Abstract
This work focused on the developmental aspects, pharmacokinetic evaluation, and pharmacological assessment of a drug inclusion complex for a novel camptothecin analog (CA) and 2-hydroxypropyl-β-cyclodextrin (HP-β-CD). Camptothecins analog belong to topoisomerase-I inhibitor class of compounds with proven anti-tumor activity but exhibit poor solubility. To enhance solubility a drug inclusion complex with cyclodextrin was developed using a spray-drying process. The powder complex characterized using DSC, XRPD, FT-IR, and 1H NMR techniques confirmed interaction of cyclodextrin with the CA indicating formation of a true complex wherein the drug is encapsulated in the cyclodextrin cavity. The saturation solubility and dissolution kinetics of drug complex evaluated in a discriminating medium showed significantly higher solubility and faster dissolution as compared to a physical mixture or powder blend comprising of drug and cyclodextrin. Pharmacokinetic (PK) studies in Wistar rats indicated a significant increase in the rate and extent of absorption for the drug complex as compared to a nanoparticulate dispersion that was used as the positive control. Pharmacological activity following peroral administration of drug complex in athymic nude mice with implanted tumors revealed that the tumor inhibition activity was equivalent to commercially available intravenous (IV) formulation with comparable safety profile. These studies demonstrated for the first instance feasibility of developing a safe and efficacious peroral formulation for a sparingly soluble camptothecin analog that may provide another viable, patient compliant, and cost effective option for the treatment of solid tumors.
Introduction
Camptothecin, a plant alkaloid, is a potent anti-cancer agent acting through the inhibition of topoisomerase I during the S-phase of the cell cycle (CitationHertzberg, 1989). CA and its derivatives have shown a wide spectrum of anti-cancer activity against human malignancies including human lung, prostate, breast, colon, stomach, ovarian carcinomas, melanoma, lymphomas, and sarcomas (CitationPotmesil, 1995; CitationDancey & Eisenhauer, 1996; CitationTakimoto, 1998). These class of drugs are usually administered either by continuous infusion or using multiple time-spaced injections, both resulting in a low patient comfort and compliance. Following parenteral administration, there has been incidence of grade 3 or higher diarrhea, thus necessitating the need for development of safer, orally effective camptothecin analogs to enable administration for a prolonged duration, conveniently and cost effectively on an outpatient basis (CitationSlichenmyer, 1993; CitationDancey & Eisenhauer, 1996).
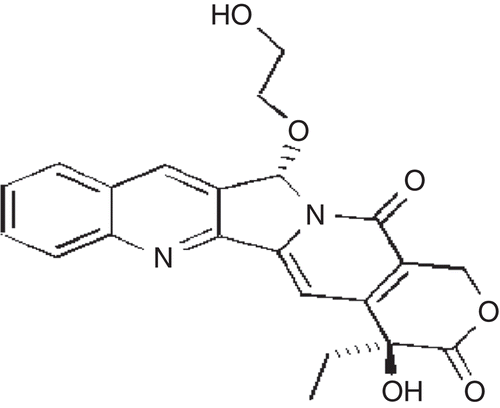
The novel camptothecin analog (CA) synthesized () for peroral administration exhibited poor wettability and low aqueous solubility. The pharmaceutical developability assessment studies indicated that the compound showed pH-dependent solubility and stability. Solubility increased in alkaline conditions (pH > 7) but at these conditions it exhibited poor chemical stability because of epimer inter-conversion (S isomer conversion to R). In solution phase the impurities increase as a function of time and temperature with pronounced conversion of S to R isomer under alkaline conditions (Patent No. W02008/148080A2).
Figure 1. Structure of CA.
There are various formulation strategies reported to enhance solubility, dissolution, and concomitantly bioavailability concerns associated with sparingly soluble compounds, these include the use of surfactants, particle size reduction including drug nanoparticles (CitationMuller et al., 2001; CitationBasa et al., 2008; CitationNekkanti et al., 2009c), solid dispersions, and self-micro-emulsifying drug delivery systems or SMEDDS (CitationNekkanti, 2009a). The majority of these approaches have been used on a case-by-case basis depending on the physico-chemical characteristics of the drug resulting in limited success. Earlier studies indicated that the drug inclusion complex with cyclodextrin is a viable approach for developing a commercially viable dosage form for sparingly soluble compounds. Currently there are ~ 30 commercialized products incorporating cyclodextrins for solubility enhancement (CitationDavis & Brewster, 2004; CitationBrewster & Loftsson, 2007).
Cyclodextrin encapsulation of a drug affects its physicochemical properties that include aqueous solubility and chemical stability. An alkylated derivative, 2-hydroxypropyl-ß-cyclodextrin (HP-ß-CD) was used because of its higher aqueous solubility, complexation ability due to its larger pore size, and better safety profile. The work detailed herein summarizes the developmental aspects, pharmacokinetic evaluation, and pharmacological assessment of the drug inclusion complex of the novel camptothecin analog and 2-hydroxypropyl-β-cyclodextrin (HP-β-CD).
Material and methods
Materials
The novel Camptothecin Analog (CA) was synthesized at Dr Reddy’s Laboratories Ltd (Hyderabad, India). 2-Hydroxy propyl-β-cyclodextrin (HP-β-CD) was purchased from Signet Laboratories (Dedham, MA), Mannitol (Pearlitol SD 200) from Signet Chemical Corporation (Mumbai, India), L-Arginine from AJI Nomoto Company., Inc. (Kanagawa, Japan), and Sodium Lauryl Sulfate (SLS) from Signet Chemical Corporation (Vanguard, NJ, USA). All other chemicals and reagents were of analytical grade. Milli Q water (Millipore, Bedford, MA) was used throughout the studies.
Phase-solubility studies
For phase-solubility studies, an excess amount of CA was mixed with aqueous solution of HP-β−CD at increasing concentrations. The samples were stirred at room temperature and an aliquot was filtered through a 0.45 µm pore size membrane filter (Millipore). The amount solubilized was determined using an HPLC method. The association constant (Ka) was determined from the slope of the linear portion of phase-solubility diagram according to equation (1) proposed by CitationHiguchi and Connors (1965).
where S0 is the aqueous solubility of CA. All experiments were carried out in triplicate. The complexation efficiency (CE) based on the drug (D):cyclodextrin (CD) ratio was determined using equations (2) and (3), as detailed below (CitationLoftson et al., 2005; Citation2007).
where [D/CD] is the concentration of drug complex, [CD] the concentration of cyclodextrin in the complex, and the slope was obtained from phase-solubility plot.
Preparation of drug cyclodextrin inclusion complex
A solution of drug–cyclodextrin complex was prepared using a laboratory-scale high shear mixer (Ultra Turrax; IKA, Wilmington, DE). The formula composition of drug complex is summarized in . In this process, CA, HP-β-CD, mannitol, SLS, and L-Arginine were dispersed in distilled water. The dispersion was stirred continuously until a clear solution was obtained. The solution was filtered using 0.45 μm pore size membrane filter (Millipore), and the filtrate was placed in the feed tank of spray-drier. A pilot-scale spray-drier (PSD01; Lab India, Mumbai, India) with a water evaporation capacity (WEC) of 5.0 kg/h was used to dry the solution feed to obtain solid intermediates. The spray-dryer was equipped with a rotary nozzle having a narrow diameter (0.5 mm) that was used for the atomization of liquid feed. The process parameters used for manufacture of spray-dried inclusion complex was optimized using design of experiments (DOE) to obtain a drug complex with pre-determined quality attributes (CitationNekkanti et al., 2009b). The drug complex was subsequently blended with excipients for capsule filling.
Table 1. Formula composition of drug complex.
Preparation of physical mixture or powder blend
CA, HP-β−CD, mannitol, SLS, and L-Arginine were separately sieved through a 60# sieve, mixed using a geometric mixing technique, and co-grinded using a planetary mono mill (Fritsch GmbH, Germany). The formula composition of powder blend is similar to the drug complex summarized in .
Differential scanning calorimetry (DSC)
Thermal properties of the drug complex, physical mixture, and CA was investigated using a Perkin-Elmer DSC-7 differential scanning calorimeter fitted with a TAC-7 thermal analysis controller and intracooler-2 cooling system (Perkin-Elmer Instruments, Massachusetts, USA). For this analysis, ~ 5 mg of sample was placed in perforated aluminum sealed 50 µL pans and the heat runs for each sample was set from 40 to 200°C at 5°C/min, an inert environment was maintained using nitrogen. The apparatus was calibrated using pure metals like Indium with known melting points and heat of fusion (ΔHfusion).
X-ray powder diffraction (XRPD)
The solid state properties of ‘as is’ drug (CA), physical mixture, and drug complex were evaluated using XRPD techniques. An Analytical Xpert Pro Diffractometer (PANalytical, Eindhoven, Netherlands) fitted with a Cu line as the source of radiation was used for analysis. The samples were run in a standard mode using a 40 kV voltage, a 40 mA current, and a scanning rate of 0.02°/min over a 2θ range of 3–40° were used.
Fourier transformed infra-red (FT-IR) spectroscopy
CA, physical mixture, HP-β-CD, and drug complex were subjected to FT-IR spectroscopic studies by potassium bromide disc method using a Perkin Elmer system 2000 FT-IR Spectrophotometer in the region of 4000 cm−1 to 400 cm−1. The data acquisition was done using spectrum software version 5.0.2 from Perkin Elmer Corporation.
Proton nuclear magnetic resonance spectroscopy (1H NMR)
1H NMR spectroscopy has been a frequently used spectroscopic analytical tool to study the inclusion of guest molecules into a host (CitationFaucci et al., 2000; CitationSun et al., 2006). In this study, we have addressed the possible inclusion/complexation of the CA in a host macrocycle (HP-β-CD) with simple 1D proton spectroscopy, combined with other complementary characterization techniques. It was possible to record proton spectra of aqueous saturated solution (in DMSO-d6) of CA–HP-β-CD. This spectrum was compared to the spectrum of an HP-β-CD solution in order to identify line shifts that could provide evidence for complexation phenomena (CitationMura et al., 1998). The 1H NMR measurements were performed using a FT-NMR spectrophotometer (Bruker, Ettlingen, Germany) operating at 500 MHz. Trimethyl Silane (TMS) was used as a reference standard.
Preparation of nanoparticulate dispersion
The nanoparticulate dispersion of the drug was prepared using a method described elsewhere (CitationMuller et al., 1999). In this process, CA was dispersed in an aqueous solution containing a specified amount (25.39% w/w) of surfactant—soya-lecithin using an Ultra Turrax stirrer T25® (IKA® - Laboratechnik, Staufen, Germany) for 3 min at 9500 rpm. The coarse dispersion was homogenized using an Avestin EmulsiFlex-B3 (Avestin, Ontario, Canada); two cycles at 2 × 104 KPa and two cycles of 5 × 104 were applied as pre-milling, and then the dispersion was processed for 15–30 cycles at 15 × 104 KPa to obtain nanoparticulate dispersion. The formula composition of nanoparticulate dispersion is summarized in .
Table 2. Formula composition of nanoparticulate dispersion.
Particle size distribution of nanoparticulate suspension
The particle size distribution of the nanoparticulate dispersion was determined using a laser diffraction method (Malvern Master Sizer 2000, Malvern Instruments, Worcestershire, United Kingdom). The particle size reported was calculated using a volume distribution. The analysis provided the mean diameter (Z-average) and polydispersity index (PI) indicative of heterogeneity in particle size.
Dissolution studies
The dissolution characteristics of physical mixture, spray-dried drug complex, and nanoparticulate dispersion of 25 mg strength were performed using a USP type II dissolution apparatus. The dissolution medium comprised of 900 mL of 0.1 N HCl (pH 1.2), that was maintained at 37°C and agitated using a paddle rotating at 75 rpm. An aliquot of 10 mL was withdrawn at pre-determined time intervals (15, 30, 45, and 60 min) and filtered through a 0.45 µm pore size membrane filter (Millipore, Bangalore, India). The amount of drug dissolved at different time intervals was determined using an HPLC method. All experiments were performed in triplicate.
Estimation of drug content using HPLC analysis
The assay of drug complex and nanoparticulate dispersion was estimated using an HPLC method. An Agilent HPLC system (Agilent, Santa Clara, CA) fitted with a Waters symmetry 5 μm C18 (250 × 4.6 mm; Waters, Los Angeles, CA) column was used for analysis. The mobile phase comprised of 0.025 M phosphate buffer (pH 6.8) and acetonitrile at a ratio of 70:30 (v/v). The flow rate was 1 mL/min, the injection volume was 10 μL, and the samples were analyzed using an UV detector at a λmax of 257 nm.
Evaluation of pharmacokinetic (PK) parameters
Male Wistar rats weighing 230 ± 20 g were fasted for 12–14 h prior to dosing, but allowed free access to water. Eight rats were assigned to two groups and they received a liquid formulation comprising either a spray-dried drug complex dissolved in sterile water or nanoparticulate dispersion, respectively, at a dose level of 5.0 mg/kg. The animals feed was returned 3 h post-dosing. Blood samples were collected from the orbital sinus at pre-determined time intervals (0, 0.5, 1.5, 3, 5, 8, 10, and 24 h) post-dosing. The plasma was separated immediately by centrifugation (13 000 rpm for 2 min at 4°C) and stored in polypropylene vials stored below −10°C until analysis. The samples were analyzed for drug content using an HPLC-Florescence method. The animal experiments were carried out in accordance with the guidelines provided by the Institutional Animal Care and Ethics Committee.
Plasma sample analysis
An aliquot of 100 µL plasma (stored at −20°C) was precipitated with 400 µL of ACM (Acidified cold Methanol) for the estimation of drug. Following mixing using a cyclo-mixer for 2 min and centrifugation for 4 min at 12,800 rpm, the clear supernatant was separated into a 300 µL auto-sampler vial and 20 µL of this was injected into ACE, 5 μm phenyl (250 × 4.6 mm; Advance Chromatography Technologies, Scotland, UK) analytical column for HPLC analysis. The plasma drug concentrations were calculated from the linearity curve plotted by spiking known concentrations of CA.
Pharmacological evaluation for anti-tumor activity
The anti-tumor activity of drug complex following peroral administration was evaluated on human tumor xenografts grown in athymic nude mice and compared with a positive control—Irinotecan Hydrochloride (commercial product) administered intravenously (IV). The formulations were evaluated on different solid tumor cell lines—NCI - H460 (human non-small cell), HCT-15 (colon), and A431 (epidermoid carcinoma). For assessment of pharmacological activity, tumor pieces measuring ~ 60 mm3 were implanted in the space between the dorsal lateral flanks of female mice to initiate tumor growth. When the tumors grew to ~ 100–300 mm3, the animals were randomized into groups of five prior to initiation of therapy. Animal care was in accordance with standard laboratory guidelines. Animals were housed in a pathogen-limited facility where ambient light was controlled automatically to produce 12-h light and dark cycles. The temperature inside the room was maintained at 23 ± 2°C. Animals were fed food and water ad libitum and used when they had achieved a pre-treatment weight of 18–23 g.
The placebo and drug complex formulations were dissolved in sterile water for peroral administration in (d × 5)2 schedules (dosing once daily for 5 days for 2 consecutive weeks followed by 9 days treatment-free period). The comparator product, Irinotecan Hydrochloride, was dissolved in 0.9% sterile saline for intravenous (IV) administration. Animals were weighed on each day of dosing and each animal was administered drug complex in solution using an oral gavage, the volume was kept constant at 10 mL per kg body weight of mice. The doses were evaluated on different solid tumor cell lines—NCI - H460 (3 mg/kg), HCT-15 (2, 4, and 8 mg/kg), and A431 (2 and 4 mg/kg). The control animals received only placebo dissolved in sterile water. All mice bearing subcutaneous tumors received the test compound and the control group received the placebo vehicle. Tumor diameters were measured twice a week using vernier calipers. Tumor volumes were calculated assuming tumors to be ellipsoid using the following formula;
where V (mm3) is tumor volume, D is longest diameter in mm, and d is shortest diameter in mm.
Changes in tumor volumes (Δ volumes) for each treated (T) and control (C) group were calculated by subtracting the mean tumor volume on the first day of treatment (starting day) from the mean tumor volume on the specified observation day. These values are used to calculate a percentage growth (%T/C) using the following formula;
%T/C = (ΔT/ΔC) × 100 where ΔT > 0 or,
= (ΔT/Ti) × 100 where ΔT < 0,
where Ti is the mean tumor volume at the start of the treatment.
Percentage tumor growth inhibition was calculated using the formula;
Percentage Tumor Growth Inhibition = 100 − %T/C.
The percentage body weight change in comparison to starting day body weight of each animal was calculated using the following formula;
Percentage Body Weight Change = [(Body weight on specified observation day − Body weight on starting day)/Body weight on starting day] × 100
The other parameters observed were mortality and clinical signs of toxicity.
Stability studies
The chemical stability and dissolution characteristics of drug complex filled in hard gelatin (HG) capsules were studied upon storage at accelerated (40°C/75%RH) and room temperature conditions (25°C/60%RH) for a period up to 3-months using a standard protocol. For the stability studies, 20 capsules of 25 mg strength were packed in a 40 cc HDPE (high density poly ethylene) container containing a molecular sieve, polystyrene filler, and induction sealed with a 33 mm child-resistant cap. The HDPE containers were loaded on stability and study monitored as per ICH conditions.
Results and discussion
The insolubility and stability of CA in most biocompatible solvents made it difficult to develop a commercially viable formulation using conventional approaches. Several delivery systems, microparticles, nanoparticles, liposomes, micelles, and mini-emulsions have been developed and investigated to overcome the solubility and stability problems of CA (CitationCortesi, 1997; CitationScenderova, 1997; CitationHatefi, 2002; CitationKunii, 2007; CitationDavis, 2009) with limited success. The use of cyclodextrins provides a feasible approach to enhance solubility and dissolution of sparingly soluble compounds like CA in the gastrointestinal (GI) fluids and concomitantly affects its bioavailability.
Phase-solubility studies
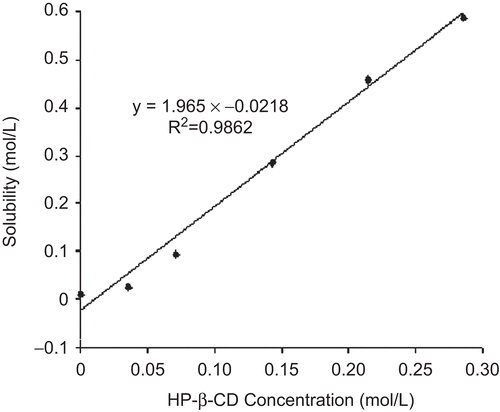
The complexation phenomenon can be described by parameters such as time to reach solubilization, association constant (ka), and complexation efficiency (CE) (CitationLoftsson et al., 2005; Citation2007). Most methods for determining the Ka values are based on the analysis of concentration dependencies, using titration experiments with the drug molecule and CD (CitationLoftsson & Brewster, 1996). There was a significant enhancement in aqueous solubility of CA in the presence of cyclodextrin that increased linearly with cyclodextrin concentration (), corresponding to an Ap-type profile as defined by CitationHiguchi and Connors (1965). This relationship suggests that the complex is first order with respect to drug and second order with respect to HP-ß-CD. The Ka value determined was 84.84 M−1 at pH 7.5, indicating formation of a stable complex (CitationLoukas et al., 1998). Using equations (2) and (3) a CE value of 2.03 was obtained which indicated that one out of every two cyclodextrin molecules form an inclusion complex with drug molecule (CA). For a drug with molecular weight of 350 Da and CD of molecular weight of 1400 Da, a CE of 0.5 will result in a 13-fold increase of the dosage bulk in the solid dosage form. Similarly, a CE of 0.1 would result in a ~ 40-fold increase in the dosage bulk (CitationBrewster & Loftsson, 2007). The high CE value for the formulated drug complex translated into reduced dosage bulk for easy processing.
Figure 2. Phase solubility plot of CA with HP-β-CD.
Differential scanning calorimetry (DSC)
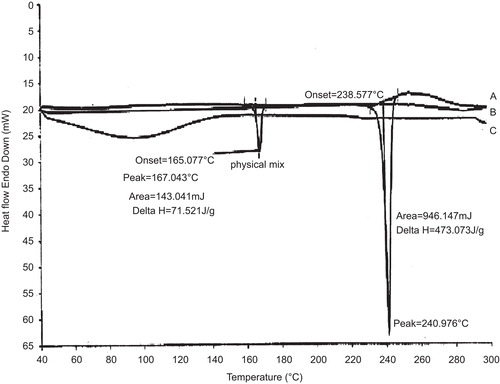
The DSC thermograms of CA, physical mixture, and drug complex are shown in . The CA has a unique behavior with a characteristic endotherm at 240.97°C. The physical mixture shows an endotherm similar to plain CA sample with less intensity. However, the drug complex completely lacks the endotherm indicating encapsulation in the cyclodextrin cavity (CitationLoukas et al., 1998).
Figure 3. DSC thermograms of A - Camptothecin Analog; B - Physical mixture and, C - drug complex.
Powder X-ray diffraction (XRPD)
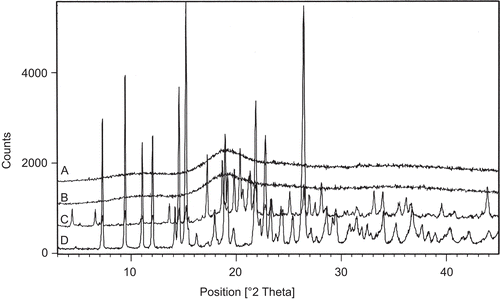
The XRPD spectra of drug complex, cyclodextrin (HP-β−CD), physical mixture, and CA shown in illustrate a significant change in the intensities of powder after complexation. The relative degree of crystallization for this reason was calculated based on the values obtained by simply subtracting the intensities of plain CA at corresponding 2θ values. Subtraction of the intensities of complex and physical mixtures from CA gives an effective picture of the effect of complex formation, as shown in . This enables us to visualize clearly the differences in crystal structure of CA and inclusion complex following complexation.
Figure 4. XRPD diffractograms for: A - drug complex; B - HP-β-CD; C - physical mixture and D - camptothecin analog.
Fourier transformed infra-red (FT-IR) spectroscopy
The FT-IR spectra of CA, HP-β-CD, physical mixture, and drug complex are shown in for comparison. The IR spectrum of CA is identified by absorption peaks at 3279 cm−1 (C-H stretch), 1743 cm−1 (Ester C=O stretch), and 1670 cm−1 (Amide C = 0 stretch). The IR spectrum of HP-β-CD shows prominent peaks at 3405 cm−1 (O-H stretch), 2929 cm−1 (C-H Stretch), and 1652 cm−1 (C=O stretch).
Figure 5. FT-IR spectrum of drug complex, HP-β-CD, physical mixture and CA.
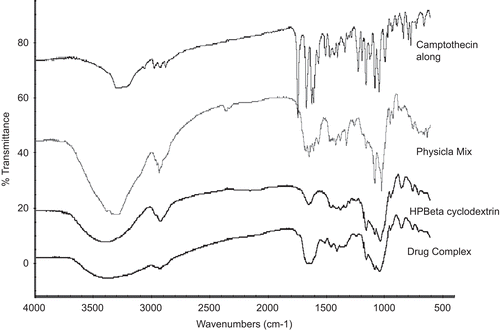
In IR spectra of physical mixture, the peaks at 1743 cm−1 and 1670 cm−1 were shifted to 1744 cm−1 and 1673 cm−1, indicating a strong physical interaction between CA and HP-β-CD. These peaks completely disappeared in IR spectra of drug complex as a result of complex formation. The broad peak of HP-β-CD at 3405 cm−1 gradually reduced in intensity for the physical mixture and drug complex, indicating an interaction between the OH groups of cyclodextrin and CA.
Proton nuclear magnetic resonance spectroscopy (1H NMR)
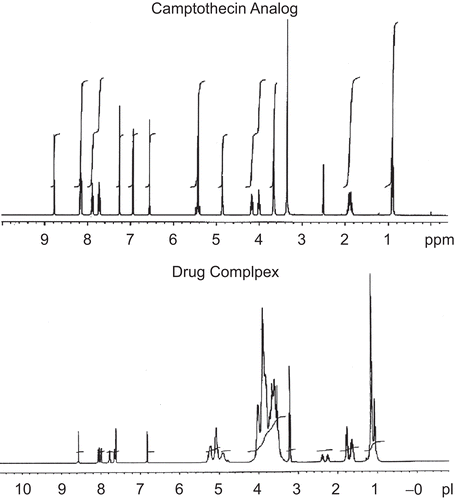
The 1H NMR spectra of CA and drug complex is shown in . The spectra show significant shifts in the protons indicating a strong inclusion complex. The delta shift values of 1H NMR spectra are summarized in . The negative delta ppm values for hydroxyl protons at 20 and 24 positions indicate a downfield shift occurring due to deshielding of the OH protons because of H bonding. The positive delta values for other drug functional groups indicates an upfield shift due to more shielding of the protons caused due to increased presence of HP-β-CD around the drug functional groups. Considering the structure of CA with four rings attached to each other with hydroxyl group at one end and nitrogen in heterocyclic ring suggests the hydroxyl moiety entering into the cyclodextrin cavity forming an inclusion complex. These observations are also supported by the results from FT-IR analysis that also confirmed the interaction of CA with the hydroxyl group of cyclodextrin in the drug complex.
Table 3. NMR delta value shift of drug complex.
Figure 6. 1HNMR spectrum of drug complex and CA.
Particle size of nanoparticulate dispersion
After 30 cycles at 15 × 104 KPa, no further decrease in particle size was observed; the final mean particle diameter is 224 ± 55 with polydispersity index 0.258 ± 0.05. The majority of drug particles following homogenization were in the submicron (nanoparticle) range, thus providing a significant increase in surface area-to-volume ratio for faster solubilization in the gastrointestinal fluids (CitationMuller et al., 2001).
Evaluation of dissolution characteristics
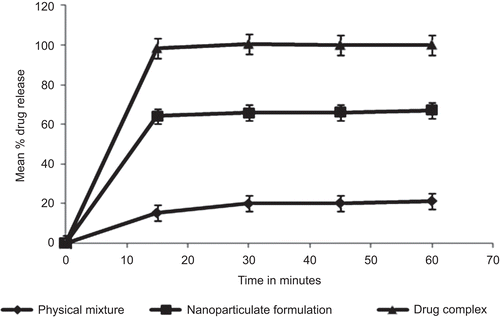
The in-vitro dissolution release kinetics of the formulated drug complex, physical mixture, and nanoparticulate dispersion in a discriminating dissolution medium (0.1 N HCl) is shown in . The faster dissolution for drug complex as compared to the physical mixture and nanoparticulate dispersion may be attributed to the formation of an inclusion complex wherein the drug is completely encapsulated in the cyclodextrin cavity. Since the drug is encapsulated in the cyclodextrin cavity, the complex behaves like a highly soluble compound and thus exhibits rapid drug release on exposure to dissolution medium.
Figure 7. Comparative dissolution profile of drug complex, physical mixture and nanoparticulate dispersion in a discriminating dissolution medium.
Evaluation of pharmacokinetic (PK) parameters
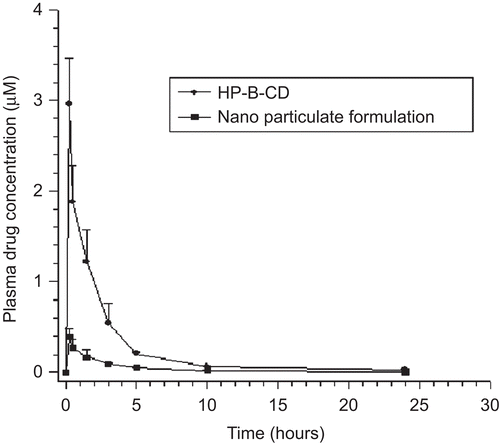
The plasma concentration–time profile after peroral administration of drug complex solution and nanoparticulate dispersion in rats is shown in . The rate and extent of drug absorption for drug complex and nanoparticulate dispersion is summarized in . The average peak plasma concentration obtained for the drug complex was 5.76 ± 0.82 as compared to 1.02 ± 0.08 µM for the nanoparticulate dispersion, indicating a 6-fold increase in the extent of absorption (AUC0–t). Similarly, there was an 8-fold increase in rate of absorption (Cmax) for drug complex as compared to the nanoparticulate dispersion (). This increase in the rate and extent of absorption for drug complex may be attributed to its higher solubility and enhanced rate of solubilization in the gastro-intestinal (GI) fluids. Based on the higher bioavailability obtained for drug complex it was evaluated for anti-tumor activity on human tumor xenograft implanted in athymic nude mice.
Table 4. In-vivo pharmacokinetic (PK) parameters following peroral administration of drug complex and nanoparticulate dispersion in Wistar rats.
Figure 8. Plasma concentration – time profiles following peroral administration of drug complex solution and nanoparticulate dispersion to Wistar rats. Each value represents the mean ± SD (n = 4).
Pharmacological evaluation for anti-tumor activity
The in-vivo anti-tumor activity of drug complex following peroral administration to NCI-H460 tumor (human non-small cell), HCT-15 tumor (colon), and A431 tumor (epidermoid carcinoma) implanted in athymic nude mice is summarized in . The results of anti-tumor activity on NCI-H460 tumor (human non-small cell) model indicated that 3 mg/kg dose of drug complex administered orally was well tolerated and inhibited tumor by 76% ± 2.72% with a mean body weight loss of 10%. In comparison, Irinotecan Hydrochloride administered intravenously at 50 mg/kg inhibited the growth of NCI-H460 tumor by 80% ± 3.58% with a mean body weight loss of 8%.
Table 5. Comparative in-vivo anti-tumor activity following peroral administration of drug complex nd intravenous (IV) administration of Irinotecan Hydrochloride in athymic nude mice with implanted tumors.
The efficacy of drug complex evaluated on HCT-15 tumor (colon) model indicated that 2, 4, and 8 mg per kg dose were well tolerated and showed a growth inhibition of 46% ± 4.24%, 62% ± 3.89%, and 70% ± 5.23%, with a mean body weight loss of 7% ± 0.83 %, 10% ± 1.25%, and 12% ± 1.78%, respectively. In comparison, Irinotecan Hydrochloride administered intravenous at 50 mg/kg, inhibited tumor growth by 60% ± 4.25%, with a mean body weight loss of 14% ± 1.98%.
The efficacy of drug complex evaluated in A431 tumor (epidermoid carcinoma) model showed that 2 mg/kg and 4 mg/kg doses were well tolerated and showed a tumor growth inhibition of A431 tumors by 38% ± 2.64% and 79% ± 1.84%, respectively, with a mean body weight loss of 0% and 15%. In comparison, Irinotecan Hydrochloride administered at 50 mg/kg intravenously inhibited the tumor growth by 88% ± 1.46%, with a mean body weight loss of 9%.
The results from these studies demonstrated that drug complex administered perorally at maximum tolerated dose (MTD) showed significant tumor regression in different tumor models (human non-small cell, colon, and epidermal carcinoma) implanted in nude mice as compared to animals in the control group. In addition, the efficacy and safety profile of drug complex administered PO at lower doses () were comparable to that obtained following intravenous administration of Irinotecan Hydrochloride (@ 50 mg/kg), the gold standard, and current treatment of care. The pharmacological activity with the drug complex following peroral administration may be attributed to its enhanced solubility and dissolution in GI fluids resulting in therapeutic drug blood level even at lower doses (2–4 mg) to elicit a therapeutic response.
Stability studies
The chemical stability and dissolution kinetics of drug complex before and after storage are summarized in . The results from these stability studies indicated a moderate increase in drug-related impurity (R-isomer) for drug product stored at accelerated (40°C/75%RH) condition after 3-months. This observed increase may be attributed to the conversion of S-isomer to R-isomer that is more pronounced at higher temperatures. However, this moderate increase in drug-related impurities at accelerated stress conditions did not impact potency and performance (dissolution profile). There was no increase in drug-related impurities or affect on drug product performance upon storage at room temperature conditions (25°C/60%RH) for a period of up to 3-months.
Table 6. Chemical stability and dissolution of drug complex filled in hard gelatin capsules following storage at accelerated and room temperature conditions.
Conclusion
A peroral dosage form for sparingly soluble CA was developed by formulating it as a drug complex. The formulation approach used herein addressed limitations of the currently marketed product that is only amenable for intravenous administration and has significant side-effects including pain, tissue damage at injection site, hemolysis, phlebitis, neutropenia, and diarrhea upon prolonged use. The drug complex following peroral administration demonstrated efficacy and safety comparable to marketed intravenous formulation on human tumor xenografts implanted in athymic nude mice. The process used for manufacture of drug complex is relatively simple and scalable. The short-term stability studies on the drug product indicated that purity, potency, and performance were unaffected upon storage. These results demonstrated the feasibility of developing a commercially viable peroral dosage form of sparingly soluble camptothecin analogs that should result in lower treatment cost, better patient compliance, and concomitantly improved therapeutic outcome for better disease management.
Acknowledgements
We acknowledge the contributions of Dr Naveen Ahuja (COE− Clinical Pharmacology and Pharmacokinetics, Dr Reddy’s Laboratories Limited, Hyderabad), Dr Khalid Ansari and Mr Ranadeep Bokalial (Integrated Product Development, Dr Reddy’s Laboratories Limited, Hyderabad) for their valuable suggestions and support.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aungst, B.J. (1993). Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J Pharm Sci. 82:979–87.
- Basa, S., Muniyappan, T., Karatgi, P., Pillai, R. (2008). Production and in vitro characterization of solid oral dosage form incorporating drug nanoparticles. Drug Dev Ind Pharm. 34:1209–18.
- Brewster, M.E., Loftsson, T. (2007). Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 59:645–66.
- Cortesi, R., Esposito, E., Maietti, A., Menegatti, E., Nastruzzi, C. (1997). Formulation study for the antitumor drug camptothecin: liposomes, micellar solutions and a microemulsion. Int J Pharm. 159:95–103.
- Dancey, J., Eisenhauer, E.A. (1996). Current perspective on campothecins in cancer treatment. Br J Cancer. 74:327–38.
- Davis, M.E. (2009). Design and development of IT-101, a cyclodextrin containing polymer conjugate of camptothecin. Adv Drug Deliv Rev. 61:1189–92.
- Davis, M.E., Brewster, M.E. (2004). Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev. 3:1023–35.
- Faucci, M.T., Melani, F., Mura, P. (2000). 1H NMR and molecular modeling techniques for the investigation of the inclusion complex of econazole with α-cyclodextrin in presence of malic acid. J Pharm Biomed Anal. 23:25–31.
- Hatefi, A., Amsden, B. (2002). Camptothecin delivery methods. Pharm Res. 19:1389–1399.
- Hertzberg, R.P., Caranfa, M.J., Hecht, S.M. (1989). On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme–DNA complex. Biochemistry. 28:4629–4638.
- Higuchi, T., Connors, K.A. (1965). Phase-solubility techniques. Adv Anal Chem Inst. 4:117–21.
- Kunii, R., Onishi, H., Machida, Y. (2007). Preparation and antitumor characteristics of PLA/(PEG–PPG–PEG) nanoparticles loaded with camptothecin. Eur J Pharm Biopharm. 67:9–17.
- Loftsson, T., Brewster, M.E. (1996). Pharmaceutical applications of cyclodextrins. Drug solubilization and stabilization. J Pharm Sci. 85:1017–25.
- Loftsson, T., Hreindóttir, D., Másson, M. (2005). Evaluation of cyclodextrins solubilization of drugs. Int J Pharm. 302:18–28.
- Loftsson, T., Hreindóttir, D., Másson, M. (2007). The complexation efficiency. J Incl Phenom Macrocycl Chem. 57:545–52.
- Loukas, Y.L., Vraka, V., Gregoriadis, G. (1998). Drugs, in cyclodextrins in liposomes: a novel approach to the chemical stability of drugs sensitive to hydrolysis. Int J Pharm. 162:137–42.
- Müller, R.H., Becker, R., Kruss, B., Peters, K. (1999). Pharmaceutical nanosuspensions for medicament administration as systems with increased saturation solubility and rate of solution. U.S. Patent 5, 858, 410.
- Muller, R.H., Jacobs, C., Kayser, O. (2001). Nanosuspensions as particulate drug formulations in therapy rationale for development and what we can expect for the future. Adv Drug Del Rev. 47:3–19.
- Mura, P., Bettinetti, G.P., Manderioli, A., Faucci, M.T., Bramanti, G., Sorrenti, M. (1998). Interactions of ketoprofen and ibuprofen with β-cyclodextrins in solution and in the solid state. Int J Pharm. 166:189–203.
- Nekkanti, V., Karatgi, P., Prabhu, R., Pillai, R. (2009a). Solid self-microemulsifying formulation for candesartan cilexetil. AAPS PharmSciTech. DOI 10.1208/s12249-009-9347-6.
- Nekkanti, V., Muniyappan, T., Karatgi, P., Hari, M., Marella, S., Pillai, R. (2009b). Spray-drying process optimization for manufacture of drug-cyclodextrin complex powder using design of experiments (DOE). Drug Dev Ind Pharm. DOI: 10.1080/03639040902882264
- Nekkanti, V., Pillai, R., Venkateswarlu, V., Harisudhan, T. (2009c). Development and characterization of solid oral dosage form incorporating candesartan nanoparticles. Pharm Dev Tech. 14:290–8.
- Potmesil, M., Pinedo, H.M. (1995). Camptothecins: new anticancer agents. Boca Raton, FL: CRC Press.
- Scenderova, A., Burke, T., Schwendeman, S. (1997). Stabilization of 10- hydroxycamptothecin in poly(lactide-co-glycolide) microsphere delivery vehicles. Pharm Res. 14(10):1406.
- Slichenmyer, W.J., Rowinsky, E.K., Donehower, R.C., Kaufmann, S.H. (1993). The current status of camptothecin analogs as antitumor agents. J Natl Cancer Inst. 85:271–291.
- Sun, D.Z., Li, L., Qiu, X.M., Liu, F., Yin, B.-L. (2006). Yin, isothermal titration calorimetry and 1H NMRstudies on host–guest interaction of paeonol and two of its isomers with β-cyclodextrin. Int J Pharm. 316:7–13.
- Takimoto, C.H., Wright, J., Arbuck, S.G. (1998). Clinical applications of the camptothecins. Biochim Biophys Acta. 1400:107–119.