Abstract
This study aimed to examine the effects of bile salts on pharmacokinetics of lovastatin, which has low bioavailability. Lovastatin solid dispersions were prepared using sodium deoxycholate (NaDC) and sodium glycholate (NaGC) at ratios of 1:19, 1:49, and 1:69. The formulated solid dispersions and control (commercial tablet) were administered to rats and plasma concentrations were determined by a validated LC-MS/MS method. Statistically significant differences were found in Cmax, AUC0–10, and AUC0–∞ values among lovastatin formulations (p < 0.05). NaDC-containing formulations revealed higher bioavailabilities than NaGC-containing solid dispersions at ratios of 1:19 and 1:49. Especially, NaDC-containing formulation at a ratio of 1:19 (NaDC19) showed the highest bioavailability. The AUC (both AUC0–10 and AUC0–∞) of NaDC19 was statistically higher than control and NaDC69 (p < 0.05). The AUC values decreased as bile salt concentrations increased. Overall, formulations containing bile salts showed higher AUC values than control, even though all formulations did not show significantly higher AUC. In conclusion, the addition of bile salts to lovastatin could enhance drug bioavailabilities. However, too high concentrations of bile salts could decrease bioavailabilities of lovastatin.
Introduction
Lovastatin is a competitive inhibitor of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase which catalyzes the conversion of HMG-CoA to mevalonate. It is synthesized from Asperigillus terreus and is an inactive prodrug that is converted to ß-hydroxylate by the liver (CitationEndo, 1981; CitationSirtori, 1990). It is highly effective in reducing apolipoprotein-B-containing lipoproteins, especially low-density lipoprotein cholesterol and, to a lesser extent, plasma triglycerides, and may cause a small increase in high-density lipoprotein cholesterol (CitationTobert, 2003; CitationPasha et al., 2006).
Lovastatin is rapidly absorbed following administration, reaching the steady-state peak plasma concentration (Tmax) within 4 h in humans (CitationPan et al., 1990). Due to the short elimination half-life of 1.1–1.7 h, lovastatin is best administered in the evening, when the rate of endogenous cholesterol synthesis is highest (CitationSchachter, 2004; CitationLacy et al., 2009). Food intake has an effect on lovastatin absorption, which is more effectively absorbed when taken along with food (CitationGarnett, 1995). Lovastatin is lipophilic, and its oral bioavailability is very low (5%) (CitationSchachter, 2004).
Bile salts have been paid attention as delivery systems for medicines and chemicals because of their solubilizing and emulsifying capacity. Bile salts have a large, rigid, and hydrophobic steroid nucleus with hydrophobic moieties of two or three hydroxyl groups and an ionic head of a carboxyl group, which provide the molecule a planar polarity with hydrophilic and hydrophobic domains. They show distinct behavior in terms of self-association and molecular solubilization, the critical micelle concentration (CMC) for a given bile salt being largely determined by its hydrophilic/hydrophobic balance. It’s reported that micelle formation can enhance aqueous solubility of otherwise slightly soluble organic substances. In addition, it can cause incorporation of hydrophobic organic substances into micelles, which increases the adsorption and transfer of materials in living tissue (CitationSubuddhi & Mishra, 2007).
The objective of this study was, therefore, to examine the effects of bile salts on the pharmacokinetic characteristics of lovastatin, which has low bioavailability.
Materials and methods
Materials
Sprague-Dawley (SD) rats weighing 250 g were obtained from Orient Bio Inc. (Gapyeong, Korea). Lovastatin and simvastatin (internal standard, IS) were kindly provided by Daewon Pharm. Co., Ltd. (Seoul, Korea). A commercial lovastatin product (Mevastin®) was purchased from Choongwae Pharm. Co., (Seoul, Korea). Sodium deoxycholate (NaDC) and sodium glycholate (NaGC) (Sigma Chemical Co., St. Louis, MO) were obtained. Ethyl acetate, hexane, and ammonium acetate were purchased from Duksan Company (Ansan, Korea). Acetonitrile used was of HPLC grade.
Instrumentation and chromatographic conditions
The LC system consisting of Pump NANOSPACE SI-2 3101, Degasser NANOSPACE SI-2 3010, Autosampler NANOSPACE SI-2 3133 and column oven NANOSPACE SI-2 3104 were from Shiseido (Tokyo, Japan). The analytical column was a Luna C18 (5 μm, 50 mm × 3.0 mm, Phenomenex, Torrance, CA). The isocratic mobile phase, a mixture of 2 mM ammonium acetate and acetonitrile mixture (80:20, v/v%) was delivered at 0.5 mL/min into the mass spectrometer’s electrospray ionization chamber. Quantitation was achieved by MS/MS detection in positive ion mode for lovastatin and IS, using an API-5000 mass spectrometer (Applied Biosystems/MDS SCIEX, Foster City, CA), equipped with a Turboionspray interface at 500°C. The ion spray voltage was set at 5000 V. The common parameters, viz., collision gas, curtain gas, ion source gas 1, and ion source gas 2 were set at 10, 20, 50, and 50 psi, respectively. The compound parameters, viz., declustering potential, collision energy, entrance potential, and collision exit potential were 110, 37, 10, and 25 V for lovastatin and 140, 25, 10, and 14 for IS, respectively. Detection of the ions was performed in the multiple reaction monitoring (MRM) mode, monitoring the transition of the m/z 405.1 precursor ion to the m/z 199.1 product ion for lovastatin and the m/z 419.2 precursor ion to the m/z 199.1 product ion for IS. In this method, both Q1 and Q3 quadrupoles were operated unit resolution. The analytical data were processed by Analyst software (Version 1.4.2, Appliedbiosystems, Carlsbad, CA).
Standard and quality control sample preparation
Stock solutions of lovastatin and IS were prepared in 50% acetonitrile at a concentration of 1 mg/mL. Prior to use, these two stock solutions were further diluted with 50% acetonitrile to obtain working solutions at a concentration of 1 μg/mL. An appropriate dilution of the working solution with drug free plasma from rats gave a concentration range between 0.039–20 ng/mL of lovastatin.
Ten calibration samples (0.039, 0.0781, 0.156, 0.313, 0.625, 1.25, 2.5, 5, 10, and 20 ng/mL) were prepared by spiking blank plasma with appropriate volumes of the working solutions. Quality control samples (0.039, 0.3125, 2.5, and 20 ng/mL) were independently prepared in the same manner.
Sample preparation and method validation
All plasma samples were stored at −30°C, thawed at room temperature, vortex-mixed and centrifuged at 3,500 rpm for 5 min at 4°C prior to analysis. Plasma samples (200 μL) were mixed with 20 μL IS (10 ng/mL) solution, and 100 μL acetonitrile was added to precipitate protein. The samples were extracted with 1 mL of ethyl acetate and hexane (9:1) mixture using a vortex mixer. The tubes were centrifuged at 13,000 rpm for 5 min. The organic layer was pooled in a conical borosilicate centrifuge tube, and evaporated to dryness at 45°C under a stream of nitrogen gas. The residue was reconstituted with 100 μL mobile phase solution and then 30 μL was injected directly into the HPLC system.
Calibration curves were constructed based on the peak area ratios of the drug-to-IS. Intra-day precision was defined by the percentage of relative standard deviation (R.S.D) of five standards at five different concentrations analyzed on the same day. Inter-day precision was estimated from the analysis of the five standards on five separate days during method validation.
Preparation of lovastatin solid dispersions
Lovastatin and bile salt mixtures at ratios of 1:19, 1:49, and 1:69, respectively, were dissolved in methanol. The solvents were evaporated under reduced pressure at 50°C. The residues were dried for 1 h and then passed through a sieve (100 mesh) to give a solid dispersion.
Animal studies
Male SD rats were anesthetized with ether (Daejung Chemicals and Metals, Siheung, Korea) and the jugular vein was cannulated using a polyethylene tube (0.76 mm i.d. × 1.22 mm o.d.; Becton Dickinson, Franklin Lakes, NJ). After surgery, each animal was housed in a separate cage. The animals fasted overnight and the fasting continued for the first 6 h of the experiment but they were allowed water ad libitum. The rats were then divided into seven groups, each group comprised of six rats. Groups 1–7 were administered with formulated solid dispersions 1–6 and commercial lovastatin tablet (Mevastin®, control), respectively, at a dose of 2.5 mg/kg as lovastatin. Solid dispersions 1–3 were composed of lovastatin and NaDC at ratios of 1:19 (NaDC19), 1:49 (NaDC49), and 1:69 (NaDC69) and 4–6 consisted of lovastatin and NaGC at ratios of 1:19 (NaGC19), 1:49 (NaGC49), and 1:69 (NaGC69), respectively. Before administration, commercial lovastatin was ground. Formulated solid dispersions and the ground commercial tablet at a dose of 2.5 mg/kg as lovastatin were separately dissolved in 1 mL water and administered to rats using syringes. Plasma samples (0.3 mL) were collected at pre-determined time intervals and analyzed by LC-MS/MS. The pharmacokinetic studies of lovastatin solid dispersion were carried out according to the Principles for Biomedical Research Involving Animals developed by the Council for International Organizations of Medical Sciences.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed using WinNonlin (Version 1.1, Scientific Consulting Inc., Cary, NC). The drug concentration–time curves were fitted to a one-compartment model with a first order absorption. The area under the plasma concentration–time profile (AUC) was calculated using the log-linear trapezoidal method. The maximum plasma concentration (Cmax) and the time to reach Cmax (Tmax) were determined directly from the individual drug concentration against the time curves.
Statistical analysis
All the values are expressed as the mean ± SE. The pharmacokinetic variables of all dosage forms were compared with a Kruskal-Wallis Test, which was followed by a posterior test with the use of the Bonferroni correction. A p-value of less than 0.05 was considered significant.
Results and discussion
Lovastatin and IS showed well resolved chromatographic peaks at 2.6 and 3.4 min, respectively, as shown in . The blank plasma after extraction consistently contains no significant interfering peaks. The relation between lovastatin concentrations and peak area ratios of lovastatin to IS was linear from 0.039–20 ng/mL (y = 0.764x + 0.00445, r2 = 0.999). The limit of quantitation (LOQ) of lovastatin was determined as the sample concentration of lovastatin resulting in peak heights of 10-times signal-to-noise ratio. The LOQ was found to be 0.039 ng/mL. The intra- and inter-day precision of the methods was determined by the assay of four samples of drug-free plasma containing known concentrations of lovastatin. As described in , the intra- and inter-day RSD (%) was within 8%, which was acceptable for all quality control samples including the LOQ. The accuracy of lovastatin detection ranged between 97.2–108.2%. All the batches met the quality control acceptance criteria (CitationKamas et al., 1991).
Table 1. Intra- and inter-day precision and accuracy of the determination of lovastatin in plasma.
Figure 1. Chromatograms of rat blank plasma (A), lovastatin (1.25 ng/mL, B), and IS (1 ng/mL, C).
shows the mean plasma concentration-time profiles after administration of formulated solid dispersions and control (Mevastin®) at a dose of 2.5 mg/kg as lovastatin, and reveals the pharmacokinetic parameters based on . Statistically significant differences were found in Cmax, AUC0–10 and AUC0–∞ values among lovastatin formulations (p < 0.05). From and , NaDC-containing formulations revealed higher bioavailabilities than NaGC-containing solid dispersions at ratios of 1:19 and 1:49. Especially, NaDC19-containing formulation showed the highest bioavailability. The AUC (both AUC0–10 and AUC0–∞) of NaDC19 was statistically higher than control and NaDC69 (p < 0.05). Overall, formulations containing bile salts showed higher AUC values than control, even though all formulations did not show significantly higher AUC.
Table 2. Pharmacokinetic parameters of formulated lovastatin dosage forms.
Figure 2. Mean pharmacokinetic profiles after administration of formulated lovastatin solid dispersions and commercial lovastatin tablet (Mevastin®) at a dose of 2.5 mg/kg as lovastatin (Mean ± SE, n = 6). Ratio between drug and sodium deoxycholate is 1:19 (NaDC19), 1:49 (NaDC49), and 1:69 (NaDC69). Ratio between drug and sodium glycholate is 1:19 (NaGC19), 1:49 (NaGC49), and 1:69 (NaGC69).
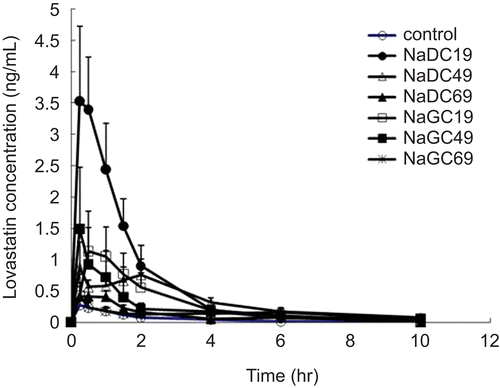
Based on the fact that lovastatin has low bioavailability due to its low solubility, the increased AUC by the addition of bile salts were attributable to the increased solubility of lovastatin. In our previous study, it was found that the addition of NaDC and NaGC at 5% to water increased solubility 553- and 365-fold, respectively; solubilities of lovastatin in water, 5% NaDC- and 5% NaGC-containing aqueous solution were 0.00085 ± 0.00004, 0.47 ± 0.03, and 0.31 ± 0.002 mg/mL, respectively (CitationChun, 2009).
Bile salts have different properties from ordinary aliphatic surfactant molecules due to their large, rigid, and planar hydrophobic moiety of a steroid nucleus with two or three hydroxyl groups. The enhanced aqueous solubility of slightly soluble drugs is caused by incorporation of hydrophobic organic substances into micelles (CitationSugioka & Moroi, 1998). In our previous study, NaDC showed the highest solubilization effect among various bile salts including NaDC, NaGC, sodium cholate, sodium taurodeoxycholate, sodium taurocholate, sodium glycodeoxycholate, and sodium ursodeoxycholate (CitationChun, 2009). The concentration of 5% used was much higher than CMC of all bile salts used. The highest solubilization effect by NaDC was thought to be due to the larger size of micelle, more hydrophobic environment, and less structural change (CitationCoello et al., 1996; CitationSubuddhi & Mishra, 2007).
Two mechanisms have been suggested for the effects of bile salts on the transfer of dissolved drugs across the absorption barrier: alteration of barrier function and decreased thermodynamic activity of the drug caused by incorporation in micelle (CitationKakemi et al., 1970; CitationYamaguchi et al., 1986). Studies have reported that bile salts enhance the epithelial transport of hydrophilic drugs through the paracellular route and that of hydrophobic compounds through both paracellular and transcellular routes (CitationSasaki et al., 1997; CitationShimazaki et al., 1998). Since lovastatin is lipophilic, the enhanced bioavailabilities by the addition of bile salts were thought to be due to the increased transport via both paracellular and transcellular routes.
Moreover, lovastain is known to be a both P-glycoprotein (P-gp) substrate and inhibitor. Many absorption enhancers such as bile salts, fatty acids, and surfactants have been shown to inhibit P-gp (CitationDudeja et al., 1995; CitationLo & Huang, 2000). These enhancers exert the P-gp inhibition activity by changing the membrane environment. The membrane perturbation might change the fluidity of cell membrane and thus inhibit the activity of membrane-spanning proteins, such as P-gp (CitationLo & Huang, 2000).
The AUC values decreased as bile salt concentrations increased regardless of which bile salt, as shown in . This result was totally different from our previous in vitro study, in which the permeation fluxes increased as the bile salt concentrations increased (CitationChun, 2009). The inverse relationship between lovastatin bioavailabilities and bile salt concentrations was partly attributed to the fact that the reduced thermodynamic activity was rate-limiting compared to barrier function change.
In a study using a lipophilic drug (ketoconazole) and a bile salt, the addition of sodium taurocholate at concentrations above the CMC resulted in a reduction of the absorption rate of the lipophilic drug, bile salt concentration-dependently. The reduction in the absorption kinetics of lipophilic drugs in the presence of sodium taurocholate was partly ascribed to the reduction of the free drug fraction due to micellar solubilization (CitationPoelma et al., 1990).
In this study, the concentration of NaDC and NaGC used at ratios of 1:19, 1:49, and 1:69 between drug and bile salts was calculated to be 115 and 97, 295 and 251, and 416 and 354 mM, respectively. It has been reported that the CMC of NaDC and NaGC at 25°C with ionic strength adjusted to 0.10 M with NaCl was 2.35 and 11.60 mM, respectively (CitationReis et al., 2004). Therefore, the used bile salt concentrations were much higher than CMC, regardless of ratio, considering that dilution is minimal in the stomach.
Another plausible explanation is that, due to the high amount of bile salts used, some bile salts could be converted into bile acids in the stomach, resulting in precipitation or aggregation. The precipitation could hinder gastric emptying of formulated lovastatin solid dispersion; thereby, reducing drug absorption.
In conclusion, the addition of bile salts to lovastatin could enhance drug bioavailabilities by several mechanisms such as solubilization by micelle formation, penetration enhancement through both paracellular and trascellular routes, and P-gp inhibition. However, too high concentrations of bile salts could rather decrease the bioavailabilities.
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Chun, I.K. (2009). Dissolution and duodenal permeation characteristics of lovastatin from bile salt solid dispersions: role of critical micelle concentration. J Kor Pharm Sci. 39:97–106.
- Coello, A., Meijide, F., Nunez, E.R., Tato, J.V. (1996). Aggregation behavior of bile salts in aqueous solution. J Pharm Sci. 85:9–15.
- Dudeja, P.K., Anderson, K.M., Harris, J.S., Buckingham, L., Coon, J.S. (1995). Reversal of multidrug resistance phenotype by surfactants: relationship to membrane lipid fluidity. Arch Biochem Biophys. 319:309–15.
- Endo, A. (1981). Biological and pharmacological activity of inhibitors pf 3-hydroxy-3-methylglutaryl coenzyme A reductase. Trends Biochem. Sci. 6:10–13.
- Garnett, W.R. (1995). Interactions with hydroxymethylglutaryl-coenzyme A reductase inhibitors. Am J Health Syst Pharm. 52:1639–45.
- Kakemi, K., Sezaki, H., Konishi, R., Kimura, T., Murakami, M. (1970). Effect of bile salts on the gastrointestinal absorption of drugs. I. Chem Pharm Bull. 18:275–80.
- Kamas, H.T., Shiu, G., Shah, V.P. (1991). Validation of bioanalytical methods. Pharm Res. 8:421–6.
- Lacy, C.F., Amstrong, L.L., Goldman, M.P., Lance, L.L. (2009). Drug information handbook. Hudson, USA: Lexi-comp.
- Lo, Y.L., Huang, J.D. (2000). Effects of sodium deoxycholate and sodium caprate on the transport of epirubicin in human intestinal epithelial Caco-2 cell layers and everted gut sacs of rats. Biochem Pharmacol. 59:665–72.
- Pan, H.Y., DeVault, A.R., Wang-Iverson, D., Ivashkiv, E., Swanson, B.N., Sugerman, A.A. (1990). Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. J Clin Pharmacol. 30:1128–35.
- Pasha, M.K., Muzeeb, S., Basha, S.J.S.B., Shashikumar, D., Mullangi, R., Srinivas, N.R. (2006). Analysis of five HMG-CoA reductase inhibitors-atorvastatin, lovastatin, pravastatin, rosuvastatin and simvastatin: pharmacological, pharmacokinetic and analytical overview and development of a new method for use in pharmaceutical formulations analysis and in vitro metabolism studies. Biomed Chromatogr. 20:282–93.
- Poelma, F.G.J., Breäs, R., Tukker, J.J. (1990). Intestinal absorption of drugs. III. The influence of taurocholate on the disappearance kinetics of hydrophilic and lipophilic drugs from the small intestine of the rat. Pharm Res. 7:392–7.
- Reis, S., Moutinho, C.G., Matos, C., de Castro, B., Gameiro, P., Lima, J.L.F.C. (2004). Noninvasive methods to determine the critical micelle concentration of some bile acid salts. Anal Biochem. 334:117–26.
- Sasaki, M., Imai, T., Ohtake, H., Azuma, H., Otagiri, M. (1997). Effects of absorption enhancers on the transport of model compounds in Caco-2 cell monolayers: assessment by confocal laser scanning microscopy. J Pharm Sci. 86:779–85.
- Schachter, M. (2004). Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 19:117–25.
- Shimazaki, T., Tomita, M., Sadahiro, S., Hayashi, M., Awazu, S. (1998). Absorption-enhancing effects of sodium caprate and palmitoyl carnitine in rat and human colons. Dig Dis Sci. 43:641–5.
- Sirtori, C.R. (1990). Pharmacology and mechanism of action of the new HMG-CoA reductase inhibitors. Pharm Res. 22:555–63.
- Subuddhi, U., Mishra, A.K. (2007). Micellization of bile salts in aqueous medium: a fluorescence study. Colloids Surf B Biointerfaces. 57:102–7.
- Sugioka, H., Moroi, Y. (1998). Micelle formation of sodium cholate and solubilization into the micelle. Biochim Biophys Acta. 1394:99–110.
- Tobert, J.A. (2003). Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2:517–26.
- Yamaguchi, T., Ikeda, C., Sekine, Y. (1986). Intestinal absorption of a beta-adrenergic blocking agents nadolol. II. Mechanism of the inhibitory effect on the intestinal absorption of nadolol by sodium cholate in rats. Chem Pharm Bull. 34:3836–43.