Abstract
The purpose of this study was to formulate a reservoir-type transdermal delivery system (TDS) for 2,3,5,6-tetramethylpyrazine (TMP) to enable the delivery of a sufficient dose through human skin to achieve an effective therapeutic plasma concentration. To improve the penetration of TMP in the reservoir-type TDS, several chemical penetration enhancers were investigated using in vitro rat dorsal skin permeation studies. Eucalyptus oil was found to enhance the permeation of TMP to the greatest extent, with the optimal concentration being 5% and the flux being 542.6 ± 49.7 μg/cm2/h, which was 4.5-fold greater than control with no enhancer (p < 0.01). The flux of the optimized reservoir-type TDS permeated through the human epidermis was 346.0 ± 27.7 μg/cm2/h. Based on the in vitro human skin permeation flux and the pharmacokinetics parameters observed, the clinical surface area of the TDS patch was predicted to be 20 cm2. The in vivo study conducted in rabbits showed that the TMP TDS patch containing 5% eucalyptus oil had a more favorable pharmacokinetic profile, with a lower Cmax and prolonged Tmax and mean residence time than that observed with the oral administration of TMP. The TMP reservoir-type TDS was shown to be a promising alternative route to oral administration or intravenous infusion of TMP.
Introduction
2,3,5,6-Tetramethylpyrazine (TMP) is a biologically active ingredient originally isolated from the traditional herbal medicine Ligusticum wallichii Franch that is widely used in China for the treatment of cardiovascular and cerebrovascular disorders (Guo et al., Citation1983). TMP was demonstrated to be a new type of calcium channel antagonist (Zou et al., Citation2001) that can inhibit platelet aggregation (Sheu et al., Citation1997; Li et al., Citation2001), improve blood viscosity (Feng et al., Citation1996) and increase cerebral blood flow in ischemic attacks (Ho et al., Citation1989). TMP was also reported to demonstrate appreciable blood–brain barrier (BBB) penetrability (Liang et al., Citation1999; Tsai et al., Citation2001).
On oral administration, TMP undergoes typical first-pass metabolism with a systemic bioavailability of 10–30% in humans (Cai et al., Citation1989), and it is rapidly eliminated with a short biological half-life of 1.6 h (Liu et al., Citation1991). Therefore, TMP must be taken orally three times per day at a high dose of 81–162 mg/day or administered by intravenous infusion at a dose of 27–54 mg for 4–6 h in the form of TMP hydrochloride or TMP phosphate (TMPP). To avoid invasive drug therapy and eliminate the need for frequent oral dosing, transdermal delivery system (TDS) may provide an alternative route for the administration of TMP. Also, TDS has the advantage of avoiding intestinal and/or hepatic first-pass metabolism, maintaining a constant drug plasma level, and improving convenience and treatment compliance.
The feasibility of transdermal delivery for TMP or TMP salts has been previously reported (Weng et al., Citation1999; Qi et al., 2003a). The permeation flux of TMP hydrochloride or TMPP was shown to be enhanced by penetration enhancers (Xu et al., Citation2001; Zhang et al., Citation2006a,Citationb, Citation2007a,Citationb) and microemulsions (Cui et al., Citation2011), and their matrix-type patches were also prepared (Zhang et al., Citation2005; Dou et al., Citation2008; Han et al., Citation2011). However, TMP was shown to be 2.5-fold more permeable than TMP hydrochloride or TMPP (Weng & Xu, Citation1999). Therefore, the use of TMP free base may be more effective for achieving a therapeutic plasma level than its salts. Various TDSs for TMP such as microemulsions (Zhao et al., Citation2011), ethosomes (Liu et al., Citation2011a,Citationb) and matrix-type (Qiu et al., Citation2006) or reservoir-type patches (Qi et al., Citation2002, 2003b) have been fabricated. Their improvement of permeation across animal skin has been observed in vitro, while prolonged and sustained plasma drug levels have been demonstrated in rats or rabbits in vivo. However, whether a therapeutic plasma level can be achieved in humans remains uncertain due to the lack of data showing sufficient permeation flux through human skin and the lack of direct pharmacokinetic data in humans.
The stratum corneum is a barrier that most drugs must permeate when delivered through human skin. The use of penetration enhancers to reversibly overcome the stratum corneum barrier is the most popular and effective method, and has been successfully applied in products such as Minitran, Nitrodisc and Deponit. We previously examined different permeation enhancers for a TMP-saturated solution and found that eucalyptus oil, azone and menthol were potent candidates among those screened (Weng & Xu, Citation1999; Shen et al., Citation2006). These permeation enhancers were shown to increase TMP flux 5- to 8-fold. At times, discrepancies are observed in the enhancing activity of penetration enhancers between solvent systems and in TDS patches.
In this study, we designed and formulated a reservoir-type TDS for TMP and investigated the effectiveness of eucalyptus oil, azone and menthol as penetration enhancers in the reservoir-type TDS by performing in vitro permeation studies through excised rat dorsal skin. The permeation flux of the optimized TDS patch across human skin was then determined. Using the human flux and pharmacokinetic parameters observed, the clinical patch area was predicted. In addition, the pharmacokinetic profile of the optimized TMP patch was determined in rabbits and compared with the profile of orally administered TMP.
Materials and methods
Materials
TMP (purity 99.8%), TMPP (purity 99.5%) and TMPP tablets (containing 50 mg TMPP, equivalent to 27 mg TMP free base per tablet) were purchased from Beijing Yan Jing Pharmaceutical Factory (Beijing, China). Other materials used included: azone (Hubei Tianmen Kejie Pharmacy Co., Ltd., Wuhan, China); eucalyptus oil and menthol (Shanghai Zhonghua Pharmaceutical Co., Ltd., Shanghai, China); CoTranTM 9728, ScotchpakTM 1022 and ScotchpakTM 9732 (donated by 3 M China Ltd., Shanghai, China); silicone pressure sensitive adhesive (Dow Corning Shanghai Co. Ltd., Shanghai, China) and Carbopol 934P (Noveon Shanghai Specialty Polymers Co., Ltd., Shanghai, China). All other chemicals used were of analytical grade.
Preparation of the reservoir gel
The formulations of the reservoir gel containing different permeation enhancers are listed in . Carbopol 934P (2.5%) was added to glycerin/water (1/1, w/w) and was stored overnight to allow for the complete swelling of the polymer. To prepare TMP from TMPP in situ, TMPP was added into a 4 M sodium hydroxide solution while stirring, and the final pH was adjusted to 7–8. The drug was then added slowly to the prepared Carbopol solution while stirring until the gel was formed. The permeation enhancers, including eucalyptus oil, azone and menthol, and 2.0% Tween 80 were premixed with 5.0% ethanol and then added to the gel and mixed well. The weight was made up to 100% by the addition of water. Each transdermal patch contained 463 mg of TMPP, equivalent to 250 mg of TMP, and 1.6 g of reservoir gel.
Table 1. Transdermal reservoir formulations of TMP containing different penetration enhancers.
Fabrication of the reservoir-type TDS
The reservoir-type TDS for TMP was fabricated by sandwiching the reservoir gel between a drug-impermeable backing laminate and an ethylene-vinyl acetate (EVA) copolymer membrane. First, the membrane/adhesive/release liner composite was prepared. The silicone adhesive solution was coated onto the fluoropolymer release liner (3 M, ScotchpakTM 1022) and was allowed to dry completely. The EVA membrane with a vinyl acetate (VA) content of 28% and a thickness of 51 μm (3 M, CoTran™ 9728) was then pressed over the adhesive layer, and thus the three-layered composite was prepared. Second, by using a syringe, the reservoir gel (1.6 g) was injected onto the membrane surface of the composite laminate, and the polyester-backing layer with an EVA heat-sealable layer (3 M, ScotchpakTM 9732) was used to cover the gel. The two layers were then heat-sealed using a mold preheated to 80 °C and cut to the appropriate sizes (20 cm2). The TDS patch was stored in a sealed aluminum pouch to minimize the loss of solvent.
In vitro permeability studies
Sprague-Dawley rats (weighing 200–220 g) were obtained from the Experimental Animal Center of Shanghai Medical College of Fudan University. All experiments involving rat and human skin samples were approved by the Animal Care and Use Committee of Fudan University. The rats were euthanized using pentobarbital sodium, and their dorsal hair was removed with a clipper. Full-thickness skin was surgically removed from each rat. Fat and connective tissues were carefully removed with ophthalmic scissors, and the samples were then frozen at −20 °C. The skin samples were thawed to room temperature before use.
Excised human skin from the chest region of a 40-year-old female cadaver was obtained within 24 h post-mortem from Zhongshan Hospital, Fudan University. The epidermis was prepared by immersing the whole skin in water at 60 °C for 90 s, followed by careful removal of the epidermis from the dermis. Samples were stored in an aluminum foil and frozen at −80 °C. On the day of the experiment, the skin was thawed and used immediately for the diffusion studies.
Modified Keshary–Chien diffusion cells were used to conduct the in vitro permeation studies. The rat skin or prepared human skin was mounted between the donor and receptor compartments with the epidermal surface facing the donor compartment. The TDS patch was fixed on the epidermal surface. The donor and receptor compartments were clamped together and placed into a water bath that was maintained at 37 ± 0.5 °C. The receptor compartment contained 15.0 mL of water, and the effective diffusion surface was 3.14 cm2. The hydrodynamics of the receptor fluid were maintained by stirring the fluid at 300 rpm with a star-head magnetic bead. At predetermined times (1, 3, 5, 7, 9, 12 and 24 h), 5 mL samples were withdrawn from the receiver compartment and replaced with an equivalent volume of water to maintain sink conditions. Three or four parallel experimental determinations were performed.
The concentration of TMP in the receptor medium was then determined using a modified, previously described HPLC method (Weng et al., Citation1997). Liquid chromatography was performed with an LC-10AD pump and an SPD-10A UV-Vis detector (Shimadzu Corp., Kyoto, Japan). The reversed column used was a Shim-Pack CLC-ODS column (150 × 4.6 mm I.D., particle size of 5 µm). The mobile phase that consisted of 80% (v/v) menthol/water was pumped at a rate of 1 mL/min. The wavelength used was 285 nm, and the injection volume was 15 μL. Calibrations were made using the external standard method.
Cumulative corrections were made to determine the total amount of TMP that permeated at each time interval. The steady-state flux (Jss) of TMP (mg/cm2/h) was calculated as the slope of the cumulative amount of drug permeated per cm2 versus time using linear regression analysis. The lag time (Tlag) was determined by extrapolating the straight-line portion of each steady-state permeation curve to the time axis. The enhancement ratio (ER) was the ratio of Jss with and without the enhancers. The cumulative amount of drug permeated per unit area (Q) versus time was plotted.
Pharmacokinetic studies
The pharmacokinetic characteristics of the TMP TDS patch were evaluated using New Zealand white rabbits (weighing 2.5–3.0 kg) obtained from the Experimental Animal Center at the Shanghai Medical College of Fudan University. All animal procedures involving animal care were approved by the Animal Care and Use Committee of Fudan University. Twelve rabbits were randomly assigned into either the transdermal application or the oral administration groups. In the transdermal application group, the dorsal side hair was carefully shaved, and no visible signs of damage on the skin surface were observed. After fasting for 24 h, the TMP TDS patch with an effective area of 20 cm2 was applied on the shaved dorsal skin of rabbits for up to 24 h with an overlay of adhesive tape. Four whole TMPP tablets (50 mg TMPP per tablet, i.e. a total of 200 mg TMPP, which is equivalent to 108 mg TMP) were administered to all rabbits in the oral group. Blood samples were taken via the marginal ear vein before and 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 20 and 24 h after transdermal administration, and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 h after oral administration.
To each of the 300 μL blood samples, 20 μL of the internal standard (16 μg/mL) and 600 μL of acetonitrile were added. The mixture was vortexed for 30 s to ensure thorough mixing, followed by centrifugation at 10 000 rpm for 5 min. A 20 μL sample of the upper layer was then injected into the HPLC apparatus for analysis. The amount of TMP in rabbit plasma was determined using a previously described HPLC method with minor modifications (Weng et al., Citation1997). Liquid chromatography was performed with an LC-10AD pump and an SPD-10A UV-Vis detector (Shimadzu Corp., Japan). The reversed column used was a Shim-Pack CLC-ODS column (150 × 4.6 mm I.D., particle size of 5 μm). The mobile phase was a binary mixture of 60% (v/v) menthol and 40% (v/v) water that was adjusted to pH 6.5 by using a 0.12% acetate solution. The wavelength was set at 294 nm. The internal standard used was carbamazepine. The flow rate was 1.0 mL/min, and the injection volume was 20 μL.
The peak plasma concentration (Cmax) and the time of its occurrence (Tmax) were determined directly from the individual plasma concentration-time profiles. The area under the time–concentration curve from time 0 to time t (AUC0–t) was calculated using the trapezoidal rule. The mean residence time (MRT) values were obtained by non-compartmental analysis. The relative bioavailability of TMP from the TDS patch was calculated by dividing its dose-adjusted AUC0–t by that of the immediate-release tablet. The steady-state plasma concentrations (Css) of TMP after the patch applications were calculated by dividing AUC0–t by the interval time. All values were reported as mean ± standard deviation (SD). The differences between various pharmacokinetic parameters were evaluated statistically by ANOVA. Differences were defined as statistically significant when p < 0.05.
Results and discussions
To prepare a reservoir-type TDS for TMP, the Carbopol gel drug reservoir was sandwiched between an impermeable backing laminate and a rate-controlling membrane. We compared gelling agents such as Carbopol, hydroxyethyl cellulose and hydroxypropyl methyl cellulose, and found that the permeation rate of TMP was highest with the Carbopol gel. The pH in the reservoir gel was adjusted to 7–8 and the pKa of TMP was 3.9. In this pH range, TMP existed predominantly in the unionized form.
To select the membrane with the fastest release rate, the effects of the amount of VA and the thickness of the membrane on the release rate of TMP were studied. We found that the release rate is greatest when the VA content is higher and the membrane thickness is thinner. Similar results were reported for bupranolol (Babu et al., Citation2005), isosorbide dinitrate (Ocak et al., Citation1999), and timolol (O’Neill et al., Citation1988). The EVA membrane (3 M, CoTranTM 9728) with a VA content of 28% and a thickness of 50.8 μm was found to have the fastest release rate. The silicone pressure-sensitive adhesive was chosen because of its good compliance, superior flexibility and favorable permeation characteristics.
Effects of penetration enhancers
Menthol, azone and eucalyptus oil were incorporated into the reservoir gel and fabricated into the TDS patch to study the effect of penetration enhancers on the permeability of TMP. The cumulative penetration amount per cm2 versus time and the penetration parameters of TMP in the reservoir-type TDS containing 2.5% azone, 2.5% eucalyptus oil and 2.5% menthol are shown in and . The permeation of TMP through excised rat dorsal skin was found to follow zero-order release kinetics, in accordance with Fick’s first diffusion law (). All three enhancers significantly improved the permeation of TMP. The highest permeation was observed with eucalyptus oil, which had a value of 410.6 ± 46.5 μg/cm2/h, providing a 3.4-fold increase in flux, followed by azone with a 2.7-fold increase. We also investigated the use of combinations of penetration enhancers, namely eucalyptus oil and menthol or eucalyptus oil and azone. However, the permeation flux observed with the combined penetration enhancers was significantly lower than that observed with eucalyptus oil alone (data not shown).
Figure 1. Mean ± SD amount of TMP permeated from the reservoir-type TDS containing different penetration enhancers across excised rat skin. The bars represent the SD (n = 3 or 4).
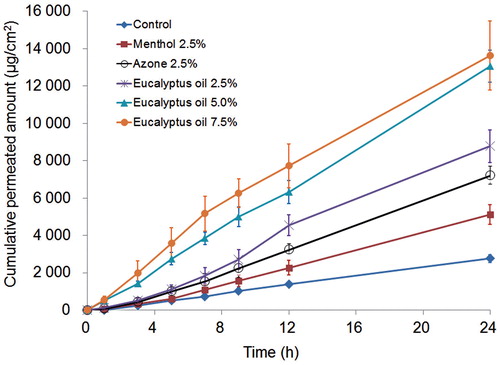
Table 2. Permeation parameters of TMP in a reservoir-type TDS across excised rat skin or human epidermis containing different penetration enhancers.
shows the permeation profile of the TMP TDS patch containing various concentrations of eucalyptus oil, with the skin permeation parameters listed in . The cumulative amount of TMP and the permeation flux increased with the concentration increasing from 2.5% to 5%, but no statistically significant difference was found between the 5% and 7.5% groups. Thus, 5.0% eucalyptus oil was determined to be a suitable concentration. A similar maximum permeation effect at a certain concentration of permeation enhancer was also reported for other enhancers such as azone and oleic acid (Williams et al., Citation2004).
Eucalyptus oil contains more than 80% cineole, a well-known skin penetration enhancer that has been shown to enhance the skin penetration of drugs such as imipramine hydrochloride (Jain et al., Citation2002), AZT (Narishetty et al., Citation2004), propranolol hydrochloride (Amnuaikit et al., Citation2005), trazodone hydrochloride (Das et al., Citation2006) and tamoxifen (Gao et al., Citation1998). Cineole is known to enhance skin permeation by disrupting the interlamellar hydrogen-bonding network of the polar head group region in the stratum corneum bilayer, facilitating the partition and permeation of small polar molecules through the skin (Narishetty et al., Citation2005; Anjos et al., Citation2007).
Permeability through human skin and estimation of TDS patch size
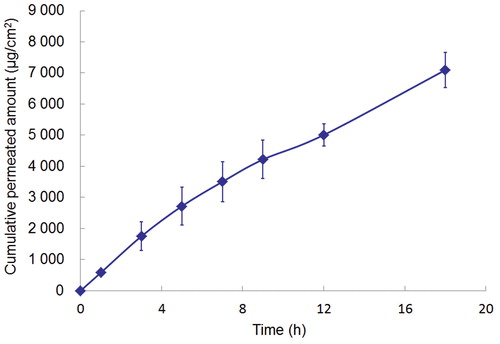
The permeability profiles and permeation parameters of TMP through excised human epidermis with the TMP TDS patch containing 5% eucalyptus oil are presented in and . The steady-state flux of TMP through human epidermis was 346.0 ± 27.7 μg/cm2/h, which was significantly lower than the steady-state flux observed through rat skin (542.6 ± 49.7 μg/cm2/h, p < 0.01). Rat skin has been extensively reported to be 2- to 11-fold more permeable than human skin (Diez et al., Citation1991; Narishetty & Panchagnula, Citation2004). Our group has reported that the flux of a saturated solution of TMP was 20–143 μg/cm2/h through different regions of the human skin (Weng & Xu, Citation1999; Shen et al., Citation2001), which is in the same order of magnitude as the flux (40–90.89 μg/cm2/h) reported by Qi et al. (2003a). In this study, the TMP flux across the human chest skin on using the TMP TDS patch with the aid of 5% eucalyptus oil was 17-fold higher (346.0 μg/cm2/h) than the flux (20.1 μg/cm2/h) of a saturated solution of TMP without enhancers across the same human chest skin (Shen et al., Citation2001).
Figure 2. Mean ± SD amount of TMP permeated from the reservoir-type TDS containing 5% eucalyptus oil across excised human epidermis (n = 4).
Based on the permeation rate through human epidermis, the area of transdermal TDS was predicted according to the following equation:
where Css is the therapeutically effective plasma concentration of the drug at steady state (200 ng/mL) (Liu et al., Citation1991; Qi et al., 2003a) and CLT is the total body clearance of TMP, which has been reported to vary from 15.27 (Cai et al., Citation1989) to 29.36 L/h (Liu et al., Citation1991). Based on a steady-state flux of TMP (Jss) through human epidermis of 346.0 μg/cm2/h and a CLT value of 29.36 L/h, the TDS patch size (A) was predicted to be approximately 17.0 cm2. For the sake of convenience, the area of the TMP patch was defined as 20 cm2. This area is half the predicted area of 40 cm2 for a reservoir-type patch, as reported by Qi et al. (2003a), which was relatively large for human use, with Jss and CLT values of 90.89 μg/cm2/h and 15.7 L/h, respectively.
Considering the flux of 346.0 μg/cm2/h through the human epidermal skin and the predicted patch area of 20 cm2, theoretically, 24 h application would deliver 166 mg of TMP, corresponding to 66% of the loading dose. The theoretical residual dose would therefore be 34%, which is required to ensure an adequate driving force in transdermal application. This is also consistent with the findings obtained for other patch products such as Duragesic (a fentanyl transdermal system for the in vivo delivery of 100 µg/h fentanyl over 72 h; each transdermal system contains 10 mg fentanyl), which has a theoretical residual dose of 28% that was calculated by labeling.
In vivo evaluation of the TDS patch
The mean plasma concentration–time profiles with transdermal and oral administration are compared in . The corresponding pharmacokinetic parameters are given in . All of the pharmacokinetic parameters of TMP (Cmax, Tmax and AUC) obtained with the TMP transdermal patches were significantly different (p < 0.05) from those obtained with oral administration.
Figure 3. Mean ± SD plasma concentration of TMP following the oral administration of an immediate release tablet (108 mg) or the application of a TMP TDS patch (250 mg) in rabbits (n = 6).
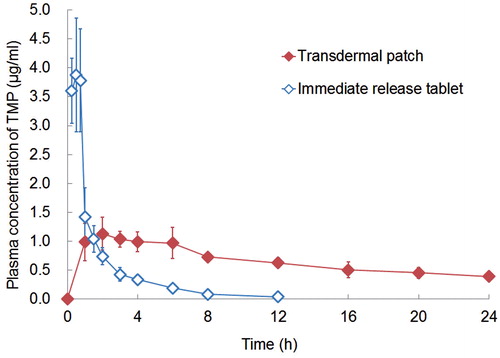
Table 3. Pharmacokinetic parameters of TMP following the oral administration of an immediate release tablet (dose 108 mg) or the application of a TMP transdermal delivery system (TDS) patch (250 mg).
The absorption of TMP after oral administration was rapid, and showed a Cmax of 3.88 ± 0.98 μg/mL at a Tmax of 0.5 ± 0.25 h. With the transdermal patches, the increase in the drug concentration was slower and a significantly lower Cmax value of 1.13 ± 0.28 μg/mL (p < 0.05) was attained at 3.5 ± 1.52 h. A measurable concentration of TMP was obtained within 1 h of patch application and relatively steady plasma concentration was observed for more than 24 h.
The AUC0–t value for transdermal administration was found to be 15.93 ± 0.83 μg·h/mL, which was approximately 2.5-fold greater (p < 0.001, Student’s t-test) than that (6.47 ± 1.83 μg·h/mL) after oral administration. Based on the adjusted dose, the relative bioavailability (%) of TMP after transdermal application compared to that of the immediate-release tablets was approximately 106.3%. Thus, the TMP reservoir-type TDS (with 5% w/w of eucalyptus oil) designed in this study was found to provide a prolonged steady-state concentration of TMP with lower Cmax, and longer Tmax and MRT.
At the end of this study, the application sites of the TDS in the rabbits were examined visually for signs of local irritation. No local irritation was observed at the application sites, indicating that the TDS device was well tolerated when applied to the skin for 1 day.
Conclusion
A reservoir-type TDS for TMP to reach an effective clinical concentration was fabricated using Carbopol gel as a gelling agent, an EVA membrane as a rate-controlling membrane and silicone adhesive as a pressure-sensitive adhesive. The penetration enhancer was chosen from azone, eucalyptus oil and menthol by performing an in vitro permeation study. Eucalyptus oil was found to be the most effective enhancer and a concentration of 5% in the reservoir-type TDS was shown to be optimal, with a flux of 542.6 ± 49.7 μg/cm2/h. The permeation rate of the TMP TDS patch through human epidermis was determined to be 346.0 ± 27.7 μg/cm2/h, which was 17-fold higher than the control without penetration enhancers. The predicted clinical surface area was 20 cm2, which was found to be sufficient to achieve the effective therapeutic plasma level. The findings from this pharmacokinetic study suggest that the transdermal delivery of TMP may be advantageous than oral administration given the more sustained plasma levels observed and the less frequent dosing required.
Declaration of interest
This work was supported by the National Natural Science Foundation of China (81072594). The authors report no declarations of interest.
References
- Amnuaikit C, Ikeuchi I, Ogawara K, et al. (2005). Skin permeation of propranolol from polymeric film containing terpene enhancers for transdermal use. Int J Pharm, 289:167–78
- Anjos JL, Neto DS, Alonso A. (2007). Effects of 1,8-cineole on the dynamics of lipids and proteins of stratum corneum. Int J Pharm, 345:81–7
- Babu RJ, Pandit JK. (2005). Effect of penetration enhancers on the release and skin permeation of bupranolol from reservoir-type transdermal delivery systems. Int J Pharm, 288:325–34
- Cai W, Dong SN, Lou YQ. (1989). HPLC determination of tetramethylpyrazine in human serum and its pharmacokinetic parameters. Acta Pharm Sin, 24:881–6
- Cui Y, Li LZ, Gu J, et al. (2011). Investigation of microemulsion system for transdermal delivery of ligustrazine phosphate. Afr J Pharm Pharmaco, 5:1674–81
- Das MK, Bhattacharya A, Ghosal SK. (2006). Effect of different terpene-containing essential oils on percutaneous absorption of trazodone hydrochloride through mouse epidermis. Drug Deliv, 13:425–31
- Diez I, Colom H, Moreno J, et al. (1991). A comparative in vitro study of transdermal absorption of a series of calcium channel antagonists. J Pharm Sci, 80:931–4
- Dou YZ, Teng H, Wang Q, et al. (2008). Preparation, in vitro release and percutaneous penetration of ligustrazine hydrochloride transdermal delivery system. Chin J Pharm, 39:745–9
- Feng J, Liu RY, Wu GZ, et al. (1996). Tetramethylpyrazine reduces cardiac-derived TXA(2) release in ischaemic arrest in isolated working rat heart. Med Sci Res, 24:121–2
- Gao S, Singh J. (1998). In vitro percutaneous absorption enhancement of a lipophilic drug tamoxifen by terpenes. J Control Release, 51:193–9
- Guo SK, Chen KJ, Qian ZH, et al. (1983). Tetramethylpyrazine in the treatment of cardiovascular and cerebrovascular diseases. Planta Med, 47:89–91
- Han HX, Ma YS, Cui LL, et al. (2011). Primarily exploration of the preparation of ligustrazine phosphate transdermal patches and the effect on permeation enhancing of volatile oil of Flos Magnoliae. Chin Pharm J, 46:1915–18
- Ho WK, Wen HL, Lee CM. (1989). Tetramethylpyrazine for treatment of experimentally induced stroke in Mongolian gerbils. Stroke, 20:96–9
- Jain AK, Thomas NS, Panchagnula R. (2002). Transdermal drug delivery of imipramine hydrochloride. I. Effect of terpenes. J Control Release, 79:93–101
- Li M, Handa S, Ikeda Y, et al. (2001). Specific inhibiting characteristics of tetramethylpyrazine, one of the active ingredients of the Chinese herbal medicine ‘Chuanxiong,’ on platelet thrombus formation under high shear rates. Thromb Res, 104:15–28
- Liang CC, Hong CY, Chen CF, et al. (1999). Measurement and pharmacokinetic study of tetramethylpyrazine in rat blood and its regional brain tissue by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl, 724:303–9
- Liu X, Liu H, Liu J, et al. (2011a). Preparation of a ligustrazine ethosome patch and its evaluation in vitro and in vivo. Int J Nanomed, 6:241–7
- Liu X, Liu H, Zeng Z, et al. (2011b). Pharmacokinetics of ligustrazine ethosome patch in rats and anti-myocardial ischemia and anti-ischemic reperfusion injury effect. Int J Nanomed, 6:1391–8
- Liu XQ, Lou YQ, Chen QT. (1991). The clinical pharmacokinetic studies of tetramethylpyrazine hydrochloride in normal volunteers and patients with acute cerebral ischemia disease. Chin J Clin Pharmacol, 7:32–6
- Narishetty ST, Panchagnula R. (2004). Transdermal delivery of zidovudine: effect of terpenes and their mechanism of action. J Control Release, 95:367–79
- Narishetty ST, Panchagnula R. (2005). Effect of l-menthol and 1,8-cineole on phase behavior and molecular organization of SC lipids and skin permeation of zidovudine. J Control Release, 102:59–70
- Ocak F, Agabeyoglu I. (1999). Development of a membrane-controlled transdermal therapeutic system containing isosorbide dinitrate. Int J Pharm, 180:177–83
- O’Neill CT, Deasy PB. (1988). Development and evaluation using hairless mouse skin of a transdermal timolol product. Int J Pharm, 48:247–54
- Qi X, Ackermann C, Sun D, et al. (2002). The prediction of plasma and brain levels of 2,3,5,6-tetramethylpyrazine following transdermal application. AAPS PharmSci, 4:E46
- Qi X, Ackermann C, Sun D, et al. (2003a). Physicochemical characterization and percutaneous delivery of 2,3,5,6-tetramethylpyrazine. Int J Pharm, 253:177–83
- Qi X, Liu RR, Sun D, et al. (2003b). Convolution method to predict drug concentration profiles of 2,3,5,6-tetramethylpyrazine following transdermal application. Int J Pharm, 259:39–45
- Qiu L, Wang Q, Zhang J, et al. (2006). Studies on inhibition of crystallization and in vitro percutaneous absorption of tetramethylpyrazine transdermal delivery system. Chin Pharm J, 41:1642–6
- Shen HX, Xu HN, Shen T, et al. (2006). Enhancing effect of eucalyptus oil on the penetration of tetramethylpyrazine through rat skin. Chin J Clin Pharm, 15:112–14
- Shen T, Xu HN, Ding GB. (2001). Variations of in vitro tetramethylpyrazine permeation through human skin of different body regions. J Fudan Unive (Med Sci), 28:354–5
- Sheu JR, Kan YC, Hung WC, et al. (1997). Mechanisms involved in the antiplatelet activity of tetramethylpyrazine in human platelets. Thromb Res, 88:259–70
- Tsai TH, Liang C. (2001). Pharmacokinetics of tetramethylpyrazine in rat blood and brain using microdialysis. Int J Pharm, 216:61–6
- Weng WY, Xu HN. (1999). Study on transdermal permeability of tetramethylpyrazine. J Shanghai Med Univ, 26:336–41
- Weng WY, Xu HN, Li GX, et al. (1997). HPLC determination of tetramethylpyrazine in serum and the permeation fluid. J Shanghai Med Univ, 24:379–80
- Williams AC, Barry BW. (2004). Penetration enhancers. Adv Drug Deliv Rev, 56:603–18
- Xu BL, Wang H, Xu WM. (2001). Enhancing effect of synthetic borneol on skin permeation of ligustrazine hydrochloride. Chin Traditional Patent Med, 23:864–7
- Zhang CF, Yang ZL, Luo JB. (2006a). Effects of d-limonene and l-limonene on transdermal absorption of ligustrazine hydrochloride. Acta Pharm Sin, 41:772–777
- Zhang CF, Yang ZL, Luo JB. (2006b). Effects of enantiomer and isomer permeation enhancers on transdermal delivery of ligustrazine hydrochloride. Pharm Dev Technol, 11:417–24
- Zhang CF, Yang ZL, Luo JB, et al. (2007a). Effects of cinnamene enhancers on transdermal delivery of ligustrazine hydrochloride. Eur J Pharm Biopharm, 67:413–19
- Zhang RT, Wang H, Chen L. (2007b). Uniform design method for optimizing proportion of transdermal penetration enhancers of ligustrazine hydrochloride. Chin Traditional Herbal Drugs, 38:50–2
- Zhang S, Lin HQ, Deng H. (2005). Preparation and in-vitro drug release of ligustrazine phosphate sustained-release patches. Drug Eval, 2:292–4
- Zhao JH, Ji L, Wang H, et al. (2011). Microemulsion-based novel transdermal delivery system of tetramethylpyrazine: preparation and evaluation in vitro and in vivo. Int J Nanomed, 6:1611–19
- Zou LY, Hao XM, Zhang GQ, et al. (2001). Effect of tetramethyl pyrazine on L-type calcium channel in rat ventricular myocytes. Can J Physiol Pharmacol, 79:621–6