Abstract
Chemotherapy in treatment of malignant tumors has many side effects due to the poor physiochemical properties and the toxicity to normal tissues. The dual-targeting drug delivery system combining two high-affinity ligands can target anticancer drug primary to the diseased tissue, then to the tumor, which provides both greater efficacy of treatment and less harm to normal tissues. In this paper, a novel dual-targeting moiety RGD7 (R-G-D-D-D-D-D-D-D; Nonapeptide for bone cancer combining D6 peptide as bone target moiety and RGD peptide as tumors target moiety was contracted. A series of bone and/or tumor targeting conjugates have been synthesized in a convergent approach and well characterized by nuclear magnetic resonance (NMR) and mass spectrometry (MS) techniques. The hydroxyapatite (HAP) binding, water solubility, the drug release and the distribution in vivo were evaluated. All the conjugates were water-soluble and able to release the parent drugs in vitro. The bone-targeting property of the dual-targeting delivery system was enhanced from the results of the HAP binding and the distribution in vivo. The experiment for verifying tumor targeting property was underway. These results provided an effective entry to the development of a new dual-targeting delivery system for bone cancer.
Keywords::
Introduction
Chemotherapy is essential in the treatment of malignant tumors; however, its efficacy is usually limited due to the poor physiochemical properties, low stability, short circulating half-life, and the toxicity to normal tissues associated with the antitumor drugs (CitationXu et al., 2012).
For meeting drug delivery needs, targeting delivery has been investigated for many years. Recently, dual-targeting delivery has been widely developed and played an important role in a variety of areas (CitationAlpizar et al., 2011; CitationPetkau et al., 2011; CitationYogev et al. 2011; CitationJackson et al., 2012), including cancer therapy (CitationShao et al., 2012; CitationXu et al., 2012). Which primary usually delivered anticancer drug to the diseased tissue, then to the tumors with some targeting moieties (CitationPetkau et al., 2011). For example, the conjugate of doxorubicin with multi-walled carbon nanotubes as a brain targeting agent and folic acid (FA) targeting as a tumor targeting agent had dual-targeting function (CitationLu et al., 2012). In addition, the conjugate of paclitaxel (PTX) with cyclic RGD and transferrin conjugated nanoparticles also had dual-targeting function (CitationXu et al., 2012).
In the treatment of bone tumor, there are many reports about the conjugates of antidrugs based on one bone-targeting moiety (tetracycline, bisphosphonate, oligopeptide, alendronate (ALN)). Such as the conjugation of ALN and the PTX with HPMA Copolymer was only based on alendronate for a bone targeting agent (CitationMiller et al., 2011). However, there are few reports about dual-targeting drug delivery used in the treatment for bone tumors. Based on the advantages of dual-targeting, a dual-targeting moiety combining bone-targeting and tumor-targeting moieties may be more effective for the drug delivery to bone tumor.
Over the years, our group has done a lot of researches on bone targeting and cancer targeting moieties specially based on peptides by active process (CitationOuyang et al., 2009; CitationOuyang et al., 2011). The advantageous property of peptides such as biocompatibility, non-cytotoxicity, and enzymatic degradation can make prodrugs more biocompatible and no unexpected long-term effects (CitationOuyang et al., 2011; CitationPan et al., 2012).
Therefore, for seeking a more effective bone tumor targeting moiety, a novel dual-targeting moiety RGD7 was designed in this manuscript, which combined D6 (L-Asp hexapeptide; Asp6) as bone-targeting moiety and RGD (arginine-glycine-aspartic tripeptide; Arg-Gly-Asp) as tumor targeting moiety.
The D6 (L-Asp hexapeptide; Asp6) is popular used in the treatment of bone diseases such as osteoporosis and osteomyelitis as well-known bone-targeting moiety, due to its remarkable affinity to hydroxyapatite (HAP), which is an inorganic component in hard tissues (bone and teeth), no in soft tissues (CitationTakahashi et al., 2008; CitationMiller et al., 2009). Moreover, unlike the traditional bone-targeting moieties, the D6 is biologically labile and enzymatically degradable, thus, it could alter the pharmacokinetic and biological properties, including the blood clearance, distribution and biological activity (CitationTakahashi-Nishioka et al., 2008). In that case, the conjugate modified with D6 could deliver drug to bone effectively as desired and be more biocompatible.
Among all the neoplastic targeting ligands that are presently under investigation, the RGD sequence is a popular one used in drug delivery systems (CitationByrne et al., 2008). The RGD sequence, an arginine-glycine-aspartic (Arg-Gly-Asp) tripeptide, has been identified as a high affinity αvβ3 selective ligand (CitationRay et al., 2011). The αvβ3, one of the integrin receptor family, is proved to be overexpressed on the angiogenic endothelium in malignant or diseased tissues (CitationTemming et al., 2007; CitationBarczyk et al., 2010), and it is also an important marker of blood vessels in the most malignant tumors, all of which make it an attractive target for antitumors strategy (CitationMurphy et al., 2008). As a result, the conjugate modified with RGD demonstrates extraordinary potential to target drug to the tumors (CitationRay et al., 2011; CitationXu et al., 2012). In addition, the αvβ3 is also highly expressed in bone metastatic cells and osteoclasts, so that the αvβ3 integrin is also an attractive target for bone (CitationTeti et al., 2002). Therefore, the RGD moiety may also have the property of bone targeting.
In present work, we aim to construct a dual-targeting moiety RGD7 combining two targeting ligands D6 and RGD. RGD7 is hoping to obtain the ability of dual targeting. Then the side effects on normal tissues and chemotherapeutic drugs administration dose could be reduced. And it can represent a novel opportunity to apply the versatile dual-targeting delivery system to applications in bone tumor treatment.
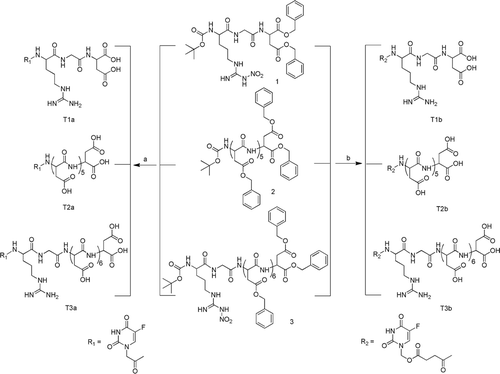
In present paper, a well-known broad spectrum anticancer drug 5-fluorouracil (5-FU) was chosen as a model drug, and modified with two linkers for exploring better release of 5-FU. The dual-targeting prodrug 5FU-linker-RGD7 was be synthesized and characterized, the same to 5FU-linker-D6 and 5FU-linker-RGD, which were used to contrast with 5FU-linker-RGD7. The full protected D6, RGD and RGD7 were shown in . A series of in vitro and in vivo evaluations of these conjugates including the HAP binding, water solubility, the drug release and the distribution in vivo were also reported, which could verify whether the dual-targeting delivery system could target to bone. The animal models with bone cancer were established, and the experiment used to verify the tumor targeting property of the dual-targeting delivery system is underway.
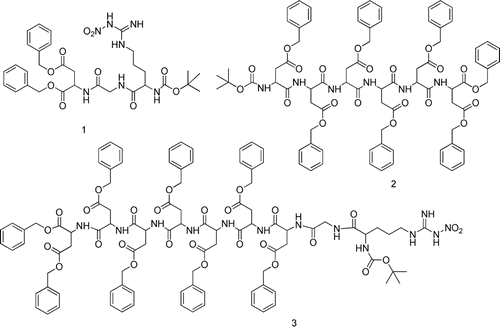
Figure 1. Structure of the fully protected RGD, D6, RGD7 (1–3).
Experimental
General: All starting materials, unless otherwise specified, were used as received. All reactions requiring anhydrous conditions were performed under an Ar or N2 atmosphere. Chemicals and solvents were either A.R. grade or purified by standard techniques. Thin layer chromatography: silica gel plates GF254; compounds were visualized by irradiation with UV light and/or by treatment with a solution of phosphomolybdic acid (20% wt. in ethanol) followed by heating. Column chromatography was performed by using silica gel with eluent given in parentheses. 1H-NMR analysis was performed using CDCl3, d6-DMSO or D2O as a solvent at room temperature. The chemical shifts are expressed in relative to TMS (= 0 ppm) and the coupling constants J in Hz. HAP were purchased from Shanghai Institute of Biochemistry with surface area 10 m2/g and average particle size 30 µm.
Chemistry
Synthetic procedure for compounds 1 and 2
The compounds 1 and 2 were synthesized according to the literature methods of our group’s work previously (CitationHe et al., 2006; CitationOuyang et al., 2009).
Synthetic procedure for compound 4
5-FU (1.95 g, 15 mmol) was dissolved in 10 mL aqueous solution of KOH (3.20 g, 57 mmol), then 5 mL aqueous solution of bromoacetic acid (3.1 g, 22 mmol) was added slowly under the temperature of 40°C while stirred smoothly. The reaction mixture was stirred for 2 h under the temperature of 40°C and then cooled by ice-bath, adjusted to pH 2 with hydrochloric acid, after filtration through a membrane, the crude product was re-crystallized by water, and compound 4 was obtained as white solid 2.14 g. Yield 76%, m.p. 253–255°C.
Data
1H-NMR (400 MHz, DMSO): δ = 4.39 (s, 2H, N-CH2), 8.07 (d, 1H, J = 6.0 Hz, 5-FU-CH), electrospray ionization-mass spectrometry (ESI-MS) (m/z): calcd. for 188.11. obsd. 189.07 ([M + H]+).
Synthetic procedure for compounds 5 and 6
The compounds 5 and 6 were synthesized according to the literature methods of our group’s work previously (CitationOuyang et al., 2011).
Data
Compounds 6: ESI-MS (m/z): calcd. for 260.04. obsd. 285.1 ([M + Na]+).
Synthetic procedure for compound 7
To the solution of trifluoroacetic acid (TFA)/anhydrous CH2Cl2 (1:1, v/v), 2 (1.44 g, 1 mmol) was added at room temperature under N2. When the protecting groups were completely removed, the solvent was rotary evaporated. (Yield: 99%). The residue dissolved in Tetrahydrofuran THF with N-methyl-morpholine (NMM) (0.11 mL, 1 mmol) was added to the solution of Boc-Asp (OBzl)-OH (0.32 g, 1 mmol) in anhydrous THF, when which had stirred for 10min after NMM (0.11 mL, 1 mmol) and isobutyl chloroformate (IBCF) (0.13 mL, 1 mmol) had been added at −10°C. The mixture was stirred for 3 h. After evaporated under reduced pressure, the residue was taken up in ethyl acetate and washed with 1 M HCl, 1 M NaHCO3, and brine each for twice. The organic layer was dried and evaporated to give the crude product. The crude product was purified by silica gel column chromatography using CHCl2 and CH3OH as an eluent to yield a white wax (1.22 g). Yield: 74% in two steps (Compound 7, fully protected D7).
Synthetic procedure for compound 8
According to the same procedure of preparation 7, the compound 8 (0.83 g, white wax) could be prepared from 7 (1.15 g, 0.7 mmol) and Boc-Gly-OH (0.12g, 0.7 mmol). Yield: 70% in two steps.
Synthetic procedure for compound 3
Similarly, according to the same procedure of preparation 7, the compound 3 (0.49 g, white wax) could be prepared from 8 (0.68 g, 0.4 mmol) and Boc-Arg(NO2)-OH (0.13 g, 0.4 mmol). Yield: 64% in two steps.
Data
1H-NMR (400 MHz,d6-DMSO): δ = 1.35 (s, 9H, Boc), 1.48–1.64 (m, 4H, Arg-β,γH), 2.50–2.55 (m, 2H, Arg-δH), 2.57–2.87 (m, 14H, Asp-βH), 3.70–3.71(br s,2H, Gly-αH), 3.92–3.93 (m, 1H, Arg-αH), 4.60–4.70 (m, 7H, Arg-αH), 5.04 (s, 16H, (Ph-CH2) × 8), 7.30–7.32 (m, 40H, C6H5 × 8). ESI-MS (m/z): calcd. for 1901.73, obsd. 1924.76 ([M + Na]+).
Synthetic procedure for compound T1a
To the solution of TFA/anhydrous CH2Cl2 (1:1, v/v), the compound 1 (0.34 g, 0.5 mmol) was added at room temperature under N2. When the protecting groups were completely removed, the solvent was rotary evaporated. The residue dissolved in THF with NMM (0.05 mL, 0.5 mmol) was added to the solution of compound 4 (0.10 g, 0.5 mmol) in anhydrous DMF, when which had stirred for 20 min after NMM (0.05 mL, 0.5 mmol) and IBCF (0.07 mL, 0.5 mmol) had been added at −10°C. The mixture was stirred for 3 h. After evaporated under reduced pressure, the residue was taken up in ethyl acetate and washed with 1 M HCl, 1 M NaHCO3, and brine each for twice. The organic layer was dried and evaporated to give the crude product. The crude product was purified by silica column chromatography using CHCl2 and CH3OH as an eluent, then deprotected the protecting groups with Pd catalyst by hydrogen in CH3OH at room temperature to afford T1a 0.16 g, yield 64% as white wax.
Data
1H-NMR (400 MHz,D2O): δ = 1.75–1.85 (m, 2H, Arg-γH), 1.86–1.94 (m, 2H, Arg-βH), 2.82–2.87 (m, 2H, Arg-δH), 2.89–2.91 (m, 2H, Asp-βH), 3.99 (s, 2H, Gly-αH), 4.00 (s, 2H, N-CH2), 4.40–4.42 (m, 1H, Arg-αH), 4.64–4.67 (m, 1H, Asp-αH), 7.87 (d, 1H, J = 6.0, 5-FU-CH). ESI-MS (m/z): calcd. for 516.17, obsd. 515.2 ([M − H]−). Anal. Calcd. for C18H25FN8O9: C, 41.86; H, 4.88; N, 21.70. Found: C, 42.06; H, 4.68; N, 21.15.
Synthetic procedure for compound T2a
The compounds T2a was synthesized according to the literature method of our group’s work previously (CitationOuyang et al., 2011). White wax (0.26 g, 59%) from compound 2 (0.72 g, 0.5 mmol).
Data
1H-NMR (400 MHz, D2O): 2.64–2.96 (br s, 12H, Asp-βCH2), 4.15 (s, 2H, N-CH2), 4.43–4.96 (m, 6H, Asp-αCH), 7.77 (d, 1H, J = 5.2 Hz, 5-FU-CH). ESI-MS (m/z): calcd. for 878.64, obsd. 903.49 ([M + Na]+). Anal. Calcd. for C30H35FN8O22: C, 41.01; H, 4.02; N, 12.75. Found: C, 41.39; H, 3.78; N, 12.28.
Synthetic procedure for compound T3a
Same procedure as described above for preparation of the compound T1a. White wax (0.32 g, 54%) from compound 3 (0.95 g, 0.5 mmol).
Data
1H-NMR (400 MHz, D2O): δ = 1.61–1.71 (m, 2H, Arg-γH), 1.80–1.93 (m, 2H, Arg-βH), 2.42–2.60 (m, 2H, Arg-δH), 2.61–2.94 (m, 14H, Asp-βH), 3.73(s, 2H, Gly-αH), 4.00 (s, 2H, N-CH2), 4.40–4.46 (m, 1H, Arg-αH), 4.54–4.86 (m, 7H, Asp-αH), 7.45 (d, 1H, J = 6.4 Hz, 5-FU-CH). ESI-MS (m/z:): calcd. for 1206.33, obsd. 1205.30 ([M − H]−) Anal. Calcd. for C42H55FN14O27: C, 41.79; H, 4.59; N, 16.25. Found: C, 41.97; H, 4.34; N, 16.04.
Synthetic procedure for compound T1b
To the solution of TFA /anhydrous CH2Cl2 (1:1, v/v), the compound 1 (0.34 g, 0.5 mmol) was added at room temperature under N2. When the protecting groups were completely removed, the solvent was rotary evaporated. The residue dissolved in THF with NMM (0.05 mL, 0.5 mmol) was added to the solution of compound 6 (0.13 g, 0.5 mmol) in anhydrous THF, when which was stirred for 20 min after NMM (0.05 mL, 0.5 mmol) and IBCF (0.07 mL, 0.5 mmol) had been added at −10°C. The mixture was stirred for 3 h. After evaporated under reduced pressure, the residue was taken up in ethyl acetate and washed with 1 M HCl, 1 M NaHCO3, and brine each for twice. The organic layer was dried and evaporated to give the crude product. The crude product was purified by silica column chromatography using CHCl2 and CH3OH as an eluent, then deprotected the protecting groups with Pd catalyst by hydrogen in CH3OH at room temperature to afford T1b (0.14 g, yield 49%) as white wax.
Data
1H-NMR (400 MHz, d6-DMSO) δ = 1.26–1.30 (m, 2H, Arg-γH), 1.36–1.50 (m, 2H, Arg-βH), 2.45–2.46 (m, 2H, Arg-δH), 2.50–2.54 (m, 2H, Asp-βH), 2.67 (br s, 4H, SA-CH2×2), 3.72 (s, 2H, Gly-αH), 4.23–4.29 (m, 1H, Arg-αH), 4.41–4.52 (m, 1H, Asp-αH), 5.53 (s, 2H, N-CH2); 8.11 (d, 1H, J = 6.4 Hz, 5-FU-CH). ESI-MS (m/z): calcd. for 588.19, obsd. 587.20 ([M − H]−). Anal. Calcd. for C21H29FN8O11: C, 42.86; H, 4.97; N, 19.04. Found: C, 43.06; H, 4.78; N, 18.89.
Synthetic procedure for compound T2b
The compounds T2b was synthesized according to the literature method of our group’s work previously (CitationOuyang et al., 2011). White wax (0.22 g, 46%) from compound 2 (0.72 g, 0.5 mmol).
1H-NMR (400 MHz, D2O): 1H-NMR (400 MHz, D2O): 2.56 (br s, 4H, SA-CH2 × 2), 2.72–3.01 (br s, 12H, Asp-βCH2), 4.64–4.99 (m, 6H, Asp-αCH), 5.74 (s, 2H, N-CH2), 7.86 (d, 1H, J = 5.2 Hz, 5-FU-CH). ESI-MS (m/z): calcd. for 950.70, obsd. 951.43 ([M + H]+). Anal. Calcd. for C33H39FN8O24: C, 41.69; H, 4.13; N, 11.79. Found: C, 40.98; H, 4.25; N, 11.28.
Synthetic procedure for compound T3b
Same procedure as described above for preparation of the compound T1a. White wax (0.26 g, 42%) from compound 3 (0.95 g, 0.5 mmol).
Data
1H-NMR (400 MHz, D2O) δ = 1.75–1.82 (m, 2H, Arg-γH), 1.83–1.94 (m, 2H, Arg-βH), 2.59 (br s, 4H, SA-CH2 × 2), 2.69–2.77 (m, 2H, Arg-δH), 2.77–2.96 (m, 14H, Asp-βH), 4.00 (s, 2H, Gly-αH), 4.36–4.89 (m, 1H, Arg-αH), 4.50–4.82 (m, 7H, Asp-αH), 5.73 (s, 2H, N-CH2), 7.46 (d, 1H, J = 6.0Hz, 5-FU-CH) ESI-MS (m/z): calcd. for 1278.36, obsd 1277.20 ([M − H]−). Anal. Calcd. for C45H59FN14O29: C, 42.26; H, 4.65; N, 15.33. Found: C, 42.63; H, 4.29; N, 15.06.
In vitro HAP binding assay
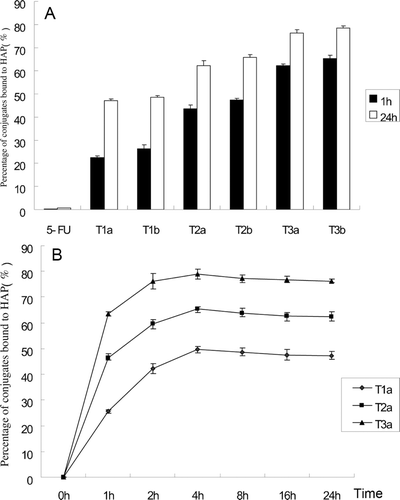
To study the binding of these conjugates to bone, an in vitro HAP binding assay was set up using in vitro HAP binding methods (CitationWang et al., 2003). The conjugates were dissolved in water with various precise concentrations and the adsorption amounts were determined by a UV spectrophotometer at 274 nm to obtain the A–C linear regression equation. Then, in tubes, conjugates were dissolved in water (concentration 100 μg/mL) and incubated with HAP (10 mg) in a shaker bath at 37°C. Each formulation was cultured for 1 h and 24 h. After the prescribed time, conjugate solutions were removed from the water bath and centrifuged for 5 min at 3000 rpm. Concentrations of non-bound conjugates were determined by UV spectrophotometry absorbance at 274 nm. The bound percentage was calculated by (C0−C)/C0×100% and the result was presented in .
Figure 2. (A) Percentage of conjugates bound to HAP at 1 and 24 h. (B) Change of percent conjugates T1a, T2a, and T3a bound to HAP. Error bars represent the mean and SD of three independent experiments.
Solubility assessment
The solubility of the target molecules was determined in water. The method used for sample preparation was similar to each system (CitationTang et al., 2008), i.e. A–C linear regression equation was obtained in vitro HAP binding assay. Excess target molecules (T1a-b, T2a-b, T3a-b) were added to 1 mL of water (pH = 7) respectively to ensure the compound solution reaching saturation. The solutions were mechanically shaken for 10 min at 37°C and centrifuged at 10 000 rpm for 10 min. 200 μL supernatant were pipetted and diluted by water (pH = 7) as proper multiple to make the concentration of the diluted solution was in the range of the obtained A–C linear regression equation. The adsorption amounts were determined by UV at 274 nm. Then the C1 (diluted solution) was obtained by the special equation and the C (saturated solution) was calculated by C1×N (diluted multiple). The data was presented in .
Table 1. Solubility of T1a-b, T2a-b, T3a-b in water (pH=7).
HPLC analysis
The HPLC system consisted of an variable UV-vis detector and a set of Model liquid chromatograph including a manometric module as well as a dynamic mixer from Waters 2695-2487 HPLC system. The mobile phase consists of pure water-acetonitrile (3:97), which was filtered through a 0.45-mm membrane filter before use. A Kromasil RP18 column (250 mm × 4.6 mm, 5 μm) was eluted with the mobile phase at flow rate of 1.0 mL/min. The Column temperature was 35°C. The eluate was monitored by measuring the absorption at 274 nm. The retention time (RT) of 5-FU is 6.432 min.
Drug release study
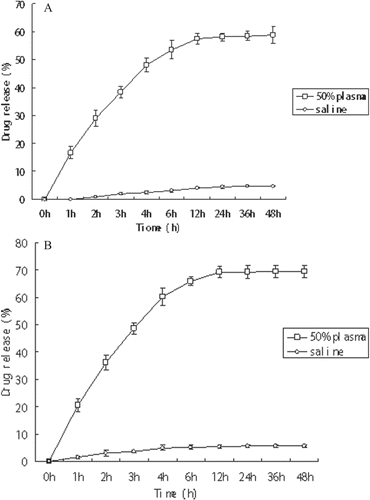
Since the conjugate contains different covalent bond between the drug and the bone-targeting peptides, it should be enzymatically and/or hydrolytically degradable to release the drug molecules to bone tissue and the different in vitro release capabilities of the two types of 5-FU prodrugs can be compared. The conjugates (compounds T3a and T3b) were incubated in saline pH 7.0 or 50% (v/v in saline) rat plasma at 37°C and the hydrolytic release of 5-FU in the solution was monitored by RP-HPLC. The concentration of 5-FU was analyzed using the HPLC conditions mentioned above. The percentage of drug release was deduced from the difference between the initial amounts of T3a or T3b and that of regenerated drug 5-FU. The data was presented in .
Figure 3. Release of 5-FU from compound T3a (A) and compound T3b (B) in physiological conditions. Error bars represented the mean and standard deviation of three independent experiments. 5-FU, 5-fluorouracil.
Biodistribution in rat tissue in vivo
According to the requirements of the National Act on the Usage of Experimental Animals (PR China), the Sichuan University Animal Ethical Experimentation Committee, approved all procedures of our in vivo studies. Tissue distribution study was performed in rats to evaluate and compare the targeting potential of T1b, T2b, and T3b. Thirty female Sprague-Dawley (SD) healthy rats were selected. These animals were kept in well-spaced ventilated cages and were maintained in healthy and fixed diets. All animals were kept on starvation 12 h with free access to water before injection. The animals were divided into five groups and every group contained six rates. The first group served as control, in which the normal saline was injected via intraperitoneal rout. The second group was injected 5-FU through the same route at a dosage of 3 mg/kg. The remaining three groups were respectively injected T1a,T2a, and T3a through the same route at a dosage equivalent to the plain drug of 3 mg/kg. At the indicated time, the animals were sacrificed and blood samples were collected from the tail vein of rats. Then the animals were dissected and each tested organ was removed, including kidney, liver, and femur. Organs were rinsed with cold normal saline, blotted dry with a paper towel, extracted with methanol, diluted, centrifuged, and dispensed in plastic sample vials. All the samples were centrifuged at 3500 rpm for 15 min. After that, 20 μL of the supernatants were removed and the concentration of 5-FU was analyzed using the HPLC conditions mentioned above.
Results and discussion
Synthesis and characteristic of the prodrugs
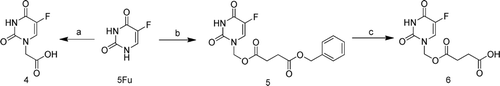
The synthetic procedures of the N-1 acetic acid modified and N-1 succinate acid modified 5-FU prodrugs were outlined in . First, the bromine acetic acid was conjugated to 5-FU through a simple substitution reaction to obtain the compound 4. Then 5-FU reacted with formaldehyde to give N, N-1, 3-dimethylol-5-FU, the N-3 substituted 5-FU derivatives were relatively unstable and C-N bond was easily cleavable, and then condensed with benzyl succinate in the presence of DCC (dicyclohexylcarbodiimide) and DMAP (dimethylamino-pyridine) to obtain 5-FU derivatives compound 5 with a labile ester bond linkage. After purified by silica gel chromatography column and N-3 substituted coproducts removed, we used the method of Pd/C catalyzed hydrogenation to obtain compound 6.
Scheme 1. The synthesis of compound 4–6. Reagents and Conditions: (a) BrCH2COOH, KOH,40°C; (b) i, HCHO, 60°C; ii, PhCH2OOCCH2CH2COOH, CH3CN, DCC, DMAP, r.t.; (c) 10% Pd/ C, CH3OH, r.t. DCC, dicyclohexylcarbodiimide; DMAP, dimethylamino-pyridine.
The synthetic procedures of the fully protected bone and/or tumor targeting peptides 1 and 2 were synthesized by a conventional liquid-phase peptide synthetic method from Boc-Asp-OBzl, Boc-Gly and Boc-Arg(NO2)-OH utilizing IBCF(IBCF) and NMM (N-methyl morpholine) (mixed anhydrides method) by a series of segment condensation which were described in our previous work. (CitationHe et al., 2006; CitationOuyang et al., 2009).
The synthetic procedure of the fully protected dual-targeting peptide compound 3 was outlined in . It is synthesized by a conventional liquid-phase peptide synthetic method utilizing IBCF and NMM (mixed anhydrides method) from compound 2 by a series of stepwise coupling.
Scheme 2. The synthesis of compound 3. Reagents and Conditions: (a) i, TFA, CH2Cl2, r.t.; ii, Boc-Asp(OBzl)-OH, IBCF, NMM, THF; (b) i, TFA, CH2Cl2,r.t.; ii, Boc-Gly-OH, IBCF, NMM, THF; (c) i, TFA, CH2Cl2,r.t.; ii, Boc-Arg(NO2)-OH, IBCF, NMM, THF. IBCF, isobutyl chloroformate; NMM, N-methyl-morpholine; TFA, trifluoroacetic acid.
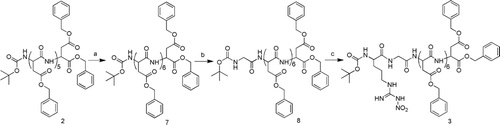
The 5-FU-peptides conjugates T1a–T3a were obtained by divergent synthesis of compound 1–3 with compound 4 in the presence of IBCF and NMM in anhydrous dimethylformamide and removal of protected groups by catalytic hydrogenolysis. And the 5-FU-peptides conjugates T1b–T3b were obtained by divergent synthesis of compound 1–3 with compound 6 in the presence of IBCF and NMM in anhydrous tetrahydrofuran and removal of protected groups by catalytic hydrogenolysis. The synthetic route was outlined in .
Scheme 3. The synthesis of compounds T1a-b, T2a-b, T3a-b. Reagents and Conditions: (a) i, TFA, CH2Cl2,r.t.; ii, 4, IBCF, NMM, THF; iii, 10% Pd/ C, CH3OH, r.t.; (b) i, TFA, CH2Cl2,r.t.; ii, 6, IBCF, NMM, THF; iii, 10% Pd/ C, CH3OH, r.t. IBCF, isobutyl chloroformate; NMM, N-methyl-morpholine; TFA, trifluoroacetic acid.
The high degree of symmetry in these molecules enabled facile confirmation of both structure and purity by NMR techniques. For example, in the 1H-NMR spectrum of compound T1a–T3a, the 5-FU linker protons observed the resonance signals at 7.69 (d) and 4.00 (s) were clearly distinguishable from the resonances arising from the Asp at 2.72 (m) and 4.90 (m) ppm, Gly at 3.90 (s) ppm and Arg at 1.85 (m), 2.64 (m) and 4.40 (m). In the 1H-NMR spectrum of compound T1b–T3b, succinate protons were observed the resonance signals at 2.56 (br s) and 5.73 (s). Integration of the respective areas of all the protons confirmed the complete coupling and the purity of the target compounds. Furthermore, the structures of these compounds were further verified by ESI-MS. All the spectra displayed a very prominent peak corresponding to the compounds complexed with protons or sodium cation. Moreover, elemental analysis was also in good agreement with those of the signed structures.
In Vitro HAP binding assay
From the data presented in , all conjugates of 5-FU exhibited HAP binding capability and the binding was fast and long. Non-modified 5-FU hardly showed any binding demonstrating that these moieties played an important role in the HAP binding process. And the HAP binding property of these conjugates based on the same moiety with different linkers had no significant difference. The binding trend of these moieties was RGD7 > D6 > RGD, which indicated the bone-targeting property of the dual-targeting moiety RGD7 was enhanced.
Solubility assessment
The data presented in clearly demonstrated the solubility of these compounds. It was observed that the solubility of these conjugates was better than 5-FU. And the solubility property of these conjugates based on the same moiety with different linkers had no significant difference. The solubility trend of these moieties was RGD7 > D6 > RGD. This was presumably due to the surface hydrophilic groups that were available to interact with water molecules (CitationPan et al., 2012). The amino group, carboxyl group and guanidine group all were hydrophilic groups, so the compound T3a and T3b, which contained more hydrophilic groups exhibited better solubility.
Drug release study
In order to ensure the parent drugs could be released from the 5-FU-oligopeptides conjugate to exhibit therapeutic effect, the hydrolyzable linker between the 5-FU and the linker should be neither too stable nor too labile. So it is important to choose an appropriate bond. In the most case of 5-FU prodrugs designing, attaching directly to the N-1 of 5-FU by the use of a carbonate or carbamate linker was chosed. For better release, a longer carbon chain was introduced to the N-1 of 5-FU: a succinate chain which was used as a simple self-immolative linker was connected between the 5-FU and moieties (CitationOuyang et al., 2011). We compared the different release characteristics of prodrugs with the two different linkers above. The results from these in vitro assays () suggest several trends. First, two different types of prodrugs were able to release the parent drugs in vitro. Specifically, the drug released rapidly from the compound T3a in 50% human plasma, reaching 48 % after 6 h and almost 60% within 48 h, whereas the drug release in saline was much slower, implying 2.5% and 4.85% in the same period, respectively. Second, in 50% human plasma, the drug released from the compound T3b reaching 60.2% after 6 h and almost 70% within 48 h. The trends observed from drug release data would suggest that the two linkages were labile under blood circulation conditions and relatively stable in saline to allow the transport of the prodrug to bone issues and effectively release the free active drug. Compound T3b with succinate ester had a more rapid activation pathway, possibly because it had a longer and more labile carbon chain or the eliminate process was accelerating by a self-immolative disassembly pathway or the ester bond was instable than the amide bond. Anyway, 5-FU prodrugs with succinate linkage were better substrates of enzymes in human plasma. The results shown in this study provided a proof of concept that succinate ester linkages modified bone-targeting 5-FU prodrugs had more effective release profiles.
Biodistribution in rats tissue in vivo
An in vivo pharmacokinetic and biodistribution assay was taken to further exhibit the targeting ability of the prodrugs. The pharmacokinetic parameters for conjugates T1a, T2a, and T3a were summarized in . The AUC value of conjugates T1a, T2a, and T3a in blood were respectively 20.33, 40.89, and 70.92 mg·h/L. The T1/2 value of these conjugates in blood were 2.12, 7.92, 8.91 h, respectively. The values above were much larger than those of 5-FU (10.21 mg × h/L and 077 h, respectivly). From this point of view, 5-FU-oligopeptides conjugates, with a longer RT and a higher therapeutic window, was better than 5-FU for bone-targeting chemotherapy.
Table 2. Pharmacokinetic parameters of 5-FU and 5-FU-oligopeptides conjugate T1a, T2a, and T3a in rats.
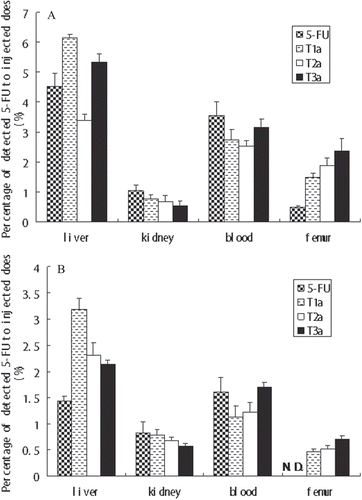
showed the accumulation of the conjugates in the femur. After 2 h, the detected 5-FU percentage of conjugates T1a, T2a, and T3a in femur were respectively 1.50, 1.89, and 2.38%, much larger than that of non-modified 5-FU (0.49%). After 12h, the detected 5-FU percentage of conjugates T1a, T2a, and T3a in femur were respectively 0.47, 0.52, and 0.70%, however, which of non-modified 5-FU could not been detected. From this point of view, all conjugates reached the femur within 2 h after administration, and the amount of deposited conjugates began to decrease after 12 h. In contrast to nontargeted (no peptides) controls, peptides containing conjugates have a tendency of targeting and accumulation to the bone. And the targeting tendency of these moieties was RGD7 > D6 > RGD. This comparison indicated that all these moieties could target to bone, and the bone-targeting property of the dual-targeting moiety RGD7 was enhanced. Overall, the moiety RGD7 was more effective to be a bone-targeting moiety, and the compounds based on RGD7 could reach a higher drug accumulation in bone tissue and reduce side effects in other organs.
Figure 4. The biodistribution of the T1b, T2b, T3b, and naked 5-FU. The biodistribution was analyzed at (A) 2 h and (B) 12 h after oral administration. Error bars represented the mean and standard deviation of three independent experiments. N.D. cannot be detected.
Conclusion
Site-specific bone drug delivery via a dual-targeting function approach has generated considerable interest for enhancing the potency and diminishing the side effects of a drug. In the present study, a series of novel bone-targeting and biodegradable conjugates were designed and prepared with amido coupling reactions. All the conjugates were water-soluble and the water solubility was increased by the structures with more hydrophilic groups. Drug release study showed that conjugates with two linkages were relatively stable in saline and labile under blood circulation conditions to allow the transport of the prodrugs to bone issues and the effectively release of the free active drug. And the succinate ester linkages modified prodrug had more effective release profiles. The in vitro HAP binding investigations and the in vivo biodistribution data obtained both highlighted the strong affinity of these conjugates to bone, as opposed to the negligible bone affinities of the parent drug. Among all the prodrugs, the conjugates based on RGD7 showed the most remarkable targeting tendency, which revealed this new kind of dual-targeting moiety RGD7 could deliver 5-FU to the bone tissue more effectively and selectively. The experiment for verifying the tumor targeting property of the dual-targeting delivery system was under way. Overall, the preliminary results seem to be very promising, these novel dual-targeting conjugates ware water-soluble and able to release the parent drugs in vitro, moreover, the bone targeting property of which was enhanced. Thus, the side effects in normal tissues and chemotherapeutic drugs administration dose could be reduced. And it can represent a novel opportunity to apply the versatile dual-targeting delivery system to applications in bone tumor treatment.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alpizar YA, Chain B, Collins MK, Greenwood J, Katz D, Stauss HJ, Mitchison NA. (2011). Ten years of progress in vaccination against cancer: the need to counteract cancer evasion by dual targeting in future therapies. Cancer Immunol Immunother 60:1127–1135.
- Barczyk M, Carracedo S, Gullberg D. (2010). Integrins. Cell Tissue Res 339:269–280.
- Byrne JD, Betancourt T, Brannon-Peppas L. (2008). Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev 60:1615–1626.
- He G, Guo L, Ma LF. (2006). Synthesis of novel dendrimers containing amino acids and peptides. Chinese Chem Lett 17:289–292.
- Jackson KE, Pham JS, Kwek M, De Silva NS, Allen SM, Goodman CD, McFadden GI, de Pouplana LR, Ralph SA. (2012). Dual targeting of aminoacyl-tRNA synthetases to the apicoplast and cytosol in Plasmodium falciparum. Int J Parasitol 42:177–186.
- Lu YJ, Wei KC, Ma CC, Yang SY, Chen JP. (2012). Dual targeted delivery of doxorubicin to cancer cells using folate-conjugated magnetic multi-walled carbon nanotubes. Colloids Surf B Biointerfaces 89:1–9.
- Miller K, Eldar-Boock A, Polyak D, Segal E, Benayoun L, Shaked Y, Satchi-Fainaro R. (2011). Antiangiogenic Antitumor Activity of HPMA Copolymer-Paclitaxel-Alendronate Conjugate on Breast Cancer Bone Metastasis Mouse Model. Mol pharm 8:1052–1062.
- Miller K, Erez R, Segal E, Shabat D, Satchi-Fainaro R. (2009). Targeting bone metastases with a bispecific anticancer and antiangiogenic polymer-alendronate-taxane conjugate. Angew Chem Int Ed Engl 48:2949–2954.
- Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K, Wrasidlo W, Cheresh DA. (2008). Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci USA 105:9343–9348.
- Ouyang L, He D, Zhang J, He G, Jiang B, Wang Q, Chen Z, Pan J, Li Y, Guo L. (2011). Selective bone targeting 5-fluorouracil prodrugs: synthesis and preliminary biological evaluation. Bioorg Med Chem 19:3750–3756.
- Ouyang L, Huang WC, He G, Guo L. (2009). Bone targeting prodrugs based on peptide dendrimers, synthesis and hydroxyapatite binding in vitro. Lett Org Chem 6:272–277.
- Ouyang L, Zhang J, Pan J, Yan L, Guo L. (2009). Synthesis and preliminary evaluation in vitro of novel naproxen-dendritic peptide conjugates. Drug Deliv 16:348–356.
- Pan JZ, Wen M, Yin DQ, Jiang B, He DS, Guo L. (2012). Design and synthesis of novel amphiphilic Janus dendrimers for bone-targeted drug delivery. Tetrahedron 68, 2943–2949.
- Petkau K, Kaeser A, Fischer I, Brunsveld L, Schenning AP. (2011). Pre- and postfunctionalized self-assembled p-conjugated fluorescent organic nanoparticles for dual targeting. J Am Chem Soc 133:17063–17071.
- Ray A, Larson N, Pike DB, Grüner M, Naik S, Bauer H, Malugin A, Greish K, Ghandehari H. (2011). Comparison of active and passive targeting of docetaxel for prostate cancer therapy by HPMA copolymer-RGDfK conjugates. Mol Pharm 8:1090–1099.
- Shao H, Gao C, Tang H, Zhang H, Roberts LR, Hylander BL, Repasky EA, Ma WW, Qiu J, Adjei AA, Dy GK, Yu C. (2012). Dual targeting of mTORC1/C2 complexes enhances histone deacetylase inhibitor-mediated anti-tumor efficacy in primary HCC cancer in vitro and in vivo. J Hepatol 56:176–183.
- Takahashi T, Yokogawa K, Sakura N, Nomura M, Kobayashi S, Miyamoto K. (2008). Bone-targeting of quinolones conjugated with an acidic oligopeptide. Pharm Res 25:2881–2888.
- Takahashi-Nishioka T, Yokogawa K, Tomatsu S, Nomura M, Kobayashi S, Miyamoto K. (2008). Targeted drug delivery to bone: pharmacokinetic and pharmacological properties of acidic oligopeptide-tagged drugs. Curr Drug Discov Technol 5:39–48.
- Tang X, Zhang P, Ye H, Zhang C, Shen W, Ping Q. (2008). Water-soluble gambogic acid PEGylated prodrugs: synthesis, characterization, physicochemical properties and in vitro hydrolysis. Pharmazie 63:711–717.
- Temming K, Meyer DL, Zabinski R, Senter PD, Poelstra K, Molema G, Kok RJ. (2007). Improved efficacy of alphavbeta3-targeted albumin conjugates by conjugation of a novel auristatin derivative. Mol Pharm 4:686–694.
- Teti A, Migliaccio S, Baron R. (2002). The role of the alphaVbeta3 integrin in the development of osteolytic bone metastases: a pharmacological target for alternative therapy? Calcif Tissue Int 71:293–299.
- Wang D, Miller S, Sima M, Kopecková P, Kopecek J. (2003). Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconjug Chem 14:853–859.
- Xu Q, Liu Y, Su S, Li W, Chen C, Wu Y. (2012). Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials 33:1627–1639.
- Yogev O, Naamati A, Pines O. (2011). Fumarase: a paradigm of dual targeting and dual localized functions. FEBS J 278:4230–4242.