Abstract
The objective of the present study was to develop a colon targeted system of meloxicam for potential application in the prophylaxis of colorectal cancer. Efficacy of selective cyclooxygenase–2 inhibitors has been proven in colorectal cancer. Meloxicam is a selective cyclooxygenase–2 inhibitor with pH-dependent solubility. To achieve pH-independent drug release of meloxicam, pH modifying agents (buffering agents) were used. Meloxicam tablets containing polyethylene oxide were dually coated with ethyl cellulose containing hydrophilic material, polyethylene glycol as an inner coating layer and methyl acrylate, methyl methacrylate, and methacrylic acid copolymer (Eudragit® FS 30D) as outer coating layer for colon targeting. Optimized tablet formulations demonstrated good potential to deliver the drug to the colon by successfully exhibiting a lag time of 5 h during in vitro drug release study. An in vivo evaluation study conducted to ascertain pharmacokinetic parameters in rabbits revealed that the onset of drug absorption from the coated tablets (Tlag time = 4.67 ± 0.58 h) was significantly delayed compared to that from the uncoated tablets. The AUC0→t and AUC0→∞ for coated tablets were lower than of uncoated tablets, although the difference was not significant (p > 0.01). The roentgenography study revealed that the tablet remained intact, until it reached the colon (5 h), which demonstrates that the system can efficiently deliver the drug to the colon. This study demonstrated that a meloxicam-loaded colon targeted system exhibited promising targeting and hence may be used for prophylaxis of colorectal cancer.
Introduction
Targeted drug delivery systems are useful for selective and efficient delivery of pharmacologically active compounds to the pre-determined targets in therapeutic concentration along with minimizing side-effects of the drug. In the last decade, colon targeted drug delivery systems (CoDDS) have attracted tremendous interest of researchers in the field of targeted drug delivery. Colonic drug delivery may be achieved by either oral or rectal administration; however, the oral route is the most preferred. CoDDS may be useful for localized treatment of colonic diseases viz. inflammatory bowel disease, colorectal cancer, amoebiasis, etc.; as well as for the systemic delivery of proteins and peptides which are degraded by the digestive enzymes of the stomach and small intestine. Various approaches have been used for oral delivery of drugs to the colon, which include a time-dependent, pH-dependent, pressure controlled system and systems that use bacteria that colonize in the colon or produce enzymes to modulate the drug release (CitationPatel et al., 2007).
Colorectal cancer is the third most common cancer in the world and the second most common cause of cancer-related deaths (CitationRicchi et al., 2003). Estimated new colorectal cancer cases and deaths by sex in the US in 2009 is 52,010 in males and 54,090 in females (CitationAhmedin et al., 2009). Thus, colorectal cancer prevention has become an important goal for health professionals, physicians, and the general public. Recent progress in molecular biology and pharmacology enhances the likelihood that cancer prevention will increasingly rely on chemoprevention. Chemoprevention, a new emerging science, utilizes agents which inhibit, delay, or reverse carcinogenesis (CitationArber & Levin, 2005). One aspect currently attracting much attention is the involvement of cyclooxygenase–2 (COX-2) products in the promotion of cancer. A number of epidemiological studies have exhibited that prolonged use of non-steroidal anti-inflammatory drugs leads to reduced (by 63%) relative risk of colorectal carcinoma (CitationGiovannucci et al., 1994; CitationGiardiello et al., 1995; CitationDuBois et al., 1996; CitationWhittle, 2000; CitationHarris et al., 2007). Studies have demonstrated that COX-2 is over-expressed in 85% of primary colorectal carcinomas and in cell lines derived from such cancers (CitationSano et al., 1995; CitationDuBois et al., 1996; CitationSheng et al., 1997). Both in vitro and in vivo studies have demonstrated that inhibition of the cyclooxygenase pathway, and particularly COX-2, results in the inhibition of tumor growth and development. Selective inhibitors of COX-2 have been reported to reduce the formation, growth, and metastasis of experimental tumors (CitationAyakawa et al., 2009). It is also important to note that recent pre-clinical studies have demonstrated stronger anti-neoplastic effects of selective COX–2 inhibitors against colon cancer (CitationHarris et al., 2007).
Meloxicam, a non-steroidal anti-inflammatory drug, has demonstrated inhibitory actions against COX-2 (CitationChurchill et al., 1996; CitationTsubouchi et al., 2000a) and anti-tumor effects in several human tumor cell lines (CitationGoldman et al., 1998; CitationNaruse et al., 2006; CitationTsubouchi et al., 2000b; CitationSeong-Hee et al., 2008). Various investigations have been reported about the relationship of meloxicam and colorectal polyps and/or cancer. It has been found that the growth of HCA-7 colorectal tumor xenografts in nude mice was significantly suppressed by meloxicam after 4 weeks of treatment (CitationGoldman et al., 1998). CitationZhang et al. (2007) studied the effect of meloxicam on the expression of vascular endothelial growth factor and angiopoietin-2 in colon carcinoma cells, at cellular level to investigate its anti-neoplastic mechanisms. It has been observed that meloxicam exhibits a cytotoxic effect on the cell proliferation of HT-29 cells in a concentration-dependant and time-dependant manner. Cell cycle analysis showed that the cells are mainly blocked in the G0/G1 phase. The authors reported that meloxicam can reduce the expression of vascular endothelial growth factor and angiopoietin-2 at the protein and mRNA level in colon carcinoma cell line. From the above discussions, it can be regarded that meloxicam is a potential drug for the prevention and treatment of colorectal polyps and/or cancer and it could be beneficial if targeted to the colon.
The objective of the present study was to develop meloxicam-loaded CoDDS for potential application in the prophylaxis of colorectal cancer. Meloxicam is an extremely poorly soluble acidic drug; having a pH-dependent solubility (solubility increases at higher pH, i.e. above pH 7.0 meloxicam exhibits free aqueous solubility) (CitationGhorab et al., 2004). The pH of the colon varies from 6.4–7.0, thus in order to maintain the micro-environmental pH of the tablet above 7.0 and hence to achieve a pH-independent drug release two classes of pH modifying agents (water-soluble and water-insoluble) were used to assess their effect on the dissolution of meloxicam. Over the last few years, many different approaches have been used for oral delivery of drugs to the colon, which include time-dependent delivery, pH-dependent delivery, and delivery systems that use bacteria which colonize in the colon or produce enzymes to affect the drug release. The major drawback associated with pH-dependent drug delivery systems is the premature release of the drug in the small intestine before attaining the targeted colonic regions. This is because of high pH variability of the gastrointestinal tract among individuals and lack of proper coating materials that would dissolve at the desired pH of the colon. The time-dependent release systems depict lack of site specificity due to variation in the gastric emptying time. Since pH- and time-dependent systems when used alone were not found suitable for achieving CoDDS, a combination of both the systems (CitationFukui et al., 2000; CitationSangalli et al., 2001; Citation2004; CitationQi et al., 2003; CitationBott et al., 2004; CitationKrishnamachari et al., 2007; CitationMastiholimath et al., 2007; CitationAsghar & Chandran, 2008; CitationPatel et al., 2009) were used for dually coating meloxicam tablets with ethyl cellulose (EC) containing polyethylene glycol (PEG) as an inner coating layer (time dependent factor) and Eudragit® FS 30D as an outer coating layer (pH-dependent polymer) ().
Figure 1. Schematic diagram of the colon-targeted formulation system.
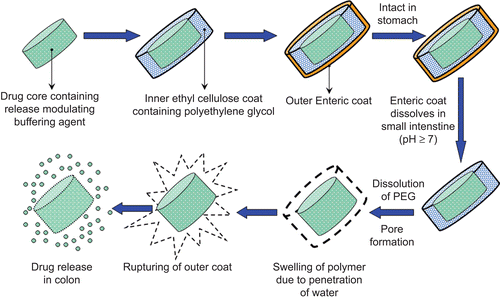
In vivo evaluation of the CoDDS of meloxicam was performed to assess the efficacy of the system. The plasma-concentration time profile of meloxicam delivered to the colon was determined in rabbits. To support the results of the pharmacokinetic evaluation of the CoDDS, roentgenography studies were also carried out using rabbits as an animal model.
Materials and methods
Materials
Meloxicam was obtained as a gift sample from Cadila Pharmaceuticals Ltd. (Gujarat, India). Eudragit® FS 30D was received as a gift sample from Evonik Röhm GmbH (Darmstadt, Germany). Polyethylene oxide (PEO, molecular weight: 900,000) was gifted from Colorcon Asia Pvt. Ltd. (India). Methanol, Chloroform, EC, and PEG 4000 were purchased from Central Drug House Pvt. Ltd. (New Delhi, India). Other excipients used were of standard pharmaceutical grade and all chemicals and reagents used were of analytical reagent grade/high-performance liquid chromatography (HPLC) grade and were used without any further purification.
Preparation of core tablets
Meloxicam and other adjuvants (PEO, pH modifiers and additives), as mentioned in were passed through a 60# sieve. Tablets were prepared by direct compression technique. Magnesium stearate was used as a lubricant and aerosil as a flow promoter. The tablets were prepared using a rotary tablet machine (Rimek, Ahmedabad, India), equipped with 9 mm punches. Average weight of tablets was 300 mg. The tablets were also evaluated for thickness, weight variation, friability, hardness, and drug content ().
Table 1. Formulation of core tablets of meloxicam.
Determination of buffering efficiency
Meloxicam exhibits free aqueous solubility above pH 7.0 and pH of the colon being 6.4–7.0, an attempt was made to increase the solubility of meloxicam by providing a buffer microenvironment. Four different buffering agents were evaluated () for their pH modifying capacity. The change in pH was determined by measuring the pH of 6.4 pH phosphate buffer (taking 0.5 ml as amount sufficient to cover 9 mm tablet to impart micro-environment, Seveneasy pH, Schwerzenbach, Switzerland), after addition of various amounts of buffering agents (equivalent to the amount used for the preparation of tablets) ().
Table 2. Effect of percentage strength of buffering agent on pH 6.4 phosphate buffer.
Swelling or water uptake studies
The core tablets were subjected to swelling studies. In order to avoid deformation during the determination of swelling, one planar base of the tablets was covered with water-insoluble coating (5% w/v EC solution in ethanol) and glued to a piece of glass slide. The glass slide and the tablets were weighed initially (W1). The samples were placed in a dissolution bowl containing 900 ml, 6.4 pH phosphate buffer at 37.0 ± 0.5°C in the USP dissolution apparatus, paddle type at 100 rpm, to simulate the in vitro dissolution process. The swollen tablets were taken out at pre-determined time intervals and the excess of water on their surface was carefully removed using tissue paper and tablets were reweighed (W2). The study was carried out over a period of 3 h and the tablets were observed for rupturing, if any. The swelling index (% weight change) was calculated using the following formula:
Preparation of meloxicam colon targeted tablets
The core tablets of meloxicam were coated with a polymeric solution of 5% w/v EC and PEG using ethanol (95%) as a solvent, for inner coating layer by pan coating technique (Kevin Process Technologies Pvt. Ltd., Ahmedabad, India). The polymeric solution also contained plasticizer triethyl citrate (20% w/w, based on dry polymer weight), an opacifier, titanium dioxide (0.5% w/v) and an anti-adherent, talc (50% w/w, based on dry polymer weight) to prevent adhering of tablets during coating. Three different compositions of EC:PEG were used for coating viz. 60:40 (solution 1), 70:30 (solution 2), and 80:20 (solution 3). Polyethylene glycol was added to the coating solution as a pore forming agent.
The outer enteric coating layer comprised of Eudragit® FS 30D polymer (15% w/v), glycerol monostearate as a glidant (5% w/w, based on dry polymer weight) and polysorbate 80 (40% w/w, based on glycerol monostearate weight) as an emulsifier. Since Eudragit® FS 30D is a ‘flexible’ polymer (higher elongation at break), only 5% w/w of plasticizer (triethyl citrate) based on dry polymer weight was required for proper coating.
The pan coating procedure was carried out by spraying the coating solution on pre-warmed tablet bed (30°C). The tablets were coated and dried using inlet air (temperature 35–40°C) at 30 rpm. The process of coating was performed till the desired weight gain was achieved. At the end of each stage of coating, the tablets were cured in the coating pan for 15 min and then dried in a tray drier at 40°C for 24 h. The percentage weight gain of the tablets after coating was assumed to be indicative of the coat thickness.
In vitro dissolution studies
In vitro dissolution studies were performed for meloxicam CoDDS using USP dissolution apparatus II (paddle method, Electrolab, TDT – 06 T, Mumbai, India) at 100 rpm, 37.0°C ± 0.5°C using 900 ml dissolution medium. The variation in the pH of the gastrointestinal tract (GIT) was mimicked by using a sequential pH change method where four dissolution media with pH 1.2, 6.0, 7.2, and 6.4 were sequentially used, as shown in (CitationAshford et al., 1993; CitationKhan et al., 1999; Citation2000; CitationGang et al., 2004; CitationPatel et al., 2009). The R2 value for calibration curve at pH 1.2, 6.0, 7.2, and 6.4 was found to be 0.9993, 0.9999, 0.9996, and 0.9998, respectively, indicating a good linearity. Samples were withdrawn at regular time intervals, filtered using Whatman filter paper (45 μ), and estimated using a UV/VIS spectrophotometer (Shimadzu UV 2450, double beam UV/VIS Spectrophotometer, Shimadzu Corporation, Japan). The cumulative percentage release for meloxicam was calculated (mean ± SD, n = 6) over the sampling times using Beer Lambert’s curve generated in the respective dissolution medium.
Table 3. Dissolution conditions and λmax of meloxicam-coated tablets.
In vivo evaluation of CoDDS
In vivo pharmacokinetic evaluation
Protocol number IPS/PCEU/PhD09/004 was approved by the Institutional Animal Ethics Committee at Nirma University (Ahmedabad, India), to perform in vivo pharmacokinetic studies of CoDDS of meloxicam. Six New Zealand white rabbits weighing 3–3.5 kg were used in this study. All rabbits were placed on overnight fasting with free access to water under 12 h light/dark cycles. After oral administration of optimized uncoated (n = 3) and coated (n = 3) tablets of meloxicam (at a dose of 2 mg/body), the ears were shaved and 2 ml of blood samples were collected from the marginal ear vein using heparinized syringes at various time intervals of 0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 24, 48, and 72 h. The plasma fraction used for meloxicam assay was obtained by centrifuging the blood samples at 4000 rpm for 20 min using refrigerated centrifuge (Biolab, Mumbai, India) at 4°C (CitationSteed et al., 1997). The plasma samples were immediately stored at −20°C until further analysis.
HPLC determination of meloxicam in plasma samples
Concentration of meloxicam in the plasma was determined using a validated HPLC method, which was equipped with a Jasco PU 2080 intelligent HPLC pump (Jasco, Japan), a Jasco UV–2075 plus intelligent UV/Visible detector (Jasco, Japan), and a Jasco Borvin™ version 1.5 software. The HPLC system consisted of a Hi-Q-Sil C18 column (5 μm, 250 mm × 4.6 mm) with a mobile phase containing methanol/water/triethylamine/phosphoric acid (650/350/0.75/0.5 v/v/v/v) at a flow rate of 1.0 ml/min. UV detection was performed at 360 nm (CitationGao et al., 2006). Calibration curve was prepared by dilution of meloxicam (100 μg/ml) stock solution in 0.05 M sodium hydroxide by addition of 20 μl of the respective dilutions to blank plasma in order to obtain seven number of plasma concentrations ranging from 0.25–12.0 μg/ml (n = 6). The correlation coefficient (R2 value) was found to be 0.9933, which indicates a strong linear relationship between the variables. The limit of detection and lower limit of quantification were found to be 0.03 and 0.25 μg/ml, respectively. The extraction efficiency was found to be greater than 80%. The intra-day and inter-day accuracy in the present study ranged from 90.43–96.29% and from 91.36–92.84%, respectively. This indicates that there was no interference from endogenous plasma components. The coefficient of variation of intra-day and inter-day precision ranged from 1.54–8.13% and 2.27–7.98%, respectively. This revealed that the proposed method is highly precise.
Analytical procedure of plasma samples
A methanol solution of piroxicam (250 μg/ml) was used as the internal standard. Twenty microliters of this solution was pipetted in a 10 ml tube and evaporated to dryness at ambient temperature under nitrogen flow. A 1 ml aliquot of rabbit plasma was added and the sample was vortexed for 20 s. On hundred microliters of HCl (1 M) was added and the sample was vortexed for another 20 s. Chloroform (5 ml) was added to extract meloxicam and piroxicam. The mixture was vortexed for 5 min and then centrifuged at 4000 rpm for 20 min. The organic layer was transferred to a new tube and evaporated to dryness at ambient temperature under nitrogen flow. The residue was dissolved in 100 μl mobile phase, vortexed, and 20 μl of this solution was injected into the column for HPLC analysis.
Pharmacokinetic parameters
The pharmacokinetic parameters were calculated by non-compartment analysis using Microsoft® Excel software. The maximum drug concentration (Cmax), the time to reach Cmax (tmax), and the time that meloxicam first appears in the plasma (Tlag time) were obtained as directly measured values from the plasma—concentration vs time profile. The AUC0→t (area under the plasma concentration-time curve) was calculated by the trapezoidal method. The AUC0→∞ was determined by the following equation:
where Ct is the plasma concentration of meloxicam at the last time point t, kel is the elimination rate constant. The pharmacokinetic parameters were statistically analyzed using their mean ± SD and performing unpaired t-test (using SPSS, SPSS Inc., Chicago, IL) (CitationPhilip & Pathak, 2008a; Citationb; CitationWei et al., 2008). A p-value < 0.01 was considered as significant.
In vivo roentgenographic study
An in vivo roentgenography study was performed in order to provide a proof of concept for the in vivo pharmacokinetic evaluation study. According to protocol number IPS/PCEU/PhD09/004 approved by the Institutional Animal Ethics Committee at Nirma University (Ahmedabad, India), three New Zealand white rabbits weighing 3–3.5 kg were used in this study. Core tablets according to the optimized meloxicam tablet formulation were prepared by partly replacing the drug with radio-opaque compound barium sulfate. The coating was carried out similarly to that of the optimized batch. Prior to administration of tablets to the rabbits for in vivo roentgenography studies the tablets were subjected to in vitro dissolution studies, to determine the intactness of the coat. Tablets were examined visually for rupturing of the coat at regular time intervals. It was observed that the coat remained intact for nearly 5 h. All rabbits were placed on overnight fasting with free access to water and were placed under 12 h light/dark cycles. After an overnight fasting, tablets were administered to rabbits with 10 ml of water. X-ray images (of abdomen) were periodically taken to trace the movement and behavior of the tablet in the GIT. In order to know the actual position of the tablet in the GIT of the rabbits, a barium meal study was conducted prior to the actual study, by administering 10 ml of standard barium meal to the rabbits. In vivo roentgenography study was performed at Mangalam Digital X-ray, Colour Doppler and Sonography Clinic (Ahmedabad, India). X-ray images were captured using a Siemens X-ray machine, with 64 MAS and 63 KV techniques. The X-ray imaging was carried out by placing the rabbits in prone position.
Stability studies
Stability studies were carried out for the optimized batch of meloxicam CoDDS by storing the tablets at 40°C ± 2°C/75% ± 5% RH for 6 months (ICH guideline; CitationMathews, 1999; CitationKrishnaiah et al., 2002). At the end of the study period the tablets were observed for change in physical appearance, color, and drug content. The tablets were also subjected to in vitro dissolution studies. The dissolution profiles of the optimized batch, subjected to the stability testing, were compared by calculating the similarity factor (f2).
Results and discussion
Optimization of matrixing and buffering agent
Core tablets of meloxicam were prepared by direct compression technique. PEO was added as a matrix forming agent to withhold the buffering agents that aid in maintaining the constant micro-environmental pH surrounding the tablets. It’s swelling property in contact with aqueous medium also helps in rupturing of the inner EC:PEG layer. Thus, swelling property of the PEO was determined using various concentrations of polymer (10–30%, batch M1 to M3, respectively). It was found that, as the amount of the PEO increases, the water absorption capacity of the polymer also increases which leads to increase in the swelling index. As shown in , tablet matrices containing 10, 20, and 30% w/w PEO showed maximum swelling for 60, 90, and 120 min, respectively. Swelling index study of batch M1 was discontinued after 60 min as tablets ruptured.
Figure 2. Percentage swelling index of batches M1 to M3 (n = 6) at pH 6.4.
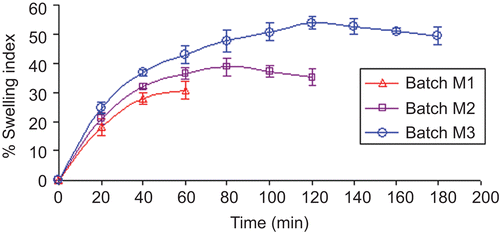
Initially to optimize the amount of PEO, batches M1 to M3 were prepared keeping the amount of buffering agent (magnesium hydroxide) constant. The cumulative percentage drug release of batches M1 to M3 (pH 6.4) at the 4th h revealed that the drug released from batch M1 containing 10% w/w PEO is low (∼ 61.81%) compared to the batches M2 and M3 containing 20% w/w (∼ 86.18%) and 30% w/w (∼ 81.17%) PEO, respectively. This might be because of inability of the tablets containing 10% w/w PEO to withstand the matrix structure, because of which buffering agent gets released in the surrounding environment and hence micro-environmental pH is not maintained, which is necessary for the pH-dependent release of meloxicam. The amount of drug released from batch M3 was lower than batch M2; due to higher swelling capacity of the tablets (). However, 100% drug release could not be achieved from batches M2 and M3 due to an insufficient amount of buffering agent. Thus, in batches M4 and M5 the amount of buffering agent was increased to 15 and 20% w/w, respectively. Since batch M3 containing 30% PEO exhibits a maximum swelling property essential for rupturing of the inner coating layer, this batch was used for further studies.
The cumulative percentage drug release of batches M4 and M5 () at the 4th h revealed that complete drug release was obtained. Since complete drug release was achieved within 4 h in batches M4 and M5, the drug release studies for all other batches, i.e. M6 to M15 was carried out for up to 4 h only for comparison purposes (). Results reveal that 15% magnesium hydroxide was optimum to achieve pH conditions for free solubility of meloxicam. This can be correlated with the buffering efficiency study shown in , which exhibited that 10% of magnesium hydroxide could not increase the pH above 7.0. Further addition of buffering agent raised the pH above 7.0, which resulted in complete drug release from the tablet.
Figure 3. Cumulative percentage drug released from batches M1 to M15 at the end of 4 h at pH 6.4.
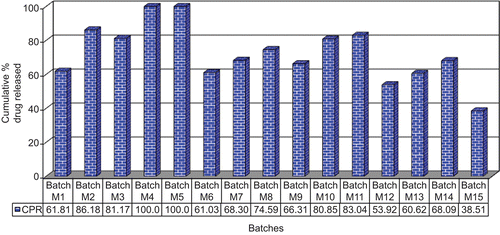
Calcium carbonate (batches M6 to M8) was studied as a water-insoluble buffering agent. The in vitro release of the batches () revealed that the increment in the drug release was observed to certain extent only, when compared to magnesium hydroxide which had exhibited a complete drug release. This can be correlated with results in which exhibit that the maximum rise in pH observed with calcium carbonate was only 6.80, which is lower than the required pH. The reason attributed could be due to the lower basic strength of calcium carbonate compared to magnesium hydroxide (CitationO’Neil, 2006).
Further, the effect of water-soluble type of buffering agents was determined by using two salts of weak acids, namely sodium carbonate and sodium citrate. Batches M9 to M14 were prepared containing 10, 15, and 20% w/w of sodium carbonate and sodium citrate, respectively (). The release of batches M9 to M14 () revealed that as the amount of sodium carbonate and sodium citrate increases the amount of drug release increases, with batches containing sodium carbonate exhibited higher release than sodium citrate. This could be due to the higher basic strength of sodium carbonate than sodium citrate (CitationO’Neil, 2006). As shown in , addition of sodium citrate to pH 6.4 phosphate buffer raises pH maximally up to 6.73, while sodium carbonate raises pH up to 8.09. Furthermore, sodium carbonate is less water-soluble than sodium citrate (solubility 1 part in 3.5 parts of water for sodium carbonate, and 1 part in 1.3 parts of water for sodium citrate at 25°C) (CitationO’Neil, 2006). Sodium citrate exhibits a faster leaching from tablets, thus leads to lower micro-environmental pH-values and results in slow meloxicam release rates. These results are in good agreement with the findings reported by CitationRiis et al. (2007).
From the dissolution profiles of the batches containing buffering agents it was observed that the release rate of meloxicam was dependent on the strength and solubility of the buffering agent. The stronger the buffering agent, the higher was the percentage drug released, and higher solubility exhibited lower percentage drug release. The low percentage drug released could also be attributed to faster leaching of water-soluble bases from the dosage form, resulting in lowering the pH of the micro environment. The faster leaching of water-soluble bases compared to water-insoluble bases showed slightly higher drug release at the initial phase of dissolution studies. Thus, batch M4 containing 15% magnesium hydroxide and 30% PEO was selected as a promising formulation for further optimization. Dissolution study of batch M15 (without buffering agent) at pH 6.4 resulted in 38.51% of drug release only, at the end of the 4th h (), which exhibited a significant influence of buffering agents on release of meloxicam.
Enteric coating of meloxicam tablets
Core tablets of meloxicam (batch M4) were enteric coated with Eudragit® FS 30D. Coatings of ∼ 10–25% w/w (batch MF1 to MF4) were applied and studied to determine the effect of concentration of Eudragit® FS 30D on drug release in the gastric fluid.
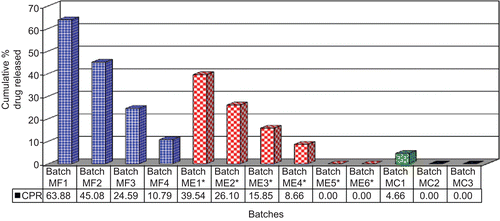
In vitro dissolution studies carried out at different pH media () exhibited that the dissolution rate was inversely proportional to the thickness of the coat. A significant difference was observed in the percentage drug released at various coating levels. At 10% w/w coating level the percentage of drug released at the end of 5th h, which is the expected time for the arrival of the dosage form in the colon, was found to be 63.88% (). Increasing the coating level from 10 to 25% w/w sequentially reduces the drug released up to 10.79% (batch MF4) (). A coating level of 10% w/w was found sufficient to impart an enteric effect. This can be explained by the fact that increasing the coat concentration made the coat more impermeable and drug release was retarded. Slowly, as the coating solubilizes, drug dissolution is facilitated (CitationPatel et al., 2009).
Figure 4. Cumulative percentage drug released from batches MF1 to MF4 (pH 1.2, 6.0, 7.2, and 6.4), ME1 to ME6 (pH 7.2), and MC1 to MC3 (pH 1.2, 6.0, 7.2, and 6.4) at the end of 5 h. (* amount of drug released after 2 h).
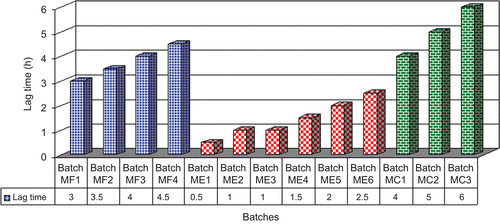
The lag time profile of batches MF1 to MF4 revealed that the lag time increases with increase in the coating level of Eudragit® FS 30D (). A coating level of 10, 15, 20, and 25% w/w showed a lag time of 3.0, 3.5, 4.0, and 4.5 h, respectively, however this lag time would not be sufficient for the tablet to reach intact to the colon. These results are not in agreement with the findings of CitationGao et al. (2006), who demonstrated that the pellets coated with 15% w/w Eudragit® FS 30D could be used for achieving colonic drug delivery. The transit time and pH of the ileum is ∼ 1.5 h (CitationHuyghebaert et al., 2005) and ≥ 7.2 (CitationNugent et al., 2001), respectively, also the threshold pH at which Eudragit® FS 30D dissolves is ≥ 7.0 (www.pharma-polymers.com). CitationGao et al. (2006) prepared meloxicam-loaded pellets, coated with 15% w/w Eudragit® FS 30D. In vitro evaluation of these pellets at pH 7.2 revealed a quick release of drug (83%) after a period of 1 h. Hence, taking into consideration the transit time and pH of the ileum, it can be concluded that the system proposed by CitationGao et al. (2006) may lead to a premature release of drug in the small intestine. It was also concluded by CitationHuyghebaert et al. (2005) that, in patients with high GI-pH profile, drug will be released in the ileum from formulations coated with Eudragit® FS 30D. These results may be correlated with reports in the literature, which demonstrate that an enteric polymer if used alone could result in a premature release of drug in the small intestine (CitationMcConnell et al., 2008; CitationPatel et al., 2009). Thus, an additional coat of EC:PEG was needed to prevent premature release of drug in the small intestine, specifically in the terminal ileum. As there was no drug release from batch MF4 at 4.5 h and also since batch MF3 showed only 7.33% drug release at the end of 4.5 h, batch MF3 was considered to be more suitable for providing an enteric effect and was used for optimizing the inner coating layer.
Figure 5. Lag time profile of batches MF1 to MF4 (pH 1.2, 6.0, 7.2, and 6.4), ME1 to ME6 (pH 7.2), and MC1 to MC3 (pH 1.2, 6.0, 7.2, and 6.4).
Delayed release coating of meloxicam tablets
Tablet cores containing meloxicam (batch M4) were coated with a mixture of EC:PEG using solutions 1, 2, and 3 at coating levels of 5 and 7.5% w/w (batches ME1 to ME6). The in vitro dissolution of batches ME1 to ME6 revealed that as the amount of EC in the mixture and the percentage coating level increases, the lag time for drug release increases. The amount of drug released from batches ME1 to ME4 at the end of 2 h (as the tablet coated with enteric polymer needs a lag time of ∼ 1–1.5 h for achieving colon targeting) was found to be 39.54, 26.10, 15.85, and 8.66, respectively, while batches ME5 and ME6 showed no drug release (). This can be explained on the basis that PEG being hydrophilic gets dissolved, which leads to the formation of pores in the coating layer that allow water to diffuse into the core tablet. The aqueous environment produced due to diffusion of water led to swelling of PEO, which ultimately resulted in rupturing of the external coat (which now comprises only of insoluble EC).
The lag time of drug release from batches ME1 to ME6 was found to increase from 0.5 h to 2.5 h, respectively (). As the tablet coated with enteric polymer needs a lag time of ∼ 1–1.5 h for achieving colon targeting and since as the batches ME2, ME3, and ME4 showed a lag time of 1, 1, and 1.5 h, respectively, these batches were selected for further optimization of CoDDS.
Formulation of time- and pH-dependent CoDDS
The CoDDS was developed by coating tablets of batches ME2, ME3, and ME4 with the enteric polymer Eudragit® FS 30D at a coating level of 20% w/w (batches MC1 to MC3). The in vitro evaluation of the batches MC1 to MC3 revealed that the amount of drug released at the end of 5 h (which is the expected time for the arrival of the dosage form in the colon) from batch MC1 was found to be 4.66%, while no drug release was observed from batches MC2 and MC3 at the end of 5 h (). Batches MC1 and MC2 showed complete drug release at the end of 10 h, while batch MC3 showed complete drug release at the end of 12 h (). It was observed that the time required for complete drug release from batches MC1 to MC3 was dependent on the amount and the composition of the inner coat applied. As batch MC3 was prepared with the highest coating level (7.5%w/w), using 70:30 ratio of EC:PEG, these batches showed maximum time for complete drug release. This can be explained by the fact that increasing the coat concentration made the coat more impermeable and drug release was retarded. Slowly as the coating solubilized, drug dissolution through it was facilitated.
Figure 6. In vitro dissolution profile of batches MC1 to MC3 (pH 1.2, 6.0, 7.2, and 6.4).
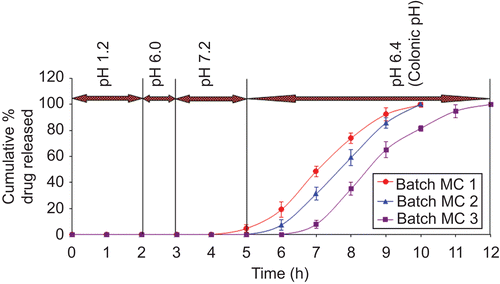
A lag time profile for drug release from batches MC1 to MC3 was found to be 4, 5, and 6 h, respectively (). As from batch MC1 a premature release of drug was observed, this formulation was not found suitable for colon targeting of meloxicam. Batch MC3 with a lag time of 6 h was not selected as the best formulation as it gives a lag time greater than that required for colon targeting, and it utilizes more coating polymer and processing time for preparation. Since the normal transit time for tablets to reach the colon was ∼ 5 h, batch MC2 was considered as a promising batch for achieving colon targeting.
From the above results we can conclude that, by keeping the coating level of Eudragit® FS 30D constant and by increasing the coat level and amount of EC, the lag time for drug release could be increased and the amount of drug release could be decreased. All the formulations could withstand the acidic pH and no drug release occurred in simulated gastric fluid.
In vivo evaluation
The performance of the CoDDS to deliver meloxicam was validated by conducting in vivo studies. The plasma-concentration vs time profile of meloxicam targeted to the colon was determined in rabbits. To support the results of the pharmacokinetic evaluation of the CoDDS, roentgenography study was also performed. Rabbits were selected as an animal model as the change in GIT pH of rabbits is similar to that of humans (pH of stomach, small intestine, and colon is reported to be 1.5–2.0, 7.2, and 6.5, respectively). Also the mean colonic arrival time in rabbits (total time: 4–6 h, stomach transit time: 2–4 h, small intestine transit time: ∼ 2 h) is almost similar to that of humans, which facilitates the in vivo evaluation of CoDDS (CitationGidenne & Lebas, 2006). As it is not advisable to administer a similar dose of drug and size of tablets for human use to the rabbits for colon targeting, a smaller size tablet (6 mm) with a corresponding dose reduction was administered to rabbits (CitationIshikawa et al., 2001; CitationGhosh, 2008; CitationVijayalakshmi et al., 2008).
Six New Zealand white rabbits weighing 3–3.5 kg were used in this study. All rabbits were placed on overnight fasting with free access to water. When optimized uncoated tablets were administered to New Zealand white rabbits (n = 3), the drug was detected in plasma at the first time point of blood sampling, i.e. at 0.5 h in all rabbits. The time to reach Cmax was found to be 5.34 ± 0.58 h (tmax). Since the half-life of meloxicam is high (15–21 h), the plasma concentration decreased very slowly and meloxicam could be quantified for 72 h after administration. shows the plasma concentration vs time profile of meloxicam in rabbits after oral administration of optimized uncoated and coated tablets. Pharmacokinetic parameters calculated from the plasma-concentration vs time profile are listed in .
Table 4. Pharmacokinetic parameters of uncoated (n = 3) and coated (n = 3) meloxicam tablets after oral administration to rabbits (each value represents the mean ± SD).
Figure 7. Plasma concentration vs time profiles of meloxicam after oral administration of uncoated and coated tablets to rabbits (mean ± SD). (←) indicates a sudden increase in the plasma concentration of meloxicam.
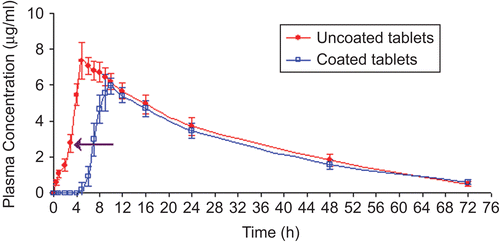
When optimized meloxicam coated tablets were administered to New Zealand white rabbits (n = 3), the onset of drug absorption was found to be significantly delayed (p < 0.01), the Tlag time was 4.67 ± 0.58 h, as well as significant (p < 0.01) delay of the tmax (9.67 ± 0.57 h) was obtained as compared with the uncoated tablets. The results are in good agreement with the in vivo roentgenography study performed in rabbits. As shown in the the colonic arrival time of tablet was ∼ 5 h, these results indicated a good correlation between the pharmacokinetic parameter, i.e. Tlag time and arrival of tablets in the colon. The drug was first detected in the plasma samples of rabbit at ∼ 4.67 ± 0.58 h (), during which the tablet was in the colon as depicted by the roentgenography study (). In addition, the AUC0→∞ and AUC0→t for coated tablets were lower than that obtained after administration of uncoated tablets (), although the difference was not significant (p > 0.01). Meloxicam released from the CoDDS might be incomplete to some extent because of considerably less water in the colon. However, considering the fact that AUC obtained after oral administration of coated tablets was not significantly decreased compared to that from the uncoated tablets, it was suggested that the coating applied had no significant influence on the relative bioavailability of meloxicam.
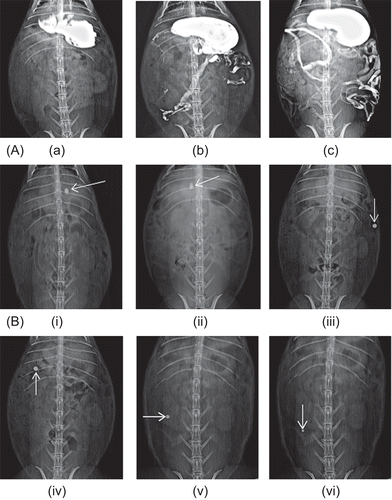
Figure 8. Roentgenography study of CoDDS in rabbits. (A) Barium meal study of rabbit GIT depicting (a) Stomach, (b) Small intestine, (c) Entire GIT along with colon. (B) Gastrointestinal transit of the colon targeted tablets in rabbits. (i) 1 h, intact tablet in stomach, (ii) 2.5 h, tablet approaching pyloric junction, (iii) 4 h, tablet intact in the small intestine, (iv) 5 h, tablet reaches in the colon, (v) 7 h, slight reduction in size of tablet in the colon, and (vi) 9 h, further reduction in size of tablet in the colon.
It was also observed that the amount of drug absorbed from the uncoated tablets was found to be 1.56 ± 0.30 μg/ml at the end of 2 h after tablet administration, while in the case of coated tablets, once the coat dissolves, the amount of drug absorbed at the end of 2 h (after a lag time of ∼ 5 h, i.e. 7 h post-dose administration), was found to be 2.96 ± 0.92 μg/ml. The reduced drug absorption from uncoated tablets compared to coated tablets might be due to poor solubility of meloxicam at the acidic pH of stomach. Also, the presence of buffering agent could not increase the solubility of meloxicam at acidic pH, since the optimization of the amount of buffering agent was carried out by using pH 6.4 (colonic pH) phosphate buffer. After 3 h of dose administration a sudden increase in the plasma concentration of meloxicam was observed (shown by arrow in ) from uncoated tablets, this might be due to increase in the solubility of the meloxicam above pH 7.0 (small intestinal pH), which leads to increase in absorption from the small intestinal region. The Cmax of uncoated tablets was found to be greater than that of the coated tablets, which revealed that the drug released from the uncoated tablets might get absorbed from the small intestinal region where the luminal pH is greater than 7.0.
Before carrying out the roentgenography study for the optimized formulation of CoDDS of meloxicam, a barium meal study was conducted to have a clear idea regarding the actual GIT of the rabbit. For this, 10 ml barium meal was administered to rabbits and X-ray images were captured at regular time intervals.
For roentgenographic evaluation, the optimized core tablet was formulated by partly replacing the drug meloxicam with radio-opaque compound barium sulfate. The coating was carried out similarly to that of the optimized batch. The results of in vitro dissolution of the optimized CoDDS containing barium sulfate were in accordance to that of the optimized meloxicam-coated tablets. The results of the roentgenography study are shown in , and the actual position of the tablet in the GIT of the rabbit could be ascertained by comparing it with the barium meal data (). represents that the tablet remains intact in the stomach and small intestine, revealing that the CoDDS could be targeted specifically to the colon, without any premature drug release in the small intestine. The time required to reach the colon was ∼ 5 h. It was observed that the size of the tablets reduces after a period of 7 h, which is a good indication of drug release in the colon. After 9 h the tablet was still detected with a reduction in the size, indicating that maximum amount (concentration) of drug was released from the tablet in the colon. This shows good agreement with the pharmacokinetic data, which revealed that the time required for achieving maximum concentration (Cmax) was 9.67 ± 0.57 h (tmax).
The in vivo test demonstrated that the CoDDS could deliver the drug to the colon successfully and highlighted the potential of this system for colonic drug delivery. Thus, we can conclude that the coated tablet remained intact in the upper part of the GIT, i.e. stomach and small intestine, and reaches the colon after a lag time of ∼ 5 h.
Stability studies
In view of the potential utility of optimized formulations for targeting meloxicam to the colon, stability studies were carried out at 40°C ± 2°C/75% ± 5% RH for 6 months (accelerated testing) to assess their long-term stability (ICH guideline, CitationMathews, 1999; CitationKrishnaiah et al., 2002). After storage, the tablets were observed for physical and color change and were subjected to drug content and in vitro drug release studies. When the optimized formulation was stored at 40°C ± 2°C/75% ± 5% RH for 6 months there appeared no change either in physical appearance, color, or in drug content (n = 6, mean ± SD) (3 months: 99.85 ± 0.53%, 6 months: 100.25 ± 0.33%). The dissolution study (n = 12) conducted in the simulated physiological environment of stomach, small intestine, and colon demonstrated no significant difference (similarity factor f2 was found to be 87.84 and 78.44 after storing for 3 months and 6 months, respectively, which is > 50) in the cumulative percentage of meloxicam released from stored tablets, when compared to the tablets of the same batch before storage. The insignificant change either in the physical appearance, color, drug content, or in dissolution profile of optimized formulation after storage indicated that developed formulation is stable and could provide a good shelf life.
Conclusions
Formulation of release modulated meloxicam CoDDS dually coated with EC containing PEG and Eudragit® FS 30D, exhibited a promising colon targeting performance during in vitro and in vivo studies. The dissolution rate of a poorly soluble acidic drug meloxicam was improved by incorporation of a buffering agent into the formulation. The in vivo pharmacokinetic study in rabbits revealed that there is a significant (p < 0.01) difference between the tmax and TLag time of uncoated and coated tablets. The Cmax and AUC parameters revealed no significant difference (p > 0.01), indicating that the coating applied did not have a critical influence on the relative bioavailability of meloxicam. The optimized batch showed a lag time of 5 h in vitro and 4.67 ± 0.58 h in vivo, demonstrating the colonic targeting of the novel pH and time dependent system. Thus, it can be concluded that the meloxicam-loaded CoDDS exhibited a promising colonic targeting and hence may be used for prophylaxis of colorectal cancer.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ahmedin, J., Rebecca, S., Elizabeth, W., Yongping, H., Jiaquan, X., Michael, J.T. (2009). Cancer Statistics. CA Cancer J Clin. 59:225–49.
- Arber, N., Levin, B. (2005). Chemoprevention of colorectal cancer: ready for routine use? Recent Results Cancer Res. 166:213–30.
- Asghar, L.F., Chandran, S. (2008). Design and evaluation of matrices of Eudragit with polycarbophil and carbopol for colon specific delivery. J Drug Target. 16:741–57.
- Ashford, M., Fell, J.T., Attwood, D., Woodhead, P.J. (1993). An in vitro investigation into the suitability of pH-dependent polymers for colonic targeting. Int J Pharm. 91:241–5.
- Ayakawa, S., Shibamoto, Y., Sugie, C., Ito, M., Ogino, H., Tomita, N., Kumagai, M., Murakami, H., Sawa, H. (2009). Antitumor effects of a cyclooxygenase-2 inhibitor, meloxicam, alone and in combination with radiation and/or 5-fluorouracil in cultured tumor cells. Mol Med Repor. 2:621–5.
- Bott, C., Rudolph, M.W., Schneide, A.R., Schirrmacher, S., Skalsky, B., Petereit, H.U., Langguth, P., Dressman, J.B., Stein, J. (2004). In vivo evaluation of a novel pH- and time-based multiunit colonic drug delivery system. Aliment Pharmacol Ther. 20:347–53.
- Churchill, L., Graham, A.G., Shih, C.K., Pauletti, D., Farina, P.R., Grob, P.M. (1996). Selective inhibition of human cyclooxygenase-2 by meloxicam. Inflammopharmacology. 4:125–35.
- DuBois, R.N., Giardiello, F.M., Smalley, W.E. (1996). Nonsteroidal anti-inflammatory drugs, eicosanoids, and colorectal cancer prevention. Gastroenterol Clin North Am. 25:773–91.
- Fukui, E., Miyamura, N., Uemura, K., Kobayashi, M. (2000). Preparation of enteric coated timed release press-coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int J Pharm. 204:7–15.
- Gang, C., Feng, A., Mei-Juan, Z., Jin, S., Xiu-Hua, H., Yun-Xia, H. (2004). Time and pH-dependent colon specific drug delivery for orally administered diclofenac sodium and 5 aminosalicylic acid. World J Gastroenterol. 10:1769–74.
- Gao, C., Huang, J., Jiao, Y., Shan, L., Liu, Y., Li, Y., Mei, X. (2006). In vitro release and in vivo absorption in beagle dogs of meloxicam from Eudragit® FS 30 D coated pellets. Int J Pharm. 322:104–12.
- Ghorab, M.M., Abdel-Salam, H.M., El-Sayad, M.A., Mekhel, M.M. (2004). Tablet formulation containing meloxicam and β-cyclodextrin: mechanical characterization and bioavailability evaluation. AAPS PharmSciTech. Available online at: http://www.aapspharmscitech.org/view.asp?art=pt050459, accessed 7 June 2008.
- Ghosh, M.N. (2008). Fundamentals of experimental pharmacology. In: Ghosh, S.K., et al., eds. Kolkata, India:1071–1076.
- Giardiello, F.M., Offerhaus, G.J.A., DuBois, R.N. (1995). The role of nonsteroidal anti-inflammatory drugs in colorectal cancer prevention. Eur J Cancer. 31:1071–6.
- Gidenne, T., Lebas, F. (2006). Feeding behavior in rabbits. In: Bels, V., ed. Feeding in domestic vertebrates: from structure to behavior. Oxfordshire: CABI Publishing, 179–94.
- Giovannucci, E., Rimm, E.B., Stampfer, M.J., Colditz, G.A., Ascherio, A., Willett, W.C. (1994). Aspirin use and the risk of colorectal cancer and adenoma in male health professionals. Ann Intern Med. 121:241–6.
- Goldman, A.P., Williams, C.S., Sheng, H., Lamps, L.W., Williams, V.P., Pairet, M., Morrow, J.D., Dubois, R.N. (1998). Meloxicam inhibits the growth of colorectal cancer cells. Carcinogenesis. 19:2195–9.
- Harris, R.E., Beebe-Donk, J., Alshafie, G.A. (2007). Cancer chemoprevention by cyclooxygenase 2 (COX-2) blockade. In: Harris, R.E., ed. Inflammation in the pathogenesis of chronic diseases: the COX-2 controversy. New York: Springer Science, 193–212.
- http://www.pharma-polymers.com, accessed 9 February 2009.
- Huyghebaert, N., Vermeire, A., Remon, J.P. (2005). In vitro evaluation of coating polymers for enteric coating and human ileal targeting. Int J Pharm. 298:26–37.
- Ishikawa, T., Koizumi, N., Mukai, B., Utoguchi, N., Fujii, M., Matsumoto, M., Endo, H., Shirotake, S., Watanabe, Y. (2001). Pharmacokinetics of acetaminophen from rapidly disintegrating compressed tablet prepared using microcrystalline cellulose (PH-M-06) and spherical sugar granules. Chem Pharm Bull. 49:230–2.
- Khan, M.Z., Prebeg, Z., Kurjaković, N. (1999). A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers. I. Manipulation of drug release using Eudragit L100-55 and Eudragit S100 combinations. J Control Rel. 58:215–22.
- Khan, M.Z.I., Helena, P.S.T., Nevenka, K. (2000). A pH-dependent colon-targeted oral drug delivery system using methacrylic acid copolymers. II. Manipulation of drug release using Eudragit® L100 and Eudragit® S100 combinations. Drug Dev Ind Pharm. 26:549–54.
- Krishnaiah, Y.S., Bhaskar Reddy, P.R., Satyanarayana, V., Karthikeyan, R.S. (2002). Studies on the development of oral colon targeted drug delivery systems for metronidazole in the treatment of amoebiasis. Int J Pharm. 236:43–55.
- Krishnamachari, Y., Madan, P., Lin, S. (2007). Development of pH- and time-dependent oral microparticles to optimize budesonide delivery to ileum and colon. Int J Pharm. 338:238–47.
- Mastiholimath, V.S., Dandagi, P.M., Jain, S.S., Gadad, A.P., Kulkarni, A.R. (2007). Time and pH dependent colon specific, pulsatile delivery of theophylline for nocturnal asthma. Int J Pharm. 328:49–56.
- Mathews, B.R. (1999). Regulatory aspects of stability testing in Europe. Drug Dev Ind Pharm. 25:831–56.
- McConnell, E.L., Short, M.D., Basit, A.W. (2008). An in vivo comparison of intestinal pH and bacteria as physiological trigger mechanisms for colonic targeting in man. J Control Rel. 130:154–60.
- Naruse, T., Nishida, Y., Hosono, K., Ishiguro, N. (2006). Meloxicam inhibits osteosarcoma growth, invasiveness and metastasis by COX-2-dependent and independent routes. Carcinogenesis. 27:584–92.
- Nugent, S.G., Kumar, D., Rampton, D.S., Evans, D.F. (2001). Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut. 48:571–7.
- O’Neil, M.J. (2006). The Merck Index: an encyclopedia of chemicals, drugs and biologicals. New Jersey: Merck and Co., INC.
- Patel, M., Shah, T., Amin, A. (2007). Therapeutic opportunities in colon specific drug delivery system. Crit Rev Ther Drug Carrier Syst. 24:147–202.
- Patel, M.M., Shah, T.J., Amin, A.F., Shah, N.N. (2009). Design, development and optimization of a novel time and pH-dependent colon targeted drug delivery system. Pharm Dev Technol. 14:62–9.
- Philip, A., Pathak, K. (2008a). Wet process-induced phase-transited drug delivery system: a means for achieving osmotics, controlled and level A IVIVC for poorly water soluble drug. Drug Dev Ind Pharm. 34:735–43.
- Philip, A.K., Pathak, K. (2008b). In situ formed phase transited drug delivery system of ketoprofen for achieving osmotic, controlled and level A in vitro in vivo correlation. Indian J Pharm Sci. 70:745–53.
- Qi, M., Wang, P., Wu, D. (2003). A novel pH- and Time-Dependent system for colonic drug delivery. Drug Dev Ind Pharm. 29:661–7.
- Ricchi, P., Zarrilli, R., Di Palma, A., Acquaviva, A.M. (2003). Nonsteroidal anti-inflammatory drugs in colorectal cancer: from prevention to therapy. Br J Cancer. 88:803–7.
- Riis, T., Bauer-Brandl, A., Wagner, T., Kranz, H. (2007). pH-independent drug release of an extremely poorly soluble weakly acidic drug from multiparticulate extended release formulations. Eur J Pharm Biopharm. 65:78–84.
- Sangalli, M.E., Maroni, A., Foppoli, A., Zema, L., Giordano, F., Gazzaniga, A. (2004). Different HPMC viscosity grades as coating agents for an oral time and/or site-controlled delivery system: a study on process parameters and in vitro performances. Eur J Pharm Sci. 22:469–76.
- Sangalli, M.E., Maroni, A., Zema, L., Busetti, C., Giordano, F., Gazzaniga, A. (2001). In vitro and in vivo evaluation of an oral system for time and/or site-specific drug delivery. J Control Rel. 73:103–10.
- Sano, H., Kawahito, Y., Wilder, R.L., Hashiramoto, A., Mukai, S., Asai, K., Kimura, S., Kato, H., Kondo, M., Hla, T. (1995). Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 55:3785–9.
- Seong-Hee, K., Choi, G.J., Lee, J.H., Yoon, A.H., Soo-Jeong, L., Kim, S.H. (2008). Differential effects of selective cyclooxygenase-2 inhibitors in inhibiting proliferation and induction of apoptosis in oral squamous cell carcinoma. Oncol Rep. 19:425–33.
- Sheng, H., Shao, J., Kirkland, S.C., Isakson, P., Coffey, R.J., Morrow, J., Beauchamp, R.D., DuBois, R.N. (1997). Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest. 99:2254–9.
- Steed, K.P., Hooper, G., Monti, N., Benedetti, M.S., Fornasini, G., Wilding, I.R. (1997). The use of pharmacoscintigraphy to focus the development strategy for a novel 5-ASA colon targeting system (“TIME CLOCK®” system). J Control Rel. 49:115–22.
- Tsubouchi, Y., Mukai, S., Kawahito, Y., Yamada, R., Kohno, M., Inoue, K., Sano, H. (2000b). Meloxicam inhibits the growth of non-small cell lung cancer. Anticancer Res. 20:2867–72.
- Tsubouchi, Y., Sano, H., Yamada, R., Hashiramoto, A., Kohno, M., Kusaka, Y., Kondo, M. (2000a). Preferential inhibition of cyclooxygenase-2 by meloxicam in human rheumatoid synoviocytes. Eur J Pharmacol. 395:255–63.
- Vijayalakshmi, P., Devi, V.K., Devi, K., Benson, M.K., Srinagesh, S. (2008). Formulation development and in vivo characterization of solubility enhanced gliclazide tablets. Curr trends Biotech Pharm. 2:456–61.
- Wei, H., Qing, D., De-Ying, C., Bai, X., Li-Fang, F. (2008). In vitro and in vivo studies of pectin/ethylcellulose film coated pellets of 5-fluorouracil for colonic targeting. J Pharm Pharmacol. 60:35–44.
- Whittle, B.J.R. (2000). COX-1 and COX-2 products in the gut: therapeutic impact of COX-2 inhibitors. Gut. 47:320–5.
- www.ich.org. Accessed 15 August 2009.
- Zhang, N., Tao, K., Huang, T. (2007). Effects of meloxicam on vascular endothelial growth factor and angiopoietin-2 expression in colon carcinoma cell line HT-29. J Huazhong Univ Sci Tech Med Sci. 27:399–402.