Abstract
In this paper, orally nanocrystal capsules were produced using nanocrystal formulations in order to optimize dissolution properties of poorly soluble drug glimepiride and improve its bioavailability. The important preparation variables, such as stabilizers, the power input and the time length of ultrasonication on the mean particle size and polydispersity index were investigated systematically, and the optimal values were 0.2% glimepiride (w/v), 1.2% Lipoid S100, 0.6% PEG 6000 (w/v), 0.6% PVPK 30 (w/v), 500 W and 2 min, respectively. Characterization of glimepiride nanocrystal was carried out by X-ray powder diffractometry, differential scanning calorimetry and scanning electron microscopy. In vitro dissolution test, the nanocrystal-loaded capsules of glimepiride showed an evident increase in dissolution rate compared to micronized and market capsules. The in vivo studies demonstrated that a marked enhancement of bioavailability of nanocrystal-loaded capsules was superior compared to the marketed formulation and microcrystal-loaded capsules, which may reduce the risk of side effect by allowing a reduction in either the dose or its frequency of administration.
Introduction
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder with serious microvascular and macrovascular complications. Therefore, T2DM is one of the major causes of morbidity and mortality worldwide (Shelesh & Swarnlata, Citation2010). Glimepiride, a new generation sulfonylurea drug, is effective in the treatment and improvement of T2DM (Massimo, Citation2003). Several preclinical and clinical studies of glimepiride demonstrated its stimulation of insulin secretion, enhancement of insulin sensitivity and additional extra-pancreatic effects (Geinsen, Citation1988; Kouichi et al., Citation2005). Unfortunately, glimepiride is a hydrophobic drug. A leading hurdle of poorly water-soluble drugs, which challenges the oral absorption process, is their lower dissolution velocity and limited solubility, and thereby suboptimal and variable bioavailability (Lipinski, Citation2002; Kipp, Citation2004).
In the recent past, some non-conventional methods, such as solid dispersions, co-solvents, micelles or cyclodextrins have been used for increasing the dissolution rate and solubility of aqueous insolubility drugs, but which have acquired limited success. Research on poorly soluble drugs have demonstrated that particle diminished to the submicron range through nanosizing and formation of a high-energy, stabilized amorphous form are two successful approaches (Kaushal et al., Citation2004; Fakes et al., Citation2009). Nanosuspensions are colloidal dispersions of pure drug nanocrystals or amorphous state in a liquid phase, which are produced by an appropriate nanosizing and stabilized form (Moschwitzer et al., Citation2004; Rabinow et al., Citation2004). According to Noyes–Whitney and Ostwald–Freundlich principles, particle size reduced to the submicron range of nanosuspensions can greatly improve its dissolution rate and saturation solubility, due to the increase in the effective particle surface area, and further enhance bioavailability (Patravale & Kulkarni, Citation2004; Kesisoglou, Citation2007). Therefore, the formulation of nanosuspensions is another very attractive concept for improving dissolution rate and bioavailability of drugs with poor aqueous solubility (Kaushal et al., Citation2004; Fakes et al., Citation2009). Compared with other nanoformulations, a remarkable advantage of the nanocrystals is that they can be applied to various administration routes, such as oral (Mauludin et al., Citation2009), intravenous injection (Ganta et al., Citation2009), ocular (Rosario et al., Citation2002) and pulmonary delivery (Jacobs & Müller, Citation2002; Yang et al., Citation2010). In addition, nanosuspensions can be altered into solid forms, which are further processed into standard dosage forms, such as capsules or tablets.
Nowadays, the different approaches used for producing nanosuspensions can be divided into two main categories, i.e. “top down” and “bottom up” technologies. The top-down processes basically employ mechanical attrition to break down of the micronized drug powder into nanoparticles, including pearl milling (Eerdenbrugh et al., Citation2008) and high-pressure homogenization (Zhang et al., Citation2007). The methods of the latter technologies are microprecipitation (Xia et al., 2010) and emulsion–diffusion (Ambrusa et al., Citation2009), and the basic challenge of these methods is that the growth of drug crystals needs to be limited by suitable surfactant addition and condition control to avoid formation of microcrystals. Sometimes, combination of the above two techniques can exploit the advantages of both processes (Keck & Müller, Citation2006).
The object of our present study was to prepare glimepiride nanosuspension and translate it to dried nanocrystals by lyophilization. The lyophilized powder of glimepiride was characterized for its particle size distribution, morphology, X-ray powder diffractometry (XRPD) and scanning electron microscopy (SEM), and its saturation solubility and dissolution rate were also determined. Finally, the bioavailability of the nanocrystal formulation was estimated in Sprague-Dawley (SD) rats.
Materials and methods
Materials and animals
Glimepiride was purchased from Yangtze River Pharmaceutical Co., Ltd (Taizhou, China). Polyethylene glycol 6000 (PEG 6000) and polyvinylpyrrolidone K-30 (PVPK30) were from Genesis Biotechnology Co., Ltd (Beijing, China). Lipoid S100 was supplied by Shanghai Tohisun Biology & Technology Co., Ltd (Shanghai, China). Sodium dodecyl sulfate (SDS) was obtained from Sigma-Aldrich (St Louis, MO). The marketed glimepiride capsules were purchased from Guangxi Beisheng Pharmaceutical Co., Ltd (Nanning, China). High-performance liquid chromatography (HPLC) grade acetonitrile and methanol were used for HPLC analysis. All other laboratory chemicals used were of analytical grade. Water, used for the preparation of all aqueous solutions, was depurated on a Milli-Q system (Millipore, Billerica, MA).
SD rats (200 ± 20 g) were supplied by the Medical Animal Test Center of Zhengzhou University. The animals were used following the National Act on the use of experimental animals.
Preparation of glimepiride nanosuspensions
Glimepiride nanosuspensions were prepared by the precipitation-ultrasonication method. Referring to the preliminary experiments, the following methods were used for production of glimepiride nanosuspensions. Briefly, glimepiride (10 mg) and Lipoid S100 (1.2%, w/v) were co-dissolved in 2 ml of methanol and acetone (1:1) by sonication and heating (60°C) to obtain organic phase, which was quickly poured into 5 ml aqueous solution of PVPK 30 (0.6 %, w/v) and PEG 6000 (0.6%, w/v). This slurry was homogenized using an ultrasonic bath at 300 W for 5 min under low temperature (1–3°C) to precipitate glimepiride particles, and then transferred to the rotary evaporator to eliminate the organic solvents. This coarse suspension was treated with an ultrasonic probe (LTD JY92-II, Scientz Biotechnology Co., Ningbo, China) at different ultrasonic power inputs (200, 300, 400, 500 and 600 W) for 2 min in an ice-water bath. Reference samples were prepared using the same compositions without the process of reduction of larger particles.
Lyophilization
Glimepiride nanosuspensions were lyophilized with various cryoprotective agents, such as mycose, mannitol, arabinose and erythrose. In brief, the freshly prepared nanosuspensions was dispensed into vials and frozen at −80°C for 4 h. Then, it was transferred to a freeze-dryer (Labconco Corporation, Kansas, MO), and dried at a pressure of 0.098 Mbar for 24 h at −80°C ± 0.5°C.
The redispersability of the lyophilized glimepiride nanosuspensions was further investigated by manual shaking until the sediment was dispersed in the aqueous phase uniformly, and the added water was equivalent to the original volume of the aqueous nanosuspension.
Particle size analysis of glimepiride nanocrystals
The average particle size (z-ave) and the polydispersity index (PI) of glimepiride nanosuspensions were determined by a laser diffractometer Nano-ZS90 (Malvern Instruments, Malvern, UK). The liquid or dried nanosuspensions were diluted with water before the measurement. All the samples were analyzed in triplicate with a scattering angle of 90°.
Scanning electron microscopy
The morphological evaluation of glimepiride nanocrystals was performed by scanning electron microscope (JEOL JSM-6700F, Tokyo, Japan). The samples were coated with platinum in a sputter coater (Polaron SC 7640, Tokyo, Japan), and then deposited on a double-sided carbon tape for examination.
X-ray powder diffractometry
Crystalline state evaluation of the lyophilized samples and other excipients were conducted by XRPD. XRPD patterns were collected using a Brucker AXS diffractometer (Model: D8 Advance) with Cu-Kα radiation operated at 40 mA and 40 kV. Diffractograms were recorded in step scan mode with a scanning rate of 0.02°/min over a 2θ range of 5–50°.
Differential scanning calorimetry
The differential scanning calorimetry (DSC) measurements performed using a Perkin-Elmer Diamond DSC (Perkin Elmer Instruments, Walthan, MA) were calibrated using indium standard. The samples were analyzed in non-hermetically sealed aluminum pans and scanned under a nitrogen purge at 10°C/min from 30°C to 270°C. The output was evaluated by the resident PYRIS Software (Norwalk, CT).
Solubility studies
The saturation solubility of glimepiride in lyophilized nanosuspensions was evaluated in hydrochloric acid solution (pH 1.2) with 0.3% SDS and phosphate buffer (PBS, pH 6.8) with 0.3% SDS. Lyophilized powder was dispersed into these media to obtain 2 mg/ml of drug suspension, and placed on a shaking water bath for 48 h to reach solubility equilibrium. The samples were centrifuged, and the resulting supernatant was diluted in mobile phase and 20 μl volume was injected into the HPLC (Agilent 1200 series equipped with a diode array detector, Kansas, MO) for analysis, and operated at 228 nm. The mobile phase was composed of methanol–acetonitrile–phosphate buffer (pH 6.8, 0.01 mol/l) (29:29:42, v/v), at a flow rate of 1.0 ml/min. Each sample was analyzed three times.
Preparation and dissolution behavior of capsule dosage forms
In order to evaluate dissolution behavior of the lyophilized glimepiride nanosuspensions, prototype capsules were prepared. Based on the HPLC analysis of lyophilized powder, an equivalent of 2 mg glimepiride was filled into marketed hard gelatin capsules to obtain the nanocrystal-loaded capsules (formulation A), and the microcrystal-loaded capsules (formulation B) were also prepared in the identical method. Briefly, the microcrystal-loaded suspensions were prepared by the precipitation-ultrasonication method without the process of reduction of larger particles, and using the same drug/surfactant ratio (w/w) of the corresponding nanocrystal-loaded suspensions. Then the processes, i.e. the condition of lyophilization and the filling of capsules were the same as that of nanocrystal capsules. Dissolution experiments were assessed by Chinese Pharmacopoeia 2010, Apparatus I (Rotating basket, ZRD6-B, Huanghai Pharmatest Apparatus Factory, Shanghai, China), using 500 ml of hydrochloric acid solution of pH 1.2 and buffer of pH 6.8 as the dissolution medium. The temperature was maintained at 37 ± 0.5°C, and the rotation rate of 75 rpm was used. At predetermined time intervals, each 1 ml sample was with drawn and replenished by an equal volume of fresh dissolution medium. The samples were immediately centrifuged and analyzed using HPLC by the above method.
Stability studies
The glimepiride nanocrystal-loaded capsules were stored in closed vials, and placed at room temperature and at 4°C in a refrigerator for up to 12 months for evaluation of physical and chemical stability. At each sampling time, samples were removed, and the average particle size and the amount of drug remained were measured.
Bioavailability studies in rats
Male SD rats weighing 200 ± 20 g were acclimatized for one week before experimentation and fed with standard diet and water. The animal experiment was evaluated and approved by the Animal Ethics Committee, Zhengzhou University, China. Eighteen SD rats were randomly and equally divided into three groups. The capsule contents of three formulations, nanocrystal-loaded capsules, microcrystal-loaded capsules and marketed capsule, were taken out and suspended in 5 ml aqueous solution, then the suspensions were i.g. administrated to three groups of rats, respectively, at a 5 mg/kg dose level. At predetermined time points (10, 20, 40 min, 1, 1.5, 2, 4, 6, 12, 24, 36 h), blood (0.3 ml) samples collected from the retro-orbital sinus into heparin with treated tubes, and centrifuged at 4000 g for 10 min for the isolation of the plasma, the collected plasma was stored at −20°C until analysis.
Plasma sample processing and data analysis
Two hundred microliters of plasma were acidized by hydrochloric acid (1.0 mol/l) for 10 min, and then extracted and homogenized with 1.2 ml mixed organic solvent of acetonitrile and dichlormethane (5:1) on a vortex mixer (XW-80 A, Shanghai, China) for 5 min. After centrifugalization at 5000 g for 10 min, the organic layer was transferred into another clean tube and evaporated under nitrogen gas flow. Then, the residue was redissolved using 50 μl of mobile phase and centrifugalization at 15 000 g for 10 min, 20 μl of the sample was injected into the HPLC for analysis as above.
Pharmacokinetic (PK) parameters of glimepiride were derived from plasma concentration–time curves using the practical PK program-version 97 (Committee of Mathematic Pharmacology of the Chinese Society of Pharmacology, Beijing, China). The various PK parameters were analyzed by SPSS (Chicago, IL).
Results
Effect of preliminary selection of solvents and stabilizers
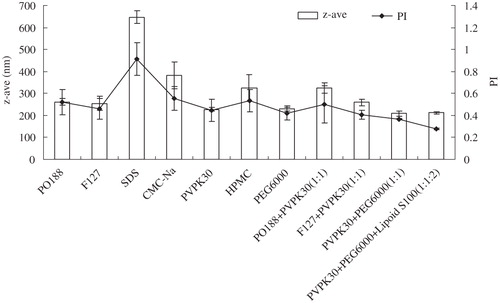
For preparation of glimepiride nanosuspensions, a co-solvent of methanol and acetone (1:1) was utilized to optimize the solubility of both drug and the stabilizers. However, simple precipitation methods have numerous drawbacks. It is very difficult to control nucleation, crystal growth and spontaneous agglomeration to obtain a narrow size distribution. Thus, small particles need to be stabilized by a layer of surfactant or/and protective polymer. A number of stabilizers were tested for their stabilization potential, and experimental data are shown in . Three stabilizers (Lipoid S100, PVPK30 and PEG6000), combination of their steric and electrostatic stabilization, were chosen to inhibit crystal growth and prevent aggregation of nanosized particles for nanosuspensions.
Figure 1. Influence of different stabilizers on z-ave and PI of glimepiride in nanosuspensions (n = 3).
Ultrasonication power and cycles
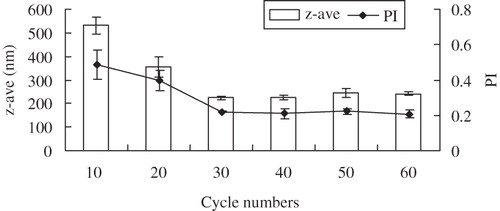
Ultrasonication is one of the most important methods to produce drug nanocrystals. The influence of the power input on the reduction of glimepiride z-ave and PI is shown in . The z-ave and PI decreased to about 220 nm and 0.27 when the ultrasonic power increased from 200 to 600 W, respectively. However, the z-ave and PI did not show obvious difference between 500 and 600 W. shows that the cycle number with the ultrasonic power of 500 W also had a pronounced effect on z-ave and PI. The z-ave and PI clearly decreased up to cycle number 40 and remained unchanged when the cycle number was raised to 100. When the cycle number was set for 60, the prepared nanosuspension was stable up to two weeks, without crystal growth and agglomeration. Therefore, the optimal values of ultrasonic power input and cycle number were selected as 500 W and 60 cycles, respectively.
Figure 2. Influence of the ultrasonic power with 60 cycle numbers on z-ave and PI of glimepiride nanosuspensions (n = 3).
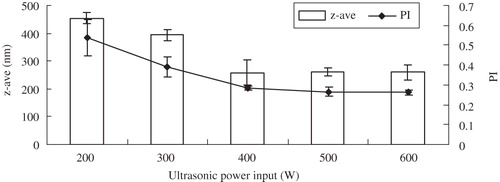
Figure 3. Influence of cycle numbers (at 500 W) on z-ave and PI of glimepiride nanosuspensions (n = 3).
Lyophilization and stability assessment
For producing solid dosage forms, the transformation of the liquid aqueous nanocrystals into a dry nanocrystal powder is a pre-requisite. Lyophilization was performed to dry the liquid glimepiride nanosuspension. Because of the hydrophobic interaction, nanocrystals are apt to aggregate during the freeze-dry process. Therefore, various cryoprotective agents at various concentrations were added to prevent the agglomeration and preserve the redispersibility of the nanocrystals upon reconstitution with water. The z-ave and PI of the reconstituted nanosuspensions are presented in . According to the tested parameters, the z-ave of the reconstituted powder with 6% xylitol and mannitol (5:1) slightly increased in comparison to those of the freshly prepared nanosuspensions (z-ave: 210 ± 15 nm), and were smaller than those with other cryoprotective agents. In the case of redispersibility, the lyophilized powder with 6% xylitol and mannitol (5:1) could be easily redispersed without aggregates or agglomerates. However, the lyophilized powder with mycose and mannitol (5:1) or arabinose and mannitol (5:1) at the same amount could not be redispersed completely. In this study, adding 3%, 6% and 8% of arabinose and mannitol at different ratios to the nanosuspensions were also investigated, and nanosuspensions with 6% xylitol and mannitol (5:1) yielded optimal results.
Table 1. The particle size, PI and appearance of the nanosuspensions.
A long-term physical and chemical stability of freeze-dried glimepiride nanosuspensions was assessed to identify its stability boundaries in support of its storage recommendation. Based on the stability studies data, the z-ave and PI did not significantly increase after 12 months of storage at room temperature (z-ave from 315 nm to 348 nm, PI from 0.265 to 0.314) and under refrigeration (4°C) (z-ave from 303 nm to 314 nm, PI from 0.241 to 0.272). Furthermore, drug contents analysis remained constant, more than 99% glimepiride with respect to the initial values was maintained, demonstrating that the lyophilized samples was stable with no drug degradation during this storage period.
Morphology analysis
The stabilizers used and drug concentration distinctly affect the morphology of particle in nanosuspensions, which was already confirmed by other authors (Zhang et al., Citation2007). In order to characterize the morphology of nanosized glimepiride particles, SEM imaging was performed and nanoparticles were irregular in shape (). These pictures reveal that the particles of nanocrystals with sizes between 200 and 400 nm were distributed homogeneously. This is in good agreement with the low PI value and z-ave determined by Zetasizer-Nano-ZS90, which shows an example of well-prepared nanosuspension. SEM micrograph of the microcrystal particles showed irregular shapes with particle size generally larger than 5 µm.
Figure 4. Morphology of nanocrystal (A) and microcrystal formulations (B) of glimepiride.

XRPD analysis
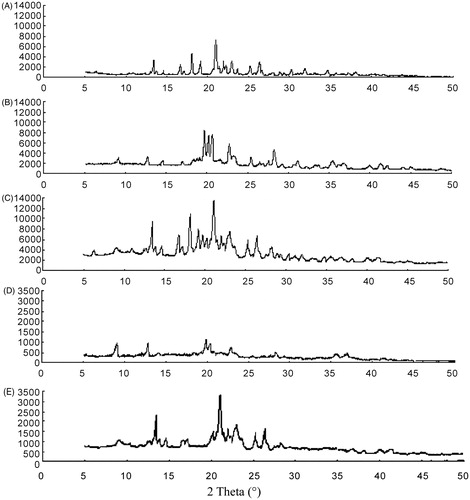
X-ray diffraction has been used to analyze potential conversion in the inner structure of glimepiride crystals. The XRPD patterns of glimepiride coarse powder, blank excipients, a physical mixture containing glimepiride and excipients, nanocrystals and microcrystals are presented in . The diffraction patterns of glimepiride coarse powders showed characteristic high-energy diffraction peaks at 2θ values between 13° and 27°, indicating the crystalline structure of glimepiride. The characteristic peaks of glimepiride for the physical mixture was observed at the same 2θ values as that of the glimepiride coarse powder, showing that glimepiride also existed in the crystalline form. In the lyophilized nanosuspensions, the sharp peaks of pure glimepiride were not observed, indicating the disappearance of crystalline structure in nanocrystals, while there was still a little drug peak in microcrystals, and the intensity was far greater than that of nanocrystals. The effect could be the result of slight changes in molecular conformation.
Figure 5. XRPD-spectra: bulk glimepiride (A), blank excipients (B), physical mixture (C), glimepiride nanocrystals (D) and microcrystals (E).
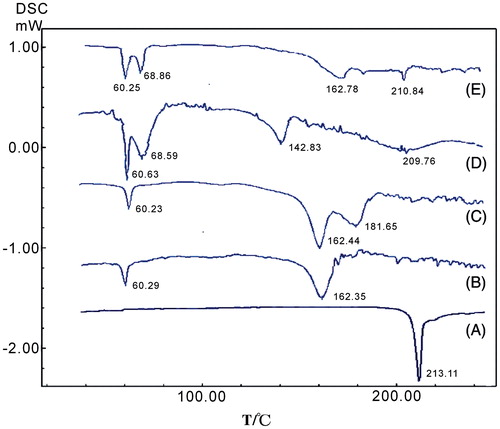
Differential scanning calorimetry
In order to further confirm the physical state, DSC was also performed to analyze the different samples. The DSC investigation () was similar to the XRPD results. The glimepiride coarse powder exhibited a single endothermic peak with a single melting point at 213.11°C. In contrast, this peak disappeared completely in the DSC thermal profile of drug particles from the nanocrystals. Only melting processes of excipients were observed, indicating that there was no glimepiride crystalline in the lyophilized nanocrystals. This finding was confirmed by comparing the XRPD spectra. However, there was the slight endothermic peak of glimepiride (at 210.84°C) in microcrystals. In the physical mixture, the endothermic peak of glimepiride drifted about 32°C (≈182°C) to the left, and this might be due to the mixing with excipients and a little amount of glimepiride. This phenomenon was also reported in previous studies (Pu et al., Citation2009; Quan et al., Citation2011) and in our prior study of about 2-methoxyestradiol nanosuspensions (Du et al., Citation2010).
Figure 6. DSC curves of the bulk glimepiride (A), blank excipients (B), physical mixture (C), glimepiride nanocrystals (D) and microcrystals (E).
Dissolution behavior of capsule dosage forms
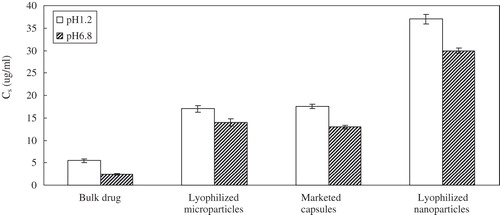
The dissolution behaviors of glimepiride nanocrystal-loaded capsules (formulation A), microcrystal-loaded capsules (formulation B) and marketed capsules are shown in . In both of different dissolution media, the nanocrystal-loaded capsules are superior compared to other formulations, with complete dissolution during the 180-min test period. Within the first 30 min almost 90% of glimepiride was dissolved from formulation A in hydrochloric acid solution of pH 1.2, while from formulation B and marketed capsules only about 40% and 50% of glimepiride were dissolved in the same conditions. Moreover, the dissolution velocity of glimepiride from the nanocrystal-loaded capsules was especially faster than the formulations with particles in the micrometer size range. The particle size down to the nanometer range will evidently increase dissolution rate of drug due to the increase in the effective particle surface area, which was ascertained by the dissolution test of three different samples. Furthermore, an increase in saturation solubility () was accomplished by the particle size reduction of glimepiride particles. Comparatively, the saturation solubility of the lyophilized microparticles was similar to that of the marketed capsules. These promising results in drug dissolution test promoted us to perform in vivo studies in rats.
Figure 7. Dissolution profiles of glimepiride: nanocrystal-loaded capsules (▴), microcrystal-loaded capsules (▪) and marketed capsules (○) in different solvents (means ± S.D., n = 3).
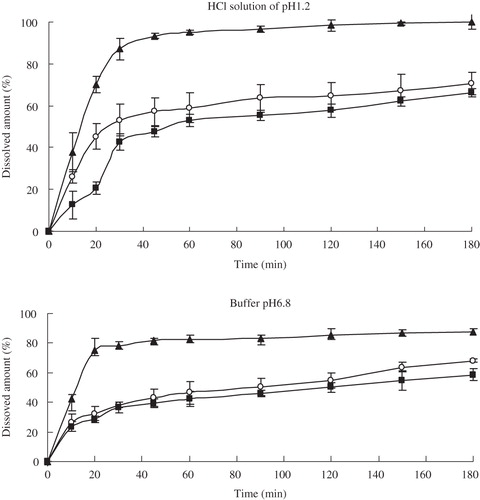
Figure 8. Saturation solubility (Cs) of different particle sizes for glimepiride (n = 3).
Bioavailability study
The plasma drug concentration–time curves after i.g. administration of different formulations of glimepiride in SD rats are shown in . The results indicated that the concentration–time curve of nanocrystal-loaded capsules presents significant improvement in drug absorption than the microcrystal-loaded capsules and marketed formulation, the plasma drug concentration at each predetermined time point was higher than that of reference formulations. The concentration–time curves for nanocrystal-loaded formulation, microcrystal-loaded formulation and marketed formulation were fitted with the two-compartment model, and the corresponding PK parameters are summarized in . The AUC0→36h values of nanocrystal-loaded formulation were approximately 1.7-fold and 1.8-fold (p < 0.05), respectively, relative to the marketed formulation and microcrystal-loaded formulation, indicating bioavailability enhancement by direct particle size reduction. The absorption phase (t1/2α) and the mean residence time (MRT) for the nanocrystal-loaded formulation were significantly (p < 0.05) longer compared to reference formulations, somewhat owing to the high adhesiveness of drug nanocrystal sticking on the epithelial gut wall.
Figure 9. Average plasma concentration–time curves after i.g. administration of glimepiride nanoparticles-loaded capsules and reference formulations at a dose of 5 mg/kg in rats (n = 5).
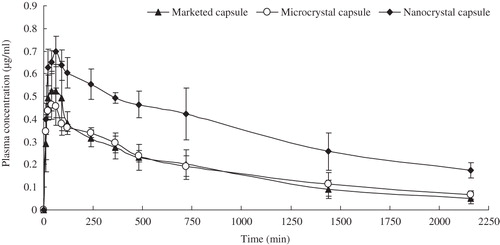
Table 2. PK parameters after i.g. administration of glimepiride nanoparticle capsules and reference formulations at a dose of 5 mg/kg (means ± S.D., n = 5).
Discussion
The preparation of nanoparticles is more complex than emulsifying processes using surfactants. It involves fracture of drug crystals, adsorption and desorption of polymers on drug surfaces. Therefore, it may be impossible to find a simple rule guiding selection of a proper stabilizer for a specific drug. The surface energy, solubility, molecular weight, type and number of functional groups, crystal structure and thermal properties of the drug crystals have an effect on nanoparticles.
In this research, we focused on the precipitation-ultrasonication method with favorable repetition to prepare glimepiride nanosuspensions, which avoided the use of toxic chlorinated organic solvents. The selection of water-miscible solvents was critical to obtain nanocrystals. In general, during precipitation process, solvents with high water-miscibility and stabilizers are able to ensure rapid supersaturation. Microprecipitation was the first size-controlling step. It could change the crystal type and physical state of drug particles and made homogenization process easier. The first homogenization process reduced drug particle size continuously, and it should help induce the absorption of surfactant onto drug particles, promoting an annealing effect which can increase stability of the nanosuspensions. Of course, this stability was also attributed to the reduced particle size. The formation of high-energy nanoparticle drug can increase Cs compared to microcrystalline, coarse drug, hence faster dissolution profiles were expected.
In order to overcome the particles growth and drug degradation during long-term storage of the nanosuspension, lyophilization was carried out to prepare dry nanoparticles and the dry nanopowders were further filled into capsules. The micronization of poorly soluble drugs could increase the dissolution rate due to the increase in surface area, but did not change the saturation solubility. When the particle size is reduced to the nanometer range, the solubility of drugs will increase significantly. In this study, the dissolution rate was markedly enhanced in the nanocrystal-loaded capsules, and nearly 90% of the drug dissolved within 30 min, as opposed to only 40–50% for the microcrystal-loaded capsules and marketed capsules. This could be attributed to the increased surface area and enhanced saturation solubility of drug in the nanoparticles. Therefore, formulating the poorly water-soluble glimepiride as nanometer-size particles had a dramatic effect on the drug solubility, dissolution rate.
In this paper, we also investigated the glimepiride release in vivo. The in vivo properties of nanoparticle formulation strongly depend on the nanoparticle size, dissolution rate, and nature and density of the coating. The nanocrystals could release slowly and stably in vivo without drug burst release. Dosing glimepiride nanoparticles-loaded capsules resulted in significantly greater Cmax, MRT and exposure (AUC), compared to two other capsule formulations and the bioavailability of the nanocrystal formulation was significantly improved, these provided important guarantee for steady therapeutic effect and low side effect. In addition, better correlation between in vitro and in vivo drug release from the nanocrystals was achieved, which indicated that the experimental method of in vitro drug release was reasonable and drug release in vivo could be inferred by drug release in vitro.
Conclusion
The dissolution behaviors of glimepiride nanocrystal-loaded capsules are superior compared to other formulations (microcrystal-loaded capsules and marketed capsules). Dissolution studies demonstrated a marked enhancement of dissolution rate was achieved by the reduction in particle size, which could account for improved absorption observed in vivo. Thus, nanocrystal-loaded capsules may reduce the risk of side effect (such as hypoglycemia) by allowing a reduction in either the dose or its frequency of administration. In our future work, we will evaluate whether this nanocrystal-loaded capsules of glimepiride will result in an increase of therapeutic efficacy in diabetic rats.
Acknowledgements
We thank Mr Tianfu Song (College of Physical Science and Engineering, Zhengzhou University) for the scanning electron micrographs of glimepiride nanoparticles.
Declaration of interest
All authors explicitly state that they all have no conflicts of interest to report.
References
- Ambrusa R, Kocbek P, Kristl J. (2009). Investigation of preparation parameters to improve the dissolution of poorly water-soluble meloxicam. Int J Pharm 381:153–9
- Du B, Li XT, Zhao Y, et al (2010). Preparation and characterization of freeze-dried 2-methoxyestradiol nanoparticle powders. Pharmazie 65:471–6
- Eerdenbrugh VB, Mooter VG, Augustijns P. (2008). Top-down production of drug nanocrystals: nanosuspension stabilization, miniaturization and transformation into solid products. Int J Pharm 364:64–75
- Fakes MG, Blisse JV, Feng Q, Sridhar D. (2009). Enhancement of oral bioavailability of an HIV-attachment inhibitor by nanosizing and amorphous formulation approaches. Int J Pharm 370:167–74
- Ganta S, Paxton JW, Baguley BC, Garg S. (2009). Formulation and pharmacokinetic evaluation of an asulacrine nanocrystalline suspension for intravenous delivery. Int J Pharm 367:179–86
- Geinsen K. (1988). Special pharmacology of the new sulfonylurea glimepiride. Drug Res 38:1120–30
- Jacobs C, Müller RH. (2002). Production and characterization of a budesonide nanosuspension for pulmonary administration. Pharm Res 19:189–94
- Kaushal AM, Gupta P, Bansal AK. (2004). Amorphous drug delivery systems: molecular aspects, design, and performance. Crit Rev Ther Drug Carrier Syst 21:133–93
- Keck CM, Müller RH. (2006). Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm 62:3–16
- Kesisoglou F. (2007). Nanosizing – oral formulation development and biopharmaceutical evaluation. Adv. Drug Dev Rev 59:631–44
- Kipp J. (2004). The role of solid nanoparticle technology in the parenteral delivery of poor water-soluble drugs. Int J Pharm 284:109–22
- Kouichi I, Masaki W, Youhei N, et al (2005). Efficacy of glimepiride in Japanese type 2 diabetic subjects. Diab Res Clin Prac 68:250–7
- Lipinski C. (2002). Poor aqueous solubility – an industry wide problem in drug discovery. Am Pharm Rev 5:82–5
- Massimo MB. (2003). Glimepiride in type 2 diabetes mellitus: a review of the worldwide therapeutic experience. Clin Ther 25:799–816
- Mauludin R, Müller RH, Kecket CM. (2009). Development of an oral rutin nanocrystal formulation. Int J Pharm 370:202–9
- Moschwitzer J, Achleitner G, Pomper H, Müller RH. (2004). Development of an intravenously injectable chemically stable aqueous omeprazole formulation using nanosuspension technology. Eur J Pharm Biopharm 58:615–19
- Patravale VB, Kulkarni RM. (2004). Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol 56:827–40
- Pu XH, Sun J, Wang Y, Wang YJ. (2009). Development of a chemically stable 10-hydroxycamptothecin nanosuspensions. Int J Pharm 379:167–73
- Quan P, Xia D, Piao HZ, et al (2011). Nitrendipine nanocrystals: its preparation, characterization, and in vitro-in vivo evaluation. AAPS PharmSciTech 12:1136–43
- Rabinow BE. (2004). Nanosuspensions in drug delivery. Nat Rev Drug Discov 3:785–96
- Rosario P, Claudio B, Piera F, et al (2002). Eudragit RS100 nanosuspensions for the ophthalmic controlled delivery of ibuprofen. Eur J Pharm Sci 16:53–61
- Shelesh J, Swarnlata S. (2010). Type 2 diabetes mellitus – its global prevalence and therapeutic strategies. Diab Metab Syndr 4:48–56
- Xia D, Quan P, Piao H, et al (2010). Preparation of stable nitrendipine nanosuspensions using the precipitation-ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm Sci 40:325–34
- Yang W, Keith P, Johnston KP, et al (2010). Comparison of bioavailability of amorphous versus crystalline itraconazole nanoparticles via pulmonary administration in rats. Eur J Pharm Biopharm 75:33–41
- Zhang DR, Tan TW, Gao L, et al (2007). Preparation of azithromycin nanosuspensions by high pressure homogenization and its physicochemical characteristics studies. Drug Dev Ind Pharm 33:569–75