Abstract
It was the aim of this study to develop a nanoparticulate oral drug delivery system for leuprolide based on polyacrylic acid (PAA). In order to achieve formation of nanoparticles in a mild, aqueous environment, two different techniques were combined, namely hydrophobic ion pairing between leuprolide and sodium dodecyl sulphate in a first step, followed by encapsulation into nanoparticles gained by interpolymer complexation between polyacrylic acid and Pluronic F68. The obtained nanoparticles were characterized regarding particle size distribution, drug encapsulation efficiency and in vitro release profile. Additionally, the pharmacokinetic profiles of leuprolide after oral administration of PAA-nanoparticulate and PAA-control tablets to male Sprague-Dawley rats were assessed and compared. It could be shown, that hydrophobic ion pairing increased encapsulation efficacy of leuprolide and leads to a slowed drug release of nanoparticulate suspensions. Relative oral bioavailability of leuprolide could be increased by nanoparticulate tablets up to 4.2-fold. Results verify that the suggested approach is a promising strategy for the design of oral delivery systems for oral administration of peptide drugs.
1. Introduction
Due to advances in the field of biotechnology, macromolecular biological drugs such as antibodies and other peptides, become more and more important and available in nowadays’ disease management. Still, their complicated way of administration via the parenteral route is a major drawback, as these drugs are usually intended for long-term treatment with patients suffering from inconvenience, pain, and risks of complications such as severe inflammation on the application site. The need for easier administration in order to benefit patient compliance is therefore strongly on demand with the oral route being the most appreciated. Therefore, high requirements have to be met by an oral drug delivery system for peptide drugs: the peptide must be protected from denaturation in the harsh acidic environment of the stomach and digestion by proteolytic enzymes. In order to reach systemic circulation an intimate contact with absorptive tissues is advantageous for uptake driven by a steep concentration gradient and longer residence time. All of those requirements can be met by formulations based on nanoparticulate systems. They can protect the encapsulated drug and provide controlled and/or prolonged drug release. It was the aim of this study to develop an oral drug delivery system for the peptide drug leuprolide, a gonadotropin-releasing hormone agonist with a molecular weight of 1209 Da, used for the treatment of hormone-responsive cancers such as prostate or breast cancer, estrogen-dependent conditions such as endometriosis or uterine fibroids, and precocious puberty. At the moment it is available as a slow-release implant or subcutaneous and intramuscular injection, but still oral application would be favourable in terms of patient compliance. Therefore, a suitable, polymeric nanoparticulate carrier system based on polyacrylic acid was established combining hydrophobic ion pairing (Citation1) of leuprolide and sodium dodecyl sulphate with nano co-precipitation of polyacrylic acid and a poloxamer block-co-polymer due to inter-polymer complexation (Citation2), which can be accomplished under mild conditions in aqueous environment. Thereby, specific interactions between polyacrylic acid and the proton accepting non-ionic polymer result in the formation of interpolymer complexes (IPC) (Citation3), based on hydrogen bonding (Citation4). Early it was already shown that precipitation of polyacrylic acid and polyethylene oxide polymers can be achieved below a critical pH value, at which carboxylic moieties are protonated, thus acting as hydrogen donors (Citation5). Hydrophobic ion pairing has already been reported to be an appropriate method for improving drug encapsulation and sustained release for leuprolide (Citation6,Citation7) and insulin (Citation8,Citation9). In contrast to the already reported studies where hydrophobic ion pairing was used in order to encapsulate hydrophilic drugs in a non-water soluble matrix under preparation in organic solvents, it was the aim of this study to develop a preparation technique employing water soluble polyacrylic acid. The gained nanoparticles were characterized by particle size distribution, zeta potential, encapsulation efficacy, drug load and in vitro drug release. For proof of concept, nanoparticles were compressed as enteric coated tablets and administered to male Sprague-Dawley rats in order to determine the dosage form’s in vivo efficacy in comparison to control tablets based on unformulated polyacrylic acid.
2. Materials and Methods
2.1. Materials
Leuprolide acetate [(Des-Gly10,D-Leu6,Pro-NHEt9)-LHRH] was purchased from Chemos, Germany. Internal standard (IS) [(Des-Gly10,D-Ala6,Pro-NHEt9)-LHRH] was obtained from Bachem AG, Switzerland. Eudragit® L 100-55 was purchased from Enovik Industries AG, Germany. Poly(acrylic acid) solution average Mw ~100 kDa (35 wt.% in H2O), Sodium dodecyl sulfate (SDS) ≥99.0%, Pluronic® F-68 (average Mw 8350 Da), ammonium acetate ≥99.99%, ethanol ≥99.5% and formic acid ≥98% were obtained from Sigma-Aldrich, St. Louis, MO. LC-MS grade water and acetonitrile were purchased from VWR International, Austria.
2.2. Methods
2.2.1. Hydrophobic ion pairing
Leuprolide acetate is readily soluble in water and therefore difficult to encapsulate into nanoparticles in aqueous media. In order to increase encapsulation efficacy, leuprolide was paired with a more hydrophobic counter ion, namely dodecyl sulphate. Therefore, a 0.55% (w/v) sodium docecyl sulphate solution in water was added to a solution of leuprolide acetate in water to give a molar ratio of 2, due to the assumption of two positive charges located one molecule of leuprolide in order to guarantee quantitative precipitation of leuprolide. The suspension of precipitated leuprolide dodecylsulfate was centrifuged for 5 minutes at 10,000 rpm, and the remaining amount of leuprolide in the supernatant was determined according to the equation via RP-HPLC analysis.
leuprolide supernatent = leuprolide total - leuprolide precipitate
2.2.2. Formation of nanoparticles
After the above described procedure and removal of the supernatant, the pellet was dissolved in ethanol containing 1% (w/v) of Pluronic F68. The resulting solution was added to a 0.5% (w/v) solution of polyacrylic acid in water at pH 3 in a weight ratio of 1:5, whereupon formation of nanoparticles occurred spontaneously. The nanosuspension was centrifuged for 5 minutes at 10,000 rpm, and the remaining amount of leuprolide in the supernatant was determined via RP-HPLC analysis in order to determine encapsulation efficacy (EE), defined as the difference between total amount of leuprolide added and the amount detected in the supernatant according to the equation:
Afterwards, nanoparticle suspensions were frozen immediately after preparation and freeze-dried at -50°C und 0.08-0.1 mbar (FreeZone 6 Liter, Benchtop Freeze Dry Systems, Labconco, USA). Nanoparticles were characterized by particle size distribution via dynamic light scattering and measurement of zeta potential (NICOMP 380™ ZLS, PSS NICOMP Particle Sizing Systems, Santa Barbara, CA). Drug load (DL) was determined after complete dissolution of a precisely weighted amount of nanoparticulate material in 0.5% sodium hydroxide solution followed by HPLC analysis, calculated according to the following formula:
2.2.3. Drug release from nanoparticle suspensions
Drug release studies were carried out with lyophylized IPC nanoparticles. Therfore, 2.5 mg of particles were suspended in 1 ml 0.5 M phosphate buffer pH 6.8 at 37°C. Aliquots of 110 µl were taken after 0, 5, 10, 15, 30, 60, 90, 120, and 180 min and immediately replaced by the same amount of buffer. The amount of released drug was determined after centrifugation of the taken samples by RP-HPLC.
2.2.4. Fabrication of tablets
Minitablets for oral application in rats were produced from lyophylized nanoparticles as test formulation and from PAA with leuprolide as control formulation. Therefore 14.3 mg of nanoparticulate material was compressed with a force of 5 kN. As control tablets 1 mg of leuprolide was mixed and ground with 13.3 mg of PAA100 and compressed as well. All tablets were enteric coated by dipping them five times into a 3% (w/v) Eudragit® L 100-55 solution in acetone. Tablets were stored at 4°C until further use.
2.2.5. Drug release from tablets
All studies were performed by placing a minitablet in 2 ml Eppendorf tubes each filled with 2 ml of release medium. According to European Pharmacopoeia 5.0 release profile from enteric coated leuprolide minitablets was first evaluated in 100 mM HCl mimicking the gastric environment for 2 h on an Eppendorf shaker at 37°C and 700 rpm in order to evaluate the stability of the formulation against gastric acidity. A control sample was drawn after 2 h of acidic incubation. Then, the medium was replaced by 150 mM phosphate buffer pH 6.8. Aliquots of 100 μL were withdrawn every 30 min up to 4 h. The withdrawn volume was substituted by release medium equilibrated to 37°C. Amount of released drug was determined by RP-HPLC.
2.2.6. Quantification of Leuprolide via RP-HPLC including reliability
Quantification of Leuprolide from in vitro experiments was performed via reversed phase HPLC on a Hitachi LaChrom Elite system equipped with a pump (L2130), autosampler (L-2200), diode array detector (L-2450-advanced) and column oven (L2300). Leuprolide was separated on a Nucleosil 100 C18 (125 × 4 mm) column equilibrated to 30°C by gradient elution from 90% eluent A (0.1% TFA)/10% eluent B (0.1% TFA in acetonitrile) to 10% A/90% B at a flow rate of 1 ml/min over 15 minutes. The method was validated regarding limits of detection and quantification, accuracy and precision, linearity and repeatability in 150 mM phosphate buffer at pH 6.8. Whereas detection limit was defined as signal-to-noise ratio of 3, quantification limit was set at 10. Linearity was assessed by measuring a calibration line ranging from 0.3 -0.00059 mg/ml with R2≥0.999. Accuracy was determined by measuring three individually prepared samples at three different concentrations (0.65, 0.325, and 0.0813 mg/ml), relating determined and theoretical concentration as follows:
Precision was determined by analyzing area under the curve (AUC) and retention time (RT) of the same sample after five repeated injections and calculating the coefficient of variation (CV) by relating mean and standard deviation (SD) according to the formula:
2.2.7. In vivo pharmacokinetic studies
The protocol for the studies on animals was approved by the Animal Ethical Committee of Vienna, Austria, and adhered to the principles of Laboratory Animal Care. Pharmacokinetic profiles of Leuprolide dosage forms were determined in male Sprague Dawley rats after oral administration of nanoparticle and control tablets in comparison to i.v. and s.c injection and oral leuprolide solution. A dose of 1 mg was administered per rat for each formulation. Composition of dosage forms are listed in detail in . Blood samples were collected from the tail vein at predetermined time points into a heparinized syringe. After centrifugation plasma was collected and stored at -80°C till analysis of leuprolide concentration via LC-MS. Therefore, an internal standard was added to withdrawn plasma samples prior to stepwise addition of 400 µl of ice-cold acetonitrile in order to precipitate plasma proteins. After centrifugation for 8 min at 12,000 rpm the supernatant was collected and evaporated to dryness (SC210A SpeedVac® Plus, coupled with RVT400 Refrigerated Vapor Trap, Thermo Savant) for 60 min at 45 °C. The residue was reconstituted in 125 μl of mobile phase for LC-MS.
Table 1. Composition of leuprolide dosage forms for in vivo studies containing 1 mg of leuprolide.
2.2.8. Quantification of Leuprolide via LC-MS
Leuprolide in blood samples from in vivo studies was performed via LC on an Agilent 1200 Series system (Agilent Technologies, Waldbrown, Germany) equipped with a G1312B SL binary pump, G1329B autosampler, vacuum degasser, and G1316B column oven. The mobile phase consisted of solvents A: 2 mM ammonium acetate with 10% acetonitrile and 0.1 % formic acid, and B: 2 mM ammonium acetate with 90% acetonitrile and 0.1% formic acid. Leuprolide and internal standard were separated on a Vydac C18 protein and peptide column (4.6 × 50 mm, 5 μm, Waters Corporation,MilfordMA, USA) using a gradient (run time 25 min) from 95 % A− 5 % B to 50 % A− 50 % B over 12 min, and 50 % A− 50 % B up to 15 min at a flow rate of 1 ml/min and injection volume of 40 μl. Mass spectrometry was performed on a Bruker MicoOTOF-Q II system operated in positive ion mode under the following conditions: end plate offset, -500 V; capillary voltage, -4500 V; nebulizer pressure, 29 psi; dry gas (nitrogen) flow rate, 6.0 L/min; dry temperature, 200°C; funnel 1 RF, 300 Vpp; funnel 2 RF, 400 Vpp; ISCID energy, 0 eV; hexapole RF, 500 Vpp; ion energy, 6.0 eV; collision energy, 100 eV; collision RF, 600 Vpp; transfer time, 85 µs; pre pulse storage, 120 µs; mass range, 50–1500 m/z. Quantification was performed in the positive mode and the extracted ion chromatogram with m/z 605.4 ± 0.5 at 14.8-14.9 min and 584.5 ± 0.5 were detected and integrated after background subtraction for the quantification of leuprolide and IS, respectively. For standards and calibration curve, stock solution of both leuprolide (1 ng/ml-100 µg/ml) and IS (75 ng/ml) were prepared in acetonitrile:water 50:50 with 0.1% of formic acid. Calibration curves (0.5 ng/ml-2000 ng/ml) were prepared in plasma by adding 20 µl of leuprolide and 20 µl of IS to 100 µl of plasma.
2.2.9. Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using OriginPro. Cmax and tmax were determined from the profiles generated by plotting the concentration of leuprolide against time. Area under the concentration time curves (AUC) was calculated according to the linear trapezoidal rule. Absolute bioavailability was calculated from the absolute dose and areas under curves (AUC) for oral against intravenous administration.
2.2.10. Statistical analysis
Statistical data analysis was performed using the student t test with p < 0.05 as the minimal level of significance unless indicated otherwise. All values were expressed as the means ± S.D.
3. Results
3.1. Hydrophobic ion pairing
Prior to nanoparticle formation leuprolide was precipitated with sodium docecyl sulphate to give a hydrophobic ion pair, which can readily be co-precipitated during nanoparticle preparation in aqueous environment. Thereby more than 99% of dissolved leuprolide could be precipitated as the corresponding hydrophobic salt, as determined by quantification of leuprolide in the supernatant via RP-HPLC after centrifugation of the precipitate. Due to quantitative precipitation, the pellet obtained after centrifugation was used directly after centrifugation without further purification.
3.2. Formation of nanoparticles
The idea of nanoparticle formation was based on interpolymer complexation due to hydrogen bonding between PAA as the hydrogen bond donor and a second polymer acting as the hydrogen bond acceptor. Therefore, PVP (Citation10,Citation11,Citation12) exhibiting a carboxylic moiety, and Pluronic F68 with polyether structure were chosen. In order to decide between the two polymers and to determine optimal ratios, titration experiments at varying ratios were performed and monitored by absorbance measurement at 400 nm. The pH was thereby set to 3, as at this pH, carboxylic acid moieties of polyacrylic acid are protonated, thus acting as hydrogen bond donor, whereas poloxamer represents the hydrogen bond acceptor. These interactions are also depicted in . Results of nanoparticle titration studies are shown In . As the plateau phase was reached sooner for Pluronic F68, PVP was abandoned for this study. For the preparation of leuprolide loaded nanoparticles, the pellet gained by hydrophobic ion pairing containing precipitated leuprolide dodecyl sulphate was dissolved in ethanol containing 1% (w/v) of Pluronic F68 and added to a 0.5% (w/v) solution of polyacrylic acid at pH 3. Drug encapsulation efficacy was calculated to be 53% (w/w), by quantifying leuprolide in the supernatant of the nanoparticle suspension after centrifugation and subsequent subtraction from the total amount of leuprolide added to polyacrylic acid solution. Drug load was determined to be 7% (w/w), by analyzing the amount of leuprolide after complete dissolution of a certain amount of lyophylized nanoparticulate material in 0.5% NaOH. Particle size distribution and zeta potential of lyoophylized nanoparticles were measured after resuspension in demineralized water. As also shown in , leuprolide loaded particles were in the size range of 200–400 nm with a zeta potential of -6 mV.
Table 2. Characterization of leuprolide loaded nanoparticles.
Figure 1. Inter polymer complexation due to formation of hydrogen bonds between polyacrylic acid (A) and co-block-polymer Pluronic (B).
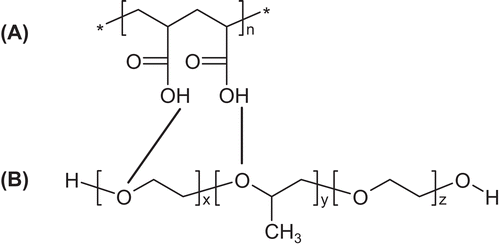
Figure 2. Absorbance monitoring during particle formation between PAA/Pluronic F68 (line) and PAA/PVP (dotted line). Abbreviations: OD (optical density).
3.3. Drug release from nanoparticle suspensions
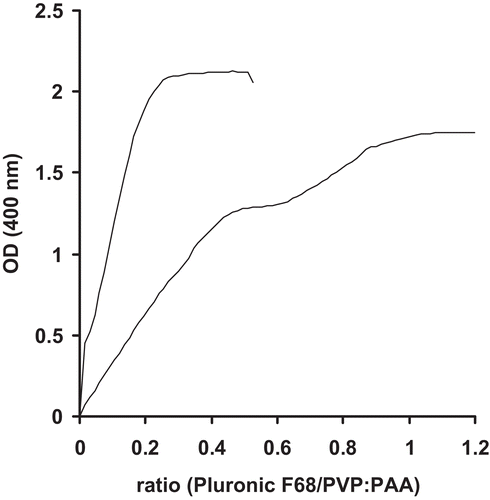
Release of leuprolide from nanoparticlulate suspensions was determined in 0.5 M phosphate buffer pH 6.8 at 37°C. Nanoparticles were found stable in 0.5 M phosphate buffer pH 6.8. as no significant change in mean particle size was observed at least after 24 hours. As illustrated in , it was shown that the drug was completely released within 4 hours, showing a sustained release of encapsulated leuprolide. The initial burst of approximately 60% can be explained by encapsulation efficacy of 50%. As the nanoparticles were not purified after formation unbound leuprolide will be detected at the beginning of the release experiments.
Figure 3. Release of leuprolide from nanoparticle suspension: Release of unbound and encapsulated leuprolide (▪) and release of encapsulated leuprolide only (▴). Indicated values represent means (± S.D.) of at least three experiments.
3.4. Drug release from tablets
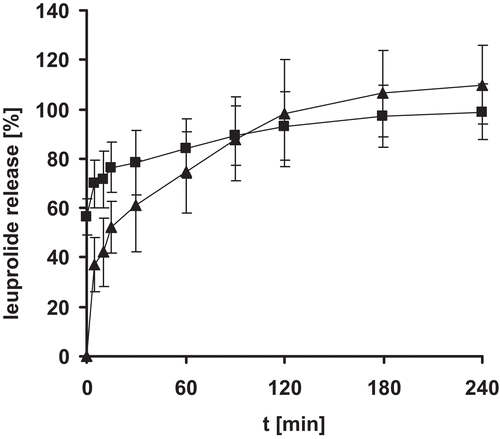
Release profile from enteric coated leuprolide nanoparticle minitablets was evaluated at 37°C in 100 mM HCl for 2 h followed by 150 mM phosphate buffer pH 6.8 for 4 h. The compressed tablets were effectively dispersed into nanoparticles with an average size of 293.1 ± 107.57 nm. From can be seen, that leuprolide was released within the first hour of incubation under intestinal conditions.
Figure 4. Release of leuprolide from nanoparticle tablets. Indicated values represent means (± S.D.) of at least three experiments.
3.5. Quantification of Leuprolide via RP-HPLC and reliability
Quantification of leuprolide from in-vitro experiments was performed by RP-HPLC. The used method was validated regarding limits of detection and quantification, accuracy and precision, linearity and repeatability in 150 mM phosphate buffer at pH 6.8. Detection limit (signal-to-noise = 3) was found to be 0.00048 mg/ml, and quantification limit (signal-to-noise = 10) 0.00144 mg/ml. Linearity criteria of a calibration line ranging from 0.3 -0.00059 mg/ml was met with R2≥0.999. Accuracy was determined by analyzing concentrations of three individually prepared samples at three different concentrations. Bias was calculated and found to be 1.63% (0.65 mg/ml), 3.90% (0.325 mg/ml), and 2.35% (0.0813 mg/ml), respectively. Precision was determined by measuring area under the curve (AUC) and retention time (RT) of the same sample after five repeated injections. The coefficient of variation served as the indicator for precision and was calculated to be 1.6 (CVAUC) and 1.3 (CVRT), respectively. According to these results the method was rendered suitable and reliable for the quantification of leuprolide.
3.6. In vivo pharmacokinetic studies
Pharmacokinetic profiles of Leuprolide dosage forms were determined in male Sprague Dawley rats after oral administration of nanoparticle and control tablets in comparison to i.v. and s.c injection and oral leuprolide solution. A dose of 1 mg was administered per rat for each formulation. Blood sampes were withdrawn from the tail vein in predetermined time points and quantified via LC-MS. Pharmacokinetic parameters and profiles are summarized in and and . Whereas maximum plasma concentrations were reaches after one hour after oral administration of leuprolide solution, maximum plasma concentrations for both tablets appear after two hours, as the drug has to be liberated from the tablets prior to absorption. Maximum plasma levels were thereby found to be 8.8 ng/ml (oral solution), 1.4 ng/ml (PAA control tablet), and 6.1 ng/ml (PAA NP tablet). Relative bioavailability of the newly developed nanoparticulate formulation could be improved 2.2-fold with an AUC of 0.025 μg*h/ml in contrast to oral solution with an AUC of 0.011 μg*h/ml. Thus tablets composed of nanoparticulate material behaved significantly better than control tablets of unformulated polyacrylic acid, namely with a 4.2-fold increase in bioavailability.
Table 3. Pharmacokinetic parameters of leuprolide dosage forms (* differs significantly from PAA control tablets with p<0.05). Abbreviations: F=absolute bioavailability, f=improvement ratio.
Figure 5. Plasma leuprolide levels [ng/ml] after administration of 1 mg of leuprolide to rats: PAA tablets (□), PAA NP tablets (▪), solution (Δ). Indicated values represent means of at least three experiments. Indicated values represent means (± S.D.) of at least three experiments. 1 differs from 2 and 3 p = 0.0001.
![Figure 5. Plasma leuprolide levels [ng/ml] after administration of 1 mg of leuprolide to rats: PAA tablets (□), PAA NP tablets (▪), solution (Δ). Indicated values represent means of at least three experiments. Indicated values represent means (± S.D.) of at least three experiments. 1 differs from 2 and 3 p = 0.0001.](/cms/asset/894883bb-da18-4dd6-a03a-58ebd1db442a/idrd_a_577108_f0005_b.gif)
Figure 6. Plasma leuprolide levels [μg/ml] after intravenous (▪) and subcutaneous (□) administration of 1 mg of leuprolide to rats. Indicated values represent means (± S.D.) of at least three experiments. 1 differs from 2 p = 0.0001.
![Figure 6. Plasma leuprolide levels [μg/ml] after intravenous (▪) and subcutaneous (□) administration of 1 mg of leuprolide to rats. Indicated values represent means (± S.D.) of at least three experiments. 1 differs from 2 p = 0.0001.](/cms/asset/9d402708-7592-4cdc-86e9-9d56908b1ffc/idrd_a_577108_f0006_b.gif)
4. Discussion
Delivery of peptide drugs still remains a challenge in pharmaceutical technology. Although most of marketed products have to be administered via parenteral routes such as subcutaneous injection, oral application would be highly beneficial not only for patient compliance, but also for lowering risks of injection complications such as pain and inflammation. Leuprolide is one of those peptide drugs, intended for long term treatment of hormone-dependent diseases in form of subcutaneous or intramuscular injection and slow-release implant. A newly developed delivery system based on mucoadhesive polymeric nanoparticles for oral delivery of leuprolide was therefore evaluated regarding in vitro drug release and in vivo pharmacokinetics in rats. Leuprolide is available as acetate salt, which is well soluble in water. This was the major obstacle met at the beginning of experiments, as it was not possible to encapsulate the soluble form of leuprolide during nanoparticle preparation in aqueous media. In order to reduce aqueous solubility of leuprolide acetate as the key parameter responsible for poor drug loading into nanoparticles and burst release, hydrophobic ion pairing was chosen to overcome these obstacles. This process involves stochiometric replacement of the polar acetate counter ion by an ionic detergent of the same charge (Citation1). In preliminary precipitation experiments, sodium dodecyl sulphate was identified as a suitable counter ion for quantitative ion pairing of leuprolide. Interactions in the presence of low SDS levels at stochiometric ratios differ from those in the presence of high SDS concentrations, as known for example from electrophoresis. At higher concentrations resolubilization of the corresponding peptide can be observed, most likely due to formation of micelles. This phenomenon has already been shown for interleukin-2: whereas no or only little precipitation was observed at SDS concentrations below stochiometric levels, concentrations above the critical micelle concentration (CMC) of SDS led to resolubilization of the precipitate gained by a stochiometric ratio (Citation13). Due to its increased lipophilicity it is assumed that leuprolide dodecyl sulphate dissolved in ethanol can be associated with nanoparticles during nanoprecipitation more readily. In addition, it should prevent the drug from being released in a burst manner and even improve its uptake and pharmacokinetics, as was already postulated elsewhere (Citation14). However, as HIP complexes are also susceptible to dissociation in the presence of further polar counter ions, general transport enhancement cannot be confirmed (Citation15,Citation16). Within the present study, it is suggested that leuprolide exhibits two positively charged basic amino acid residues when dissolved in water: arginine, containing a strongly basic guanidine function, and histidine with a weakly basic imodazole moiety. Its chemical structure is shown in .
Figure 7. Chemical structure of leuprolide.
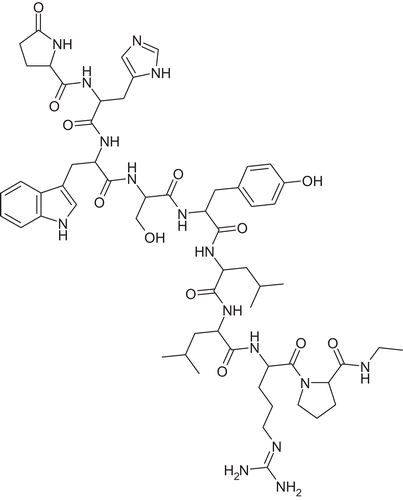
Below the peptides iso-electric point, both moieties will be protonated, and leuprolide will interact with sodium dodecyl sulphate in a stochiometric ratio of 1:2. Due to the formation of the hydrophobic ion pair, encapsulation efficacy could be raised to 53%, whereas leuprolide could not be encapsulated at all in form of the acetate salt. Nanoparticle formation via complexation was achieved by hydrogen bonding between the two used polymers, polyacrylic acid and Pluronic F68. The latter is block-co-polymer, consisting of a polypropylene oxide unit flanked by polyethylene oxide units (80%), the hydrophobic part exhibiting a molecular weight of 1800. As the pH at the time of nanoparticle preparation was kept below the pKa value of polyacrylic acid (4.75), the majority of carboxylic moieties are protonated and available as hydrogen donors. Increase of pH during intestinal transit will cause the system to disintegrate and release the incorporated drug. In vitro release studies of leuprolide from nanoparticles in suspensions at physiological intestinal pH of 6.8 indicate a sustained release of encapsulated leuprolide togehter with a burst of around 60% of leuprolide within the first minutes of incubation. This can be explained by the ratio of encapsulated to total leuprolide added during particle formation: encapsulation efficacy of 53% was reached, which means, around half of the amount of leuprolide remains dissolved. A purification step was omitted, due to the assumption that release of encapsulated leuprolide will already take place during procedures such as dialysis. The dissolved amount is thus detected in release experiments in the beginning. The total amount of encapsulated leuprolide was released from nanoparticulate suspensions within 4 hours of incubation, whereas release from tablets was found to be 80% of the initial amount. The loss can be explained by unintended release of leuprolide during incubation of tablets in simulated gastric juice (100 mM HCl) due to imperfect enteric coating with Eudragit®. Therefore, it has not been taken into account for evaluation of in vivo data.
Results from in vivo pharmacokinetic studies indicate a 2.2-fold improved oral bioavailability of leuprolide when administered in form of nanoparticulate tablets in comparison to leuprolide solution, and a 4.2-fold improvement in contrast to unmodified polyacrylic acid control tablets. This can be due to the advances offered by nanoparticulate formulations such as protection of the drug towards degradation and enzymatic cleavage. The major obstacle for orally administered peptide drugs is the enzymatic barrier before reaching the enterocytes as the site of absorption. It is therefore the aim to develop formulations which can on one hand protect the drug from degradation and the other hand enhance its absorption. Nanoparticulate delivery systems can isolate the encapsulated drug from the gastrointestinal environment and deliver them to the epithelium in high concentrations. One of the possible uptake sites of nanoparticles is assumed to be the so-called Peyer’s patches, an accumulation of lymphoid follicles located below the gastrointestinal epithelium comprised mainly of enterocytes, M cells and few mucus secreting goblet cells. As M cells are responsible for uptake and presenting of antigens form the gastrointestinal lumen, the patches were an interesting target for mucosal vaccination. Apart from Peyer’s patches, which were thought to be the only site of nanoparticle absorption for a long time, other absorption areas have been identified and documented (Citation17). In order to deliver the drug to the absorption site intact and in high concentrations, the use of nanoparticulate formulations based on mucoadhesive polymers, such as polyacrylic acid, is an interesting approach. The polymer’s mucoadhesive properties play an important role, as the delivery system can provide an intimate contact with the absorptive tissue, thus providing a steep concentration gradient of the incorporated drug, driving its absorption. It has also been reported that systems gained by chemical combination of pluronics and polyacrylic acid exhibit enhanced adhesive properties (Citation18). In addition to prolonged residence time of the drug delivery system on mucosal surfaces, nanoparticles provide a large surface area to be exploited for contact with the epithelium. Further, negative zeta potential of nanoparticles might contribute to permeation enhancement (Citation19).
5. Conclusion
Within the present study a suitable oral drug delivery system based on nanoparticles formed by interpolymer complexation based on hydrogen bonding between polyacrylic acid and poloxamer F68 for leuprolide, a peptide drug used for therapy of hormone-responsive cancers such as prostate or breast cancer, endometriosis or uterine fibroids, and precocious puberty, was developed. In order to improve encapsulation efficacy, leuprolide acetate was hydrophobically modified by substituting acetate with dodecyl sulphate via hydrophobic ion pairing. The delivery system was characterized in vitro regarding size distribution, zeta potential and drug release profile, with findings that encapsulated leuprolide was released in a sustained manner. Further the developed formulation was tested in vivo in rats in form of enteric coated tablets, and bioavailability was compared to intravenous and subcutaneous administration as well as oral administration of leuprolide solution and unmodified polyacrylic acid tablets. Bioavailability was thereby found to be improved more than 2-fold in case of nanoparticulate tablets versus oral leuprolide solution, and even 4.2-fold in comparison to control polyacrylic acid tablets.
Declaration of interest
The authors report no declaration of interest.
References
- Meyer JD, Manning MC. (1998). Hydrophobic ion pairing: altering the solubility properties of biomolecules. Pharm Res, 15, 188–93.
- Khutoryanskiy VV (2007). Hydrogen-bonded interpolymer complexes as materials for pharmaceutical applications. Int J Pharm, 334, 15–26.
- Khutoryanskiy VV, Dubolazov AV, Nurkeeva ZS, Mun GA. (2004). PH effects in the complex formation and blending of poly(acrylic acid) with poly(ethylene oxide). Langmuir, 20, 3785–90.
- Bailey FE, Lundberg RD, Callard RW. (1964). Some factors affecting the molecular association of poly(ethylene oxide) and poly(acrylic acid) in aqueous solution. J Polym Sci Part A, 2, 845–51.
- Ikawa T, Abe K., Honda K, Tsuchida E. (1975). Interpolymer complex between poly (ethylene oxide) and poly (acrylic acid). J Polym Sci Chem A, 13, 1505–14.
- Choi SH, Park TG. (2000). Hydrophobic ion pair formation between leuprolide and sodium oleate for sustained release from biodegradable polymeric microspheres. Int J Pharm, 203, 193–202.
- Yuan H, Jiang SP, Du YZ, Miao J, Zhang XG, Hu FQ. (2009). Strategic approaches for improving entrapment of hydrophilic peptide drugs by lipid nanoparticles - Colloids Surf. B. Biointerfaces, 70, 248–53.
- Shi K, Cui F, Yamamoto H, Kawashima Y. (2008). Investigation of drug loading and in vitro release mechanisms of insulin-lauryl sulfate complex loaded PLGA nanoparticles. Pharmazie, 63, 866–71.
- Shi K, Cui F, Yamamoto H, Kawashima Y. (2008). Optimized preparation of insulin-lauryl sulfate complex loaded poly (lactide-co-glycolide) nanoparticles using response surface methodology. Pharmazie, 63, 721–25.
- Deutel B, Greindl M, Thaurer M, Bernkop-Schnurch A. (2008). Novel insulin thiomer nanoparticles: in vivo evaluation of an oral drug delivery system. Biomacromolecules, 9, 278–85.
- Perera G, Greindl M, Palmberger TF, Bernkop-Schnurch A. (2009). Insulin-loaded poly(acrylic acid)-cysteine nanoparticles: stability studies towards digestive enzymes of the intestine. Drug Deliv, 16, 254–60.
- Chun MK, Cho CS, Choi HK. (2002). Mucoadhesive drug carrier based on interpolymer complex of poly(vinyl pyrrolidone) and poly(acrylic acid) prepared by template polymerization. J Control Release, 81, 327–34.
- Arakawa T, Philo J, Kenney WC. (1994). Structure and solubility of interleukin-2 in sodium dodecyl sulfate. Int J Pept Protein Res, 43, 583–87.
- Aungst BJ, Hussain MA. (1992). Sustained propranolol delivery and increased oral bioavailability in dogs given a propranolol Laurate salt. Pharm Res, 9, 1507–09.
- Mcshane MM, Kruske CA, Davio SR, Wilkinson KF, Rush BD, Ruwart MJ. (1994). The effect of aqueous solubility on the absorption of peptide-like drugs from the subcutaneous (SC) site. Pharm Res, 11, S–246.
- Trimble K, Wan J, Floy B. (1993). Effects of ion pairing agents on partition coefficient, tissue uptake, and membrane permeability of LHRH analogues - Pharm Res, 10, S–178.
- Delie F, Blanco-Prieto MJ. (2005). Polymeric particulates to improve oral bioavailability of peptide drugs. Molecules. 10, 65–80.
- Chun MK, Cho CS, Choi HK. (2001). A novel mucoadhesive polymer prepared by template polymerization of acrylic acid in the presence of poloxamer. J App Polym Sci, 79, 1525–30.
- Younessi P, Avadi MR, Shamimi K, Sadeghi AM, Moezi L, Nahid E, Bayati K, Dehpour AR, Rafiee-Tehrani M. (2004). Preparation and ex vivo evaluation of TEC as an absorption enhancer for poorly absorbable compounds in colon specific drug delivery. Acta Pharm, 54, 339–45.