive multi-particulate system. In vitro dissolution studies of the coated pellets performed following the pH progression method showed that the drug release was dependent on the coat weights applied and pH of the dissolution media (CitationShivakumar et al., 2006).
CitationDittigen et al. (2000) developed a formulation comprising four compressed compositions filled in a capsule, each with different coating compositions and soluble at different pH throughout the GIT. This system was developed with the aim of providing varying release profiles of effective ingredients or their combination. Most of the time-controlled systems or delayed-release systems suffer from the disadvantage that they are not suitable for delivering weakly basic drugs as they are insoluble at intestinal pH. To overcome this problem of pH-dependent solubility of weakly basic drugs, pH adjusters, which can provide local acidic environment within the system, can be admixtured. This type of system where a capsule containing a multitude of multi-coated particulates is expected to deliver therapeutic agents into the body in a time-controlled pulsatile release fashion. One of the coating membrane barriers is an enteric polymer and the second membrane barrier is a mixture of a water-insoluble polymer and an enteric polymer. An organic acid, such as fumaric acid, citric acid, succinic acid, tartaric acid, or malic acid, or a maleic-acid-containing membrane may be provided between the first and second membrane layers to provide for the time-separated pulses. The acids in between the membranes may delay the dissolution of the enteric polymer in the inner layer, thereby increasing the lag time as well as decreasing the rate of release of the active ingredient from the coated micro-particulates.
CitationLiu et al. (2009) designed and evaluated pH-independent pulsatile release pellets containing Isosorbide-5-mononitrate (ISMN). The process of the heart disease such as angina has a close relationship to the chronobiology which gives rise to the need of a PDDS for the anti-anginal drug. In this study pellets containing ISMN were first prepared as the core and then layered with a swelling layer followed by a water-insoluble control layer. The core pellets formulated with MCC and lactose were prepared by extrusion-spheronization. The preparation was optimized by Box-Behnken experimental design, when taking the MCC-lactose ratio as well as the operating conditions of extrusion-spheronization as variables. The core pellets were coated by a fluidized bed coater and pellets with various coating types and coating levels were studied by in vitro dissolution tests. They also studied effects of both swelling layer and control layer on the lag time and the drug release time in order to pre-determine the lag time and release time, and the pellets were evaluated in vivo by studying the pharmacokinetics after oral administration in beagle dogs. The pellets achieved a lag time of 4.1 h in vivo and had a good consistency with the in vitro results.
CitationKrogel and Bodmeier (1999) developed and evaluated floating PDDS based on a reservoir system consisting of a drug-containing effervescent core and a polymeric coating. For the floating system, a polymer coating with a high elongation value and high water and low CO2 permeabilities was selected (Eudragit RL/acetyltributyl citrate 20%, w/w) in order to initiate the effervescent reaction and the floating process rapidly while, for the PDDS, a weak semi-permeable film was selected, which ruptured after a certain lag time. With the floating system, the polymeric coating did not retard the drug release. A polymer (cellulose acetate or HPMC) was added to the core to control the drug release. The time to flotation could be controlled by the composition (type of filler, concentration of effervescent agents), hardness of the tablet core, the composition (type of polymer and plasticizer), and thickness of the coating. For the PDDS a quick releasing core was formulated in order to obtain a rapid drug release after the rupture of the polymer coating. It was clear from the outcome of their study that the lag time prior to the rapid drug release phase increased with increasing core hardness and coating level.
Systems with change membrane permeability.
Sigmoidal release pattern is therapeutically beneficial for timed release and colon-specific drug delivery and is observed in coated systems. A sigmoidal release pattern is reported based on the permeability and water uptake of Eudragit RS or RL, influenced by the presence of different counter ions in the release medium (CitationBodmeier et al., 1996). Based on this concept, CitationNarisawa et al. (1993; Citation1996) developed a device capable of giving pulse release depending on the change in diffusion properties of Eudragit RS. They found that a theophylline core coated with Eudragit RS showed very slow release rates in pure water which increases significantly when the microcapsules were immersed in an organic acid solution containing succinic, acetic, glutaric, tartaric, malic, or citric acid. They opined that this could be because of higher hydration of the film containing quaternary ammonium groups on interaction with the acids. The drug release rate from the beads coated with Eudragit NE 30D, which has no quaternary ammonium groups in the polymer chain, was not affected by succinic acid, suggesting that the quaternary ammonium groups of Eudragit RS are essential to produce the unique drug release profile. When succinic acid was incorporated into the core of Eudragit RS-coated theophylline beads, the drug release profile showed a typical sigmoidal pattern.
In another similar system, theophylline and sodium acetate, acting as the permeability modifying salt, were layered on sugar pellets followed by coating with Eudragit RS. The lag time increased with increasing thickness of the outer membrane. However, the slope of the drug release phase was independent of the thickness but was influenced by the amount of salt in the system, proving that the release mechanism is dependent on the amount of salt or the permeability modifier (CitationBeckert et al., 1999).
Low density floating multi-particulate pulsatile systems.
Conventional multi-particulate PDDS mentioned above shows longer residence time in the GI tract and may result in in vivo variability and bioavailability problems due to the highly variable nature of the gastric emptying process. In contrast, low density floating multi-particulate PDDS reside specifically in the stomach and are not affected by variability of pH, local environment, or the gastric emptying rate. These dosage forms are also specifically advantageous for drugs either absorbed from the stomach or requiring local delivery in the stomach. Overall, these considerations led to the development of multi-particulate floating PDDS possessing maximum gastric retention capabilities.
CitationSharma and Pawar (2006) developed a multi-particulate floating PDDS of meloxicam using porous calcium silicate (Florite RE) and sodium alginate. Meloxicam was adsorbed on the Florite RE (FLR) by fast evaporation of solvent from a drug solution containing dispersed FLR. Drug adsorbed FLR powder was then used to prepare calcium alginate beads by ionotropic gelation method using 32 factorial designs. The floating time for this system was controlled by density of the beads and hydrophobic character of drug. It was found that all formulations showed a lag period ranging from 1.9–7.8 h in acidic medium followed by rapid release of meloxicam in SIF USP, without enzymes. Complete drug release in SIF occurred in less than 1 h from all nine formulations. The size of beads varied from 2.0–2.7 mm for different batches. Floating time was controlled by density of beads and hydrophobic character of the drug. A pulsatile release of meloxicam was demonstrated by a simple drug delivery system which could be useful in chronopharmacotherapy of rheumatoid arthritis.
CitationBadve et al. (2007) developed hollow calcium pectinate beads for floating-pulsatile release of diclofenac sodium intended for chronopharmacotherapy of rheumatoid arthritis. To overcome limitations of various approaches for imparting buoyancy, hollow/porous calcium pectinate beads were prepared by a simple process of acid-base reaction during ionotropic cross-linking. The floating beads provided two phase release patterns with an initial lag time during floating in acidic medium followed by rapid pulse release in phosphate buffer. This approach suggested the use of hollow calcium pectinate microparticles as promising floating PDDS for site- and time-specific release of drugs. Further, combined floating and pulsatile principles were achieved using a specific technology, in which low density microporous polypropylene, Accurel MP® 1000 were used as a multi-particulate carrier for ibuprofen (CitationSher et al. 2007). Ibuprofen was adsorbed on the polymer by solvent evaporation technique; a single step method resulted in different porous particles. This drug delivery system showed distinct behavior from other approaches in chronotherapy, with desired low drug release in acidic medium, reduced process time consumption due to a single step process, and it even overcame the limitations of process variables caused by multiple formulation steps.
The major mechanism by which the drug is released from pellets depends on the type of coating, insoluble coating under all physiological conditions, pH-dependent coating whose solubility changes dramatically at some point in GI tract, and slowly erodible coating. Water-soluble drugs are found to be released mainly by diffusion, while for water-insoluble drugs the release is dependent on dissolution of drug.
Stimuli induced systems
Several polymeric delivery systems undergo phase transitions and demonstrate marked swelling–de-swelling changes in response to environmental changes including solvent composition, ionic strength, and temperature (CitationLee & Chen, 2000). Responsive drug release from those systems results from the stimuli-induced changes in the gels or in the micelles, which may de-swell, swell, or erode in response to the respective stimuli. They further classified into:
Temperature-induced system
Temperature is the most widely utilized triggering signal for a variety of triggered or PDDS. The use of temperature as a signal has been justified by the fact that the body temperature often deviates from the physiological temperature (37°C) in the presence of pathogens or pyrogens. This deviation sometimes can be a useful stimulus to activate the release of therapeutic agents from various temperature-responsive drug delivery systems.
Thermoresponsive hydrogel system.
Thermoresponsive hydrogel have been investigated as possible drug delivery carriers for stimuli responsive drug delivery systems (CitationOkano et al., 1990; Citation1994). PIPAAm cross-linked gels have shown thermoresponsive, discontinuous swelling/de-swelling phases, i.e. swelling at temperatures below 32°C, while shrinking above this temperature. A sudden temperature increase above the transition temperature of these gels results in the formation of a dense, shrunken layer on the gel surface ‘skin layer’, which hinders the water permeation from inside the gel into the environment. Drug release from the PIPAAm hydrogels at temperatures below 32°C was governed by diffusion, while above this temperature drug release stops completely due to the ‘skin layer’ formation on the gel surface (on–off drug release regulation). Swelling–de-swelling kinetics of conventional cross-linked hydrogels is normally reciprocal of the square of gel dimension (CitationBae et al., 1991).
A new method reported to accelerate gel swelling/de-swelling kinetics is based on the molecular design of the gel structure. Free mobile linear PIPAAm chains were grafted within the cross-linked PIPAAm hydrogels (CitationKishi et al., 1997; CitationChen et al., 1999), as shown in . These novel graft-type PIPAAm gels had the same transition temperature as the conventional cross-linked PIPAAm gels and existed in the swollen state below the transition temperature, while above this temperature they shrink. The molecular weight of the graft chains had a significant effect on the gel de-swelling kinetics and also on the drug release profiles, especially for large molecular weight drugs. A dense skin layer is formed on the conventional PIPAAm gels upon temperature change above the transition temperature, which limit the complete shrinkage of the gel. In contrast, the PIPAAm-grafted gels show the rapid de-swelling kinetics without the formation of a skin layer on the gel surface. Probably this could be because of the rapid dehydration of the graft chains formed by hydrophobic aggregation on the three-dimensional cross-linked gel chains. The hydrophobic cores might accelerate the entire dehydration and shrinking processes of the gel. Cylinder-shaped gels showed fast and large changes in length when subjected to repetitive temperature cycles between 20–40°C. These results proved the feasibility of the concept that the introduction of freely mobile, linear PIPAAm chains into PIPAAm cross-linked gels is effective for fast gel de-swelling kinetics (CitationYoshida et al., 1995).
Figure 6. Model structure of freely mobile linear PIPAAm-grafted PIPAAm hydrogels.
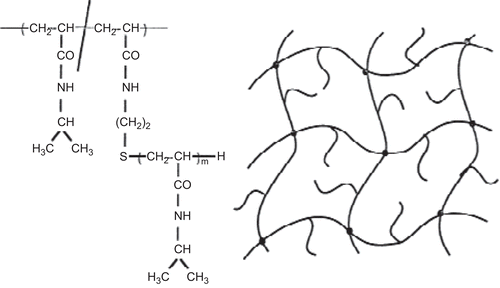
A similarly rapid de-swelling phase was achieved by incorporating PEG graft chains into PIPAAm cross-linked hydrogels (CitationKaneko et al., 1995; Citation1996; Citation1998). The introduction of PEG chains did not alter the transition temperature as it was seen in the gels with hydrophilic co-monomers, such as acrylamide and acrylic acid, which may be due to the structural independence of the PEG chains from the cross-linked PIPAAm main chains. In this case, however, the de-swelling mechanism is different from PIPAAm graft-type hydrogels. During the shrinking process, the graft PEG chains form hydrophilic channels for water molecules, most likely due to a phase separation within the shrinking gels leading to a rapid de-swelling. The majority of the drugs in the gels were released through the PEG formed channels with water molecules. As mentioned above, accelerated de-swelling of cross-linked hydrogels is achieved by the introduction of graft chains independent from gel main chains. As these gels can be activated by external stimuli they are utilized for drug release at targeted areas.
Thermoresponsive polymeric micelle system.
Several polymeric delivery systems undergo phase transitions and demonstrate marked swelling–de-swelling changes in response to change in temperature. Responsive drug release from those systems results from the stimuli-induced changes in the gels or in the micelles, which may de-swell, swell, or erode in response to the respective stimuli.
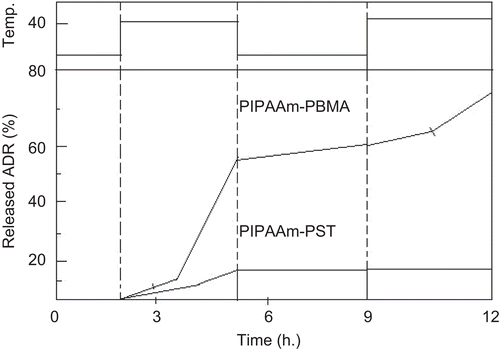
CitationKataoka et al. (2001) comprehensively reviewed the properties and biological interests of polymeric micelles to make them a most noteworthy candidate as a drug carrier for the treatment of cancer. The polymeric micelle is composed of amphiphilic block copolymers exhibiting a hydrophobic core with a hydrophilic corona. There is use of an end-functionalized PIPAAm to prepare block copolymers with hydrophobic polymers, such as PBMA, PST, or PLA (CitationKohori et al., 1999). Block copolymers formed a micellar structure (with core–shell structure) in aqueous solution below PIPAAm’s transition temperature (). The shell was made from thermoresponsive PIPAAm, while the core consisted of hydrophobic polymer aggregates. The PIPAAm corona showed an alteration in its hydration/dehydration properties with changing temperature. The hydrated corona acts as an inert material toward all biological entities, such as proteins and cells below the PIPAAm’s LCST. However, upon temperature increase above 32°C due to the dehydration of polymer chains hydrated PIPAAm chains become hydrophobic resulting in aggregation and precipitation. This core acts as a reservoir for hydrophobic drug molecules. The hydrophobic anti-cancer drug, adriamycin, was incorporated into the either PBMA or PST micelle cores via dialysis. The loaded micelles were then evaluated with regard to their thermoresponsive drug release profiles (CitationChung et al., 1999; Citation2000). The micelle formation was confirmed by dynamic light scattering measurements and LCST changes. When PIPAAm–PST was used for micelle formation, the PST core was relatively stable against temperature changes. It was observed that the drug release was suppressed at all temperatures examined. In contrast, PIPAAm–PBMA micelles showed different thermosensitive drug release profiles with temperature. At temperatures below PIPAAm’s LCST, drug release was at a minimum, with a value less than 10%. However, upon temperature increase above the PIPAAm’s LCST, accelerated release of adriamycin was observed. The application of a temperature gradient induced an on–off drug release regulation from PIPAAm–PBMA micelles between 4–37°C ().
Figure 7. Structure of block copolymer micelles.
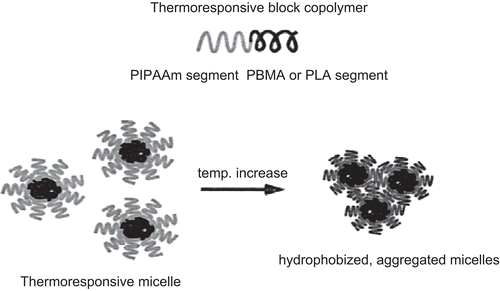
Figure 8. On–off release regulation of adriamycin from PIPAAm–PBMA micelles and PIPAAm–PST micelles in response to temperature switching at 4 and 40°C.
The thermoresponsive polymeric micelles were then added to endothelial cell culture media and the effect of thermoresponsive drug release on the cell survival rate was investigated. Addition of 0.1 mg/mL free adriamycin induced a 10% cell death rate during the experimental period, while almost all cells treated with adriamycin-containing polymeric micelles were viable at 29°C. This indicated that the PIPAAm micelles displayed no cytotoxicity and no drug release from the micelle core was evident below PIPAAm’s LCST. By increasing culture temperature to 37°C, however, PIPAAm dehydration and precipitation was induced, which destabilized the PBMA core structure. An enhanced interaction of hydrophobized micelles with the cells was observed, resulting in adriamycin release and cell death. Thus, a higher cytotoxic effect could be elicited from the thermoresponsive micelle system than from that of free adriamycin.
When the hydrophobic core forming polymers are biodegradable, the micelle structure is destabilized via core segment degradation. A PIPAAm–PLA block copolymer was prepared via ring opening polymerization of d,l-lactide, initiated by hydroxyl-terminal PIPAAm and using stannous octoate as a catalyst. Block copolymer formation was confirmed by the difference in elution profiles using gel permeation chromatography. Polymer micelles were prepared by dialysis of the polymer solution against water. Atomic force microscopic observation revealed that colloidal spheres with average diameters of 40 nm were formed by this procedure. Thermal transition phenomena were completely reversible, with micelles maintaining their original size and shape throughout the temperature cycle below and above the LCST. In order to effectively utilize this system in the body, the hydrophilic/hydrophobic transition temperature of the outer shell polymer should be just above the body temperature. To achieve this, PIPAAm-co-DMAAm with a hydroxyl terminal end and a transition temperature of 44.6°C was prepared in saline phosphate buffer (CitationKohori et al., 1998). The end hydroxyl groups were used to polymerize d,l-lactide to form block copolymers. The copolymer micelles showed an LCST of 40°C and a uniform diameter of 69 nm. The micelle’s transition temperature could be useful for active targeting in vivo, when combined with an induced hyperthermia at 42.5°C.
Chemical stimuli induced system
Chemical stress is one of the most widely utilized triggering signals for a variety of triggered or PDDS. A biochemical change in the physiology of human beings is utilized as a stimulus for triggering drug release from the pulsatile system. They further classified into:
Glucose responsive insulin release devices.
A decrease in or the absence of insulin secretion from pancreatic islets is the cause of diabetes mellitus. Diabetes mellitus patients suffer long-term from a gradual decline in the efficiency of various organs and in very severe cases the condition may lead to death. The injection of insulin at the proper time is thus necessary for the treatment of diabetic patients. Self-injection is painful and has sometimes led to the development of a hypoglycemic coma, due to an overdose of insulin. In other cases, an insufficient amount of injected insulin has led to hyperglycemia and an insufficient therapeutic effect. Thus, a great demand has arisen for the precise, effective delivery of insulin to insure normal blood glucose levels. Several systems have already been developed which are able to respond to glucose concentration changes. GOD catalyzes glucose oxidation and opens the gate for insulin transport. In this section, a novel approach for the release of insulin is reviewed using stimuli-responsive hydrogels which respond to glucose concentration changes.
CitationObaidat and Park (1996; Citation1997) prepared a copolymer of acrylamide and allyl glucose. The side-chain glucose units in the copolymer were bound to Con A. These hydrogels showed a glucose-responsive, sol–gel phase transition based on the external glucose concentration. The non-linear dependence of this sol–gel phase transition with regard to the glucose concentration was not only due to the increased binding affinity of allyl glucose to Con A compared to native glucose, but also due to the cooperative interaction between glucose containing copolymer and Con A. They further investigated the release kinetics of insulin using two chamber cells separated with glucose responsive hydrogels. The insulin concentration in the receptor chamber increased with the glucose concentration, although the slow response was apparent. This suggested the optimization of the system to achieve a simultaneous response and release of insulin sensitive to varying glucose concentrations. The systems above-mentioned use proteins of a natural origin, such as GOD and Con A, the exposure of these proteins to the body may cause an undesirable immune response once applied, and hence it was necessary to separate these systems from the environment with semi-permeable membranes, which are only permeable to glucose and insulin, but not to Con A, GOD, and the soluble polymers. Thus, a different approach using a totally synthetic polymer system has been investigated.
CitationKataoka et al. (1998) developed glucose and thermo-responsive hydrogels using acrylamidophenylboronic acid and PIPAAm. The obtained gels, containing 10 mol% phenylboronic acid moieties, showed a transition temperature of 22°C in the absence of glucose. Below this temperature the gels existed in a swollen state. The introduction of glucose to the medium altered the transition temperature of the gels in such a way that the transition temperature increases with increasing glucose concentration to reach 36°C at 5.0 g/l glucose concentration. Boronic acid was in equilibrium between the undissociated (uncharged) and the dissociated (anionically charged) form. With increasing glucose concentration, the equilibrium shifted to increase the amount of dissociated boronate groups and the gels become more hydrophilic. This indicates that the gel swelling/de-swelling was regulated by glucose concentration at a fixed temperature. Insulin was loaded into the gels and the time course of insulin release was evaluated with regard to changing glucose concentrations in the medium at pH 9.0 and 28°C. At 0–1.0 g/l glucose, the insulin release over 24 h was suppressed to below 10 and 20%, respectively. However, a more rapid insulin release was observed at a glucose concentration of 3.0 g/l, in which 80% of insulin was released within the first 10 h. At glucose concentration lower than 1.0 g/l, gels existed in a shrunken state (). Above this concentration, the gels became hydrophilic due to the increased anionic boronate units within the gels. Electrostatic repulsion may be an additional factor which influences the increase in swelling at higher glucose concentrations in the medium.
Figure 9. (a) Equilibria for alkylamidephenylboronic acid. (b) Repeated on–off release of FITC–insulin from PIPAAm–acrylamidephenylboronic acid copolymer gel beads at 28°C, pH 9.0 in response to external glucose concentration changes.
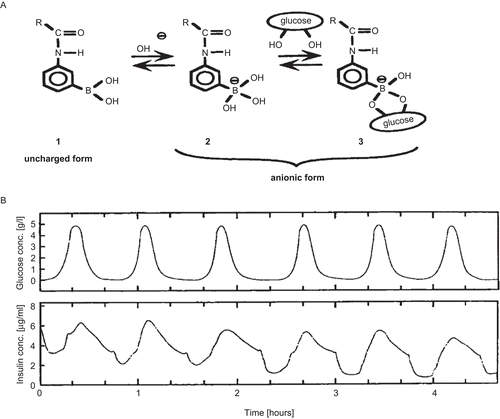
The result obtained in the above study clearly indicated that there is a threshold concentration of glucose necessary to induce loaded insulin release from the hydrated, swollen gels. Using these glucose responsive hydrogels, repeated insulin release was achieved varying the external glucose concentration at constant temperature, as shown in . When glucose concentration decreased, a dense shrunken polymer layer was formed on the gel surface, which stopped insulin diffusion from the gels. Although the system needs to be optimized to achieve release at a suitable temperature, pH range, and response rate, it can be a promising approach for the treatment of diabetes mellitus using totally synthetic copolymer systems.
Inflammation-induced system.
When human beings receive physical or chemical stress, such as injury, broken bones, etc., inflammation reactions take place at the injured sites. At the inflammatory sites, inflammation-phagocytic cells, such as macrophages and polymorphonuclear cells, play a role in the healing process of the injury. During inflammation, hydroxyl radicals are produced from these inflammation-responsive cells. CitationYui et al. (1992)., focused on these inflammatory-induced hydroxyl radicals and designed drug delivery systems, which responded to the hydroxyl radicals and degraded in a limited manner. They used HA, a linear mucopolysaccharides, composed of repeating disaccharide sub-units of N-acetyl-D-glucosamine and D-guluronic acid. In the body, HA is mainly degraded either by a specific enzyme, hyaluronidase, or hydroxyl radicals. Degradation of HA via the hyaluronidase is very low in a normal state of health, whereas degradation via hydroxyl radicals is usually dominant and rapid when HA is injected at inflammatory sites. Thus, they prepared cross-linked HA gel with ethylene glycol diglycidylether or polyglycerol polyglycidylether. These HA gels degraded only when the hydroxyl radicals were generated through the Fenton reaction between Fe2+ ions and hydrogen peroxide in vitro. Thus, a surface erosion type of degradation was achieved. Furthermore, in vivo tests of HA hydrogel degradation showed that HA gels were degraded only when inflammation at the implanted site was induced by surgical incision. Control HA gels implanted in the animals were relatively stable over a period of 100 days. Thus, it is possible to use anti-inflammatory drug incorporated HA gels as new implantable drug delivery systems for the patients with inflammatory diseases, such as rheumatoid arthritis (CitationYuk et al., 1992; CitationYui et al., 1993).
Drug release from gels responding to antibody concentration.
There are numerous kinds of bioactive compounds which exist in the body. Recently, novel gels were developed which responded to the change in the concentration of bioactive compounds to alter their swelling/de-swelling characteristics.
CitationMiyata et al. (1999a; Citationb) focused on the introduction of stimuli-responsive cross-linking structures into hydrogels. Special attention was given to antigen-antibody complex formation as the cross-linking units in the gel as specific antigen recognition of an antibody can provide the basis for a new device fabrication. Utilizing the difference in association constants between polymerized antibodies and naturally derived antibodies towards specific antigens, reversible gel swelling/de-swelling and drug permeation changes can be obtained. This study proved that the biological stimuli-responsive hydrogels can be successfully created.
pH sensitive drug delivery system.
In the case of pH-dependent system advantage has been taken of the fact that there exists a different pH environment at different parts of the GI tract. Thus, by selecting the pH-dependent polymers, drug release at specific locations can be obtained. Such a type of PDDS contains two components; one is of immediate release type and the other one is pulse released, which releases the drug in response to change in pH. Examples of pH-dependent polymers include Cellulose acetate phthalate, Eudragit L100, Eudragit S100, HPMC phthalate, HPMC trimellitate, HPMC acetate maleate, polyvinyl acetate phthalate, and Shellac. These polymers are used as enteric-coating materials so as to provide release of drug in the small intestine.
CitationSchellekens et al. (2008) described a new system for site-specific pulsatile delivery in the ileo-colonic regions. This system is based on the non-percolating incorporation of disintegrants in a coating which consists further of a continuous matrix of pH-responsive polymer (Eudragit S). Extensive in vitro release studies performed by CitationSchellekens et al. (2008) involved coatings with different concentrations and disintegrants and comparing it with non-disintegrant containing coatings. In vitro data showed that the incorporation of swelling agents in a Eudragit S coating still allows delayed release in the simulated terminal ileum. The pulse time and the robustness could be improved compared to conventional Eudragit S coatings. The augmented pH-responsiveness of the new coating was related to the swelling index of the applied disintegrant. Based on the in vitro data comparing different swelling agents they concluded that Ac-di-sol® is the best swelling agent.
CitationMastiholimath et al. (2007) studied time- and pH-dependent colon-specific pulsatile delivery of theophylline. The basic design consists of an insoluble hard gelatin capsule body filled with Eudragit microcapsules of theophylline and sealed with a hydrogel plug. The entire device was enteric-coated to overcome variability in gastric emptying time and to achieve a colon-specific release. The theophylline microcapsules were prepared in four batches with Eudragit L100 and S100 (1:2) by varying drug-to-polymer ratio and evaluated for the particle size, drug content, and in vitro release profile. One better formulation was selected based on the data obtained from the results of four batches for further fabrication of pulsatile capsules. Different hydrogel polymers were used as plugs to maintain a suitable lag period and it was found that the drug release was controlled by the proportion of polymers used. In vitro release studies of pulsatile device revealed that increasing the hydrophilic polymer content resulted in delayed release of theophylline from microcapsules. Programmable pulsatile, colon-specific release was achieved from a capsule device over a 2–24 h period, consistent with the demands of chronotherapeutic drug delivery.
CitationKadam and Gattani (2009) formulated and evaluated fast release enteric-coated tablets for pulsatile drug release to the colon. The novelty of this work was the combination of pH- and time-dependant enteric polymers as a single coating agent. Eudragit S100 was used as a pH-dependant polymer, Eudragit RL100 was used as a time-dependant polymer, and theophylline was chosen as a model drug. Dissolution studies of enteric-coated tablets were performed with different media having a pH of 1.2, 6.8, and 7.4. Results of the dissolution data showed that drug release in the colon could be controlled by using Eudragit RL100 and Eudragit S100. The lag time prior to the drug release was highly affected by a combination of two factors, namely the percentage of Eudragit RL100 and coating level. The optimum formulation was found to be one containing Eudragit RL100 and Eudragit S100, with a ratio of 60:40 of polymer and coating level of 4.66% w/w. The present study demonstrated that the theophylline enteric-coated tablets could be successfully formulated as a PDDS by the design of a time- and pH-dependant modified chronopharmaceutical formulation.
Externally regulated system
Externally regulated systems are not self-regulating, instead they require externally generated environmental changes like magnetic field, ultrasound exposure, electrical signal, light to initiate the drug release. There are four major types of PDDS based on external stimuli induced drug release. They are further classified into:
Magnetically stimulated system
Use of an oscillating magnetic field to modulate the rates of drug delivery from a polymer matrix was one of the first methodologies investigated to achieve an externally controlled drug delivery system (CitationHsieh et al., 1981). Magnetic carriers receive their magnetic response to a magnetic field from incorporated materials such as magnetite, iron, nickel, cobalt, etc. However, one must remember that magnetic carriers used for biomedical applications must be water-based, biocompatible, non-toxic, and non-immunogenic.
The mechanistic approach based on magnetic attraction is useful in the slowing down of oral drugs in the GI tract. This can be made possible by filling an additional magnetic component into capsules or tablets. The speed of travel through the stomach and intestines can then be slowed down at specific positions by an external magnet, thus changing the timing and/or extent of drug absorption into stomach or intestines. This approach was successfully utilized by CitationChen and Langer (1997) in slowing down the passage of magnetic liposomes with a magnet to increase the blood levels of drug.
CitationBabincova et al. (1999) developed magneto-liposomes for triggered release of drug. In their delivery systems, they entrapped dextran-megnetite and model drug, 6-carboxyfluorscein in the liposomes and used a laser to trigger the release of the drug. The magnetite absorbs the laser light energy to heat the lipid bilayer above the gel-liquid crystal phase transition temperature Tc, which is 41°C for dipalmitoylphosphatidylcholine. Liposomes made from this lipid release their content as soon as temperature is reached to this level. They also suggested that the absorption of laser energy by magnetite particles provide a means for much localized heating and controlled release of liposome with a single laser pulse. This may have potential applications for selective drug delivery, especially to the eye and skin. Even though the magnetic modulated therapeutic approach is one of the promising approaches for PDDS, it still needs very careful attention for a number of physical and magnetism-related properties. The magnetic force, which is defined by its field and field gradient needs to be large and carefully shaped to activate the delivery system within the target area and the magnetic materials, should be tissue stable and compatible.
Ultrasonically stimulated system
Ultrasound is mostly used as an enhancer for the improvement of drug permeation through biological barriers, such as the skin, lungs, intestinal wall, and blood vessels (CitationLevy et al., 1989; CitationKost, 1993; CitationMachluf & Kost, 1993; CitationByl, 1995; CitationMitragotri et al., 1995; Citation1996; CitationMitragotri, 2000). In many of these works where an ultrasound technique is used as a triggering mechanism, the matrix that is used for delivery of the drug was not composed of a hydrogel but of degradable polymers.
CitationKwok et al. (2000; Citation2001) prepared novel surface-modified hydrogels for ultrasound-responsive pulsatile protein delivery. On the PEG-grafted PHEMA surface, dodecyl isocyanate reacted to form self-assembling crystalline methylene chain layers, which acted as a barrier membrane against peptide leakage from the hydrogel systems. Within the PEG domains insulin was preferentially partitioned and became a reservoir for the protein (CitationMoriyama et al. 1999a; b). With the methylene chain coating around hydrogel surfaces, insulin leaching was not observed without the influence of ultrasound. In sharp contrast, a pulsatile, transient insulin release was observed after ultrasonic exposure for 1 min. During ultrasonic exposure, perturbation of the self-assembled methylene chains occurred, leading to the release of insulin from the hydrogels. Bioavailability of released insulin was also confirmed using cultured cells. After the development of new hydrogels, pulsatile release of insulin could be extended for up to 4 or 5 days. Although the biological response to the hydrogel surface is not clear, this system would be useful to deliver biologically beneficial peptide drugs in a pulsatile manner.
Electrically stimulated system
An electric field as an external stimulus for triggering the drug release is advantageous due to the availability of equipment which allows precise control with regards to the magnitude of current, duration of electric pulses, interval between pulses, etc. Electrically responsive delivery systems are prepared from polyelectrolyte’s (polymers which contain relatively high concentrations of ionizable groups along the backbone chain) and are, thus, pH-responsive as well as electro-responsive. Under the influence of electric field, electro-responsive hydrogels generally de-swell or bend dependent on the shape of the gel lies parallel to the electrodes, whereas de-swelling occurs when the hydrogel lies perpendicular to the electrodes. Synthetic as well as naturally occurring polymers separately or in combination have been used. Examples of naturally occurring polymers include hyaluronic acid, chondroitin sulfate, agarose, carbomer, xanthan gum, and calcium alginate. The synthetic polymers are generally acrylate and methacrylate derivatives such as partially hydrolyzed polyacrylamide, polydimethylaminopropyl acrylamide. Many of the gels are prepared either by cross-linking the water-soluble polymers using radiation or chemical agents such as Ca2+, Ethylene diglycidylether, N,N-methylene bisacrylamide, Ethylene glycol dimethacrylate, or by free-radical polymerization of monomers. Complex multi-component gels or interpenetrating networks have been prepared in order to enhance the gels or interpenetrating networks in order to enhance the gel’s electroresponsiveness (CitationYui et al., 1992). They prepared calcium alginate/PAA composites, where the PAA chains were expected to be entangled through the calcium alginate matrix. The increased proportion of PAA, which contains a large number of free carboxylic groups, was included to increase the gel’s sensitivity to pH and electrical stimuli. Such enhanced electro-response upon increasing the proportion of the ionizable groups in the gels has also been demonstrated in interpenetrating networks of PVA and PAA (CitationKim & Lee, 1999).
CitationKiser et al. (2000) designed lipid-coated microgels for the triggered release of drugs. Ionic microgels synthesized from the monomers of MBAM, MAA, and 4-NPMA were coated with a lipid bilayer. The release of drug is triggered from the gels using either lipid solubilizing surfactants or electroporation. The swelling and release of drugs occurs in three stages:
| i. | The permeability of the membrane might be sufficiently compromised (e.g. by electroporation or membrane dissolution or other permeabilizing species), but only to an extent that allows proton efflux from the microgel and a sodium ion influx into the gel particle, | ||||
| ii. | Microgel begins to swell due to occurrence of an exchange process, allowing additional ions to be transported across the membrane and so that disruption of membranes causes un-coating of microgel, and | ||||
| iii. | Drug is exchanged from the hydrogel by Na+ ions and diffuses down its concentration gradient out of the expanded polymer network into the surrounding medium over a period of time, resulting in a triggered release. | ||||
Electronic micro-electromechanical devices are manufactured using standard micro-fabrication techniques, the same processing techniques used to make microprocessors for computers and other microelectronic devices have been used increasingly to produce micro-scale devices whose primary functions are mechanical, chemical, and optical in nature; such devices are commonly referred to as MEMS. Even though this type of device may improve patient’s mobility and reduce infections by eliminating transcutaneous catheters, they may still be hampered by their size, cost, ability to deliver only drugs in solution, and the limited stability of some drugs in solution at 37°C. Complex release patterns (such as simultaneous constant and pulsatile release) can be achieved from the microchips. Microchip has the ability to control both release time and release rate. The rate of release from a reservoir is a function of the dissolution rate of the materials in the reservoir, the diffusion rate of these materials out of the reservoir, or both. Therefore, the release rate from an individual reservoir can be tailored to a particular application by the proper selection of the materials placed inside the reservoir (e.g. pure drug, drugs with polymers, etc.).
The pulsatile release can be achieved by using materials that quickly dissolve when the reservoir is opened. Microchip delivery devices, described by CitationSantini et al. (1998; Citation2000), provide a means to control both the rate and time of release of a variety of bioactive molecules, in either a continuous or pulsatile manner. Hundreds or thousands of reservoirs can be fabricated on a single microchip. The molecules to be delivered are inserted into the reservoirs by injection or spin coating methods in their pure form or in a release system. The release systems include polymers and polymeric matrices, non-polymeric matrices and other common excipients or diluents. The physical properties of the release system control the rate of the release of molecules. The reservoirs can contain multiple drugs or other molecules in variable doses. The filled reservoirs are capped with materials that either degrade or allow the molecules to diffuse passively out of the reservoir over time or materials that oxidize and dissolve with the use of electric potentials. Release from an active device is controlled by a pre-programmed microprocessor, remote control, or by biosensors. These devices have also been found a means for storing the bioactive compounds in their most stable form. These patents describe, for example, implanting the microchip devices by themselves into a patient for delivery of bioactive molecules. Still it needs to adapt the precise control of molecule release from these microchips into a variety of other applications.
The microchip devices include, (i) a substrate, (ii) at least two reservoirs in the substrate containing the molecules for release, and (iii) a reservoir cap positioned on or within a portion of the reservoir and over the molecules, so that the molecules are controllably released from the device by diffusion through or upon disintegration or rupture of the reservoir caps. Each of the reservoirs of a single microchip can contain different molecules and/or different amounts and concentrations which can be released independently. The filled reservoirs can be capped with materials that passively or actively disintegrate. Passive release reservoir caps can be fabricated using materials like gold or platinum that allow the molecules to diffuse passively out of the reservoir over certain time. Active release reservoir caps can be fabricated using materials like silicon carbide, silicon dioxide that disintegrate upon application of electrical, mechanical, or thermal energy. Release from an active device is also controlled by a pre-programmed microprocessor, remote control, or by biosensors (CitationMaloney et al., 2006).
Photo-stimulated system
The mechanism of photo-stimulated pulsatile release of the drug from the device involves a photoresponsive polymer consisting of a photoreceptor, usually a photochromic chromophore and a function part. The optical signal from artificial light source is captured by photochromic molecule that results in isomerization of the chromophore in the photoreceptor converting it to the electric signal which ultimately provide trigger for the release of the drug.
Various chronopharmaceutical technologies have been developed based on the principals of the above-mentioned approaches. reveals the marketed technologies for chronotherapy of diseases which shows circadian rhythm in their pathophysiology.
Table 1. Diseases requiring PDDS.
Table 2. Marketed technologies of PDDS.
Conclusion
Experts forecast a continuously rising demand for dosage forms with pulsatile drug release, since circadian rhythms have been extensively described for many diseases. Thus, more and more attempts are being made to adjust drug delivery systems accurately to patient requirements, both in terms of therapeutic efficacy and compliance. In the present review, we have described various methodologies for developing PDDS like time-controlled, Stimuli-induced, and Externally Regulated Systems. There is a constant need of new delivery system that can provide increased therapeutic benefits to the patients and PDDS is one such system that delivers the drug at the right time, at the right place, and in the right amount. PDDS also hold good promises of benefit to the patients suffering from chronic problems like arthritis, asthma, hypertension, etc.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Babincova, M., Sourivong, P., Chorvát, D., Babinec, P. (1999). Laser triggered drug release from magnetoliposomes. J Magnetism Magn Mater.:163–6.
- Badve, S.S., Sher, P., Korde, A., Pawar, A.P. (2007). Development of hollow/porous calcium pectinate beads for floating-pulsatile drug delivery. Eur J Pharm Biopharm 65:85–93.
- Bae, Y.H., Okano, T., Kim, S.W. (1991). ‘On–off’ thermocontrol of solute transport. I. Temperature dependence of swelling of N-isopropylacrylamide networks modified with hydrophobic components in water. Pharm Res 8:531–7.
- Beckert, T.E., Pogarell, K., Hack, I., Petereit, H.U. (1999). Pulsed drug release with film coatings of Eudragit RS30D. Proceedings of the International Symposium on Controlled Release of Bioactive Materials, Controlled Release Society, Minneapolis, Boston, MA, 5219.
- Bodmeier, R., Guo, X., Sarabia, R.E., Skultety, P. (1996). The influence of buffer species and strength on Diltiazem HCl release from beads coated with aqueous cationic polymeric dispersions Eudragit RS, RL30D. Pharm Res 13:52–6.
- Bussemer, T., Dashevsky, A., Bodmeier, R. (2003a). A pulsatile drug delivery system based on rupturable coated hard gelatin capsules. J Control Release 93:331–9.
- Bussemer, T., Peppas, N.A., Bodmeier, R. (2003b). Evaluation of the swelling, hydration and rupturing properties of the swelling layer of a rupturable pulsatile drug delivery system. Eur. J Pharm Biopharm 56:261–70.
- Byl, N.N. (1995). The use of ultrasound as an enhancer for transcutaneous drug delivery: phonophoresis. Phys Ther 75:539–53.
- Chen, C.M. (1993a). Pulsatile particles drug delivery system. US Patent 5,260,069.
- Chen, C.M. (1993b). Multiparticulate pulsatile drug delivery system. US Patent 5,260,068.
- Chen, C.M. (1995). Pulsatile particles drug delivery system. US Patent 5,472,708.
- Chen, H., Langer, R. (1997). Magnetically-responsive polymerized liposomes as potential oral delivery vehicles. Pharm Res 14:537–40.
- Chen, J., Park, H., Park, K. (1999). Synthesis of superporous hydrogels: hydrogels with fast swelling and superabsorbent properties. J Biomed Mater Res 44:53–62.
- Chung, J.E., Yokoyama, M., Okano, T. (2000). Inner core segment design for drug delivery control of thermo-responsive polymeric micelles. J Control Release 65:93–103.
- Chung, J.E., Yokoyama, M., Yamato, M., Aoyagi, T., Shiino, Y., Sakurai, Y., Okano, T. (1999). Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly(N-isopropylacrylamide) and poly(butyl methacrylate). J Control Release 62:115–27.
- Dashevsky, A., Mohamad, A. (2006). Development of pulsatile multiparticulate drug delivery system coated with aqueous dispersion Aquacoat® ECD. Int J Pharm 318:124–31.
- Dittigen, M., Fricke, S., Timpe, C., Gercke, H., Eichardt, A. (2000). Method of making a preorally administered solid drug with controlled effective ingredient delivery. US Patent 6, 117,450.
- Guo, T., Zheng, C.L., Song, H.T., Sui, Y., Dang, D.S., Sun, X.H. (2003). Studies on Diclofenac sodium pulsatile release pellets. Yao Xue Xue Bao 38:707–10.
- Hartman, P.J.A., Vonk, P., Kossen, N.W.F. (2000). A particulate pulse-release system and mathematical description with the Maxwell–Stefan theory. J Control Release 66:293–306.
- Hsieh, D.S.T., Langer, R., Folkman, J. (1981). Controlled release of polypeptides and other macromolecules. Proc Natl Acad Sci 78:1863–7.
- Jimoh, A.G., Wise, D.L., Gresser, J.D., Trantolo, D.J. (1995). Pulsed FSH release from an implantable capsule system. J Control Release 34:87–95.
- Kadam, V.D., Gattani, S.G. (2009). Formulation and evaluation of a pulsatile drug delivery system using time- and pH-dependant polymers. Pharm Dev Technol 14:27–36.
- Kaneko, Y., Nakamura, S., Sakai, K., Aoyagi, T., Kikuchi, A., Sakaurai, Y., Okano, T. (1998). Rapid deswelling response of poly (N isopropylacrylamide) hydrogels by the formation of water release channels using poly (ethylene oxide) graft chains. Macromolecules 31:6099–105.
- Kaneko, Y., Sakai, K., Kikuchi, A., Sakurai, Y., Okano, T. (1996). Fast swelling /deswelling kinetics of comb-type grafted poly(N-isopropylacrylamide) hydrogels. Macromol Chem Macromol Symp 109:41–53.
- Kaneko, Y., Sakai, K., Kikuchi, A., Yoshida, R., Sakurai, Y., Okano, T. (1995). Influence of freely mobile grafted chain length on dynamic properties of comb-type grafted poly(N-iso propylacrylamide) hydrogels. Macromolecules 28:7717–23.
- Kataoka, K., Harada, A., Nagasaki, Y. (2001). Block copolymer micelles for drug delivery: design, characterization andbiological significance. Adv Drug Deliv Rev 47:113–31.
- Kataoka, K., Miyazaki, H., Bunya, M., Okano, T., Sakurai, Y. (1998). Totally synthetic polymer gels responding to external glucose concentration: their preparation and application to on-Off regulation of insulin release. J Am Chem Soc 120:12694–5.
- Kikuchi, A., Okano, T. (2002). Pulsatile drug release control using hydrogels. Adv Drug Deliv Rev 54:53–77.
- Kim, S.Y., Lee, Y.M. (1999). Drug release behaviour of electrical responsive poly(vinyl alcohol)/ poly(acrylic acid) IPN hydrogels under an electric stimulus. J Appl Polym Sci 73:1752–61.
- Kiser, P., Wilson, G., Needham, D. (2000). Lipid-coated microgels for the triggered release of doxorubicin. J Control Release 68:9–22.
- Kishi, R., Hirasa, O., Ichijo, H. (1997). Fast responsive poly (N-isopropylacrylamide) hydrogels prepared by g-ray irradiation. Polym Gels Networks 5:145–51.
- Kohori, F., Sakai, K., Aoyagi, T., Yokoyama, M., Sakurai, Y., Okano, T. (1998). Preparation and characterization of thermally responsive block copolymer micelles comprising poly(N isopropylacrylamide-β-DL-lactide). J Control Release 55:87–98.
- Kohori, F., Sakai, K., Aoyagi, T., Yokoyama, M., Yamato, M., Sakurai, Y., Okano, T. (1999). Control of adriamycin cytotoxic activity using thermally responsive polymeric micelles com posed of poly(N-isopropylacrylamide-co-N,N-dimethylacrylamide)-β-poly(D,L-lactide), Colloids Surfaces. Biointerfaces 16:195–205.
- Kost, J. (1993). Ultrasound for controlled delivery of therapeutics. Clin Mater 13:155–61.
- Krogel, I., Bodmeier, R. (1999). Pulsatile drug delivery system. Int J Pharm 187:175–84.
- Kwok, C.S., Mourad, P.D., Crum, L.A., Ratner, B.D. (2000). Surface modification of polymers with self-assembled molecular structures: multitechnique surface characterization. Biomacromolecules 1:139–48.
- Kwok, C.S., Mourad, P.D., Crum, L.A., Ratner, B.D. (2001). Self- assembled molecular structures as ultrasonically-responsive barrier membranes for pulsatile drug delivery. J Biomed Mater Res 57:151–64.
- Langer, R., Igari, Y., Kibat, P.G. (1990). Optimization of a microencapsulated liposome system for enzymatically controlled release of macromolecules. J Control Release 14:263–7.
- Lee, D.Y., Chen, C.M. (2000). Delayed pulse release hydrogel matrix tablet. US Patent 6,103,263.
- Levy, D., Kost, J., Meshulam, Y., Langer, R. (1989). Effect of ultrasound on transdermal drug delivery to rats and guinea pigs. J Clin Invest 83:2074–8.
- Li, B., Zhu, J., Zheng, C., Gong, W. (2008). A novel system for three-pulse drug release based on “tablets in capsule” device. Int J Pharm 352:159–64.
- Liu, Y., Liu, S., Dai, Q. (2009). Design and evaluation of pH-independent pulsatile release pellets containing isosorbide-5-mononitrate. Chem Pharm Bull 57:55–60.
- Machluf, M., Kost, J. (1993). Ultrasonically enhanced transdermal drug delivery. Experimental approaches to elucidate the mechanism. J Biomater Sci Polym 5:147–56.
- Maloney, J.M., Sbiaa, Z., Santini, J.T., Sheppard, N.F., Uhland, S.A. (2006). Fabrication methods & structures for microreservior devices. US Patent 0, 105,275,A1.
- Mastiholimath, V.S., Dandagi, P.M., Jain, S.S., Gadad, A.P., Kulkarni, A.R. (2007). Time and pH dependent colon specific, pulsatile delivery of theophylline for nocturnal asthma. Int J Pharm 328:49–56.
- Mitragotri, S. (2000). Synergistic effect of enhancers for transdermal drug delivery. Pharm Res 17:1354–9.
- Mitragotri, S., Blankschtein, D., Langer, R. (1995). Ultrasound-mediated transdermal protein delivery. Science 269:850–3.
- Mitragotri, S., Blanlschtein, D., Langer, R. (1996). Transdermal drug delivery using low frequency sonophoresis. Pharm Res 13:411–20.
- Miyata, T., Asami, N., Uragami, T. (1999a). A reversibly antigen responsive hydrogel. Nature 399:766–9.
- Miyata, T., Asami, N., Uragami, T. (1999b). Preparation of an antigen sensitive hydrogel using antigen-antibody bindings. Macromolecules 32:2082–4.
- Moriyama, K., Ooya, T., Yui, H. (1999a). Pulsatile peptide release from multi-layered hydrogel formulations consisting of poly-(ethylene glycol)-grafted and ungrafted dextrans. J Biomater Sci Polym 10:1251–64.
- Moriyama, K., Ooya, T., Yui, N. (1999b). Hyaluronic acid grafted with poly (ethylene glycol) as a novel peptide formulation. J Control Release 59:77–86.
- Narisawa, S., Nagata, M., Danyoshi, C., Yoshino, H., Murata, K., Hirakawa, Y., Noda, K. (1993). An organic acid induced sigmoidal release system for oral controlled release preparation. Pharm Res 11:111–6.
- Narisawa, S., Nagata, M., Hirakawa, Y., Kobayashi, M., Yoshino, H. (1996). An organic acid induced sigmoidal release system for oral controlled release preparations. 2. Permeability enhancement of Eudragit RS coating led by the physico-chemical interactions with organic acid. J Pharm Sci 85:184–8.
- Neill, M.E., Rashid, A., Stevens, H.N.E. (1993). Drug dispensing device. GB Patent 2,230,442.
- Niwa, K., Takaya, T., Morimoto, T., Takada, K. (1995). Preparation and evaluation of a time-controlled release capsule made of ethylcellulose for colon delivery of drugs. J Drug Target 3:83–9.
- Obaidat, A.A., Park, K. (1996). Characterization of glucose dependent gel–sol phase transition of the polymeric glucose-concanavalin A hydrogel system. Pharm Res 13:989–95.
- Obaidat, A.A., Park, K. (1997). Characterization of protein release, through glucose-sensitive hydrogel membranes. Biomaterials 18:801–6.
- Okano, T., Bae, Y.H., Jacobs, H., Kim, S.W. (1990). Thermally on-off switching polymers for drug permeation and release. J Control Release 11:255–65.
- Okano, T., Yui, N., Yokoyama, M., Yoshida, R. (1994). Advances in polymeric systems for drug delivery. Gordon and Breach Publisher and Distributor, Yverdon, Switzerland.
- Rahemba, T.R., Bell, S., Connolly, E., Waterman, K.C. (2009). Use of scoring to induce reproducible drug delivery from osmotic pulsatile tablets. Pharm Dev Technol 14:548–55.
- Rubinstein, A., Tirosh, B., Baluom, M., Nassar, T., David, A. (1995). The rationale for peptide drug delivery to the colon and the potential of polymeric carriers as effective tools. J Control Release 46:59–73.
- Santini, J.T., Cima, M.J., Langer, R.S. (1998). Microchip drug delivery devices. US Patent 5,797,898.
- Santini, J.T., Cima, M.J., Langer, R.S. (2000). Fabrication of microchip drug delivery devices. US Patent 6,123,861.
- Schellekens, R.C.A., Stellaard, F., Mitrovic, D., Stuurman, F.E., Kosterink, J.G.W., Frijlink, H.W. (2008). Pulsatile drug delivery to ileo-colonic segments by structured incorporation of disintegrants in pH-responsive polymer coatings. J Control Release 132:91–8.
- Sharma, S., Pawar, A. (2006). Low density multiparticulate system for pulsatile release of Meloxicam. Int J Pharm 313:150–8.
- Sher, P., Ingavle, G., Ponrathnam, S., Pawar, A.P. (2007). Low density porous carrier based conceptual drug delivery system. Microporous Mesoporous Mater 102:290–8.
- Shivakumar, H.N., Suresh, S., Desai, B.G. (2006). Design and evaluation of pH sensitive multi-particulate systems for chronotherapeutic delivery of diltiazem hydrochloride. Ind J Pharm Sci 68:781–7.
- Ueda, S., Hata, T., Yamaguchi, H., Kotani, M., Ueda, Y. (1994a). Development of a novel drug release system, timecontrolled explosion system (TES): I. Concept and design. J Drug Target 2:35–44.
- Ueda, S., Yamaguchi, H., Kotani, M., Kimura, S., Tokunaga, Y., Kagayama, A., Hata, T. (1994b). Development of a novel drug release system, time-controlled explosion system (TES): II. Design of multiparticulate TES and in vitro drug release properties. Chem Pharm Bull 42:359–63.
- Ueda, S., Yamaguchi, H., Kotani, M., Kimura, S., Tokunaga, Y., Kagayama, A., Hata, T. (1994c). Development of a novel drug release system, time-controlled explosion system (TES): III. Relation between lag time and membrane thickness. Chem Pharm Bull 42:364–7.
- Ueda, Y., Hata, T., Hisami, Y., Ueda, S., Kodani, M. (1989). Time controlled explosion systems and process for preparing the same. US Patent 4, 871,549.
- Yoshida, R., Uchida, K., Kaneko, Y., Sakai, K., Kikuchi, A., Sakurai, Y., Okano, T. (1995). Comb-type grafted hydrogels with rapid temperature responses. Nature 374:240–2.
- Yui, N., Nihira, J., Okano, T., Sakurai, Y. (1993). Regulated release of drug microspheres from inflammation responsive degradable matrices of crosslinked hyaluronic acid. J Control Release 25:133–43.
- Yui, N., Okano, T., Sakurai, Y. (1992). Inflammation responsive degradation of crosslinked hyaluronic acid gels. J Control Release 22:105–16.
- Yuk, S.H., Cho, S.H., Lee, H.B. (1992). Electric current-sensitive drug delivery systems using sodium-alginate/ polyacrylic acid composites. Pharm Res:955–7.