
Abstract
Context: Designing a sustained release system for Carvedilol to increase its residence time in the stomach.
Objective: Preparation of floating microsphere by the emulsion solvent diffusion method, studying the effect of various process parameters and optimize the formulation using full factorial design.
Methods: Different microsphere formulations were prepared by varying the ratio ethanol:dichloromethane (1:0 to 1:1.5), ethyl cellulose:hydroxypropyl methyl cellulose and stirring speed (800–1600 rpm). The effect of these variables on particle size, encapsulation parameters, surface topography, in vitro floatability and drug release were evaluated.
Results: 32 full factorial design was used for the optimization of the formulation. Drug entrapment efficiency, particle size and in vitro drug release were dependent on concentration of ethyl cellulose and stirring speed. Microspheres remained buoyant for more than 10 h and showed sustained release of the drug.
Conclusion: Floating microspheres of Carvedilol with good floating ability and sustained release were developed.
Introduction
Among the various important approaches developed so far to increase the gastric retention time of dosage form, gastro retentive drug-delivery systems (GRDDS) is one among them. (Singh & Kim, Citation2000; Roy & Shahiwala, Citation2009; Sathish et al., Citation2011; Gurnany et al., Citation2012). The release of drug from the dosage form is significantly extended over a long period of time by GRDDSs. They prolong the dosing intervals, and also increase patient compliance when compared with the existing controlled release dosage forms. Gastric emptying of dosage form is an extremely variable process and the ability of GRDDSs to prolong and also to control the emptying time is an important asset for dosage forms. The GRDDS includes single unit dosage forms and multiple unit dosage forms (Streubel et al., Citation2006). Although extensive studies have been done on the single unit sustained dosage forms, they have a disadvantage of a release, all or nothing emptying process from the stomach, as a result, the absorption of drug shows a high inter-individual variability. To overcome these drawbacks multiple unit systems have been developed. Moreover, the active ingredients are released at a sustained rate and avoid dose dumping (Roy & Shahiwala, Citation2009).
The gastric retention of solid dosage forms in a controlled way may be achieved by the mechanisms of modified shape system, floatation, expansion, mucoadhesion, sedimentation or by the simultaneous administration of pharmacological agent that delay gastric emptying (Gurnany et al., Citation2012). Among the various approaches, floating drug delivery systems (FDDS) achieves gastric retention by the mechanism of floatation. These systems have a bulk density less than gastric fluids and hence they remain buoyant in the stomach for a longer period of time without affecting the gastric emptying rate (Sathish et al., Citation2011). The drug is released slowly when the system floats in the gastric fluid at the desired rate. The residual system is then emptied from the stomach after the drug release. This results in an increased gastric retention time and better control of fluctuations in the plasma drug concentration.
Among the several gastro-retentive drug delivery systems, floating microspheres are working on non-effervescent approach. These microspheres are found as free flowing powders consisting of proteins or synthetic polymers. Their size is ideally less than 300 µm (Jain et al., Citation2008; Bhadouriya et al., Citation2012). Solid biodegradable microspheres incorporating a drug dispersed or dissolved throughout particle matrix have the potential for controlled release of drugs. In case of sparingly soluble and insoluble drugs, as the solubility of drug decreases, the time available for drug dissolution becomes less adequate, hence the transit time becomes a significant factor affecting drug absorption. Floating microspheres are especially useful in the delivery of such drugs and provides continuous, controlled administration of drug at the absorption site. For drugs with relatively short half life, sustained release of the drug into the gastrointestinal tract maintain an effective concentration of drug in the systemic circulation for a long time and result in a flip-flop pharmacokinetics. So, formulating floating microspheres for short half-life drugs shows good therapeutic effect (Hoffman et al., Citation2004). Carvedilol is a dual action cardiovascular agent (Chakraborty et al., Citation2010). It is a vasodilating, non-selective beta adrenergic blocking agent with alpha-1 blocking activity and has a short biological half-life of 2–6 h and its solubility in water is very low. The oral absolute bioavailability of carvedilol is only 25%–35% (Yedurkar et al., Citation2012).
Materials and methods
Materials
Carvedilol was kindly obtained as a gift sample from Anugraha Chemicals, Bangalore, India. Ethyl cellulose and hydroxypropyl methyl cellulose was supplied by Nice Chemicals (Cochin, Kerala, India). All other reagents and chemicals used in this study were of analytical grade.
Preparation of floating microspheres
Floating microspheres with a hollow cavity at the centre were prepared by using solvent diffusion–evaporation technique. In the pre-optimization trials, a mixture of ethanol and dichloromethane in varying ratio were prepared and required quantities of drug (0.25 g), ethyl cellulose and hydroxypropyl methyl cellulose were weighed and dissolved in the solvent mixture at room temperature. This solution was then poured into distilled water (100 ml) containing 0.1% of the surfactant, Tween-80 which was maintained at a temperature of 30 °C–40 °C. The emulsion thus obtained was then stirred at 800 rpm using a propeller type agitator for a period of 45 min. During this period, the volatile solvent gets evaporated and microspheres were obtained. The microspheres were then filtered, washed with water and dried overnight at room temperature (El-Kamel et al., Citation2001; Ramachandran et al., Citation2010; Goyal et al., Citation2011).
Design of experiments
A 32 full factorial was used to design the experiments (Dave et al., Citation2004). The independent variables selected were concentration of EC and stirring speed, whereas percentage drug entrapment efficiency, percentage drug release at 10th hour and particle size were selected as the dependent variables (Awasthi et al., Citation2012; Tinny et al., Citation2013). In the pre-optimization studies, the solvent (dichloromethane:ethanol) ratio and the concentration of HPMC were optimized based on the results obtained from particle size measurement, shape and surface morphological studies, floating ability, percentage drug entrapment efficiency and percentage drug release in 10 h. Formulations B1 to B3 () were prepared by differing the solvent ratios and formulations C1 to C12 () were prepared using different concentrations of EC and HPMC. In the optimization studies, formulations F1 to F13 were prepared by differing the levels of EC concentration and stirring speed. The responses of the dependent variables were then evaluated. For each response, the polynomial equations were generated using Design Expert Software (8.0.3.1) (Statease, Minneapolis, MN), and from the results obtained the best formulation was selected, and the in vitro drug release data of the optimized formulation was treated to kinetic models such as zero order, first order, Higuchi model and Peppa’s model in order to determine the drug release mechanism (Li et al., Citation2003; Dave et al., Citation2004; Nagarwal et al., Citation2009).
Table 1. Formulation composition of carvedilol floating microspheres subjected to pre-optimization studies.
Evaluation of floating microspheres of carvedilol
Particle size and surface morphology analysis
The particle sizes of the floating microspheres were evaluated using an optical microscope fitted with a calibrated eyepiece micrometer. It randomly measured the particle diameters of about 100 microspheres and the average particle size was determined using the Edmondson’s equation:
where “n” stands for the number of counted microspheres, and “d” for the mean size range. Scanning electron microscopy (SEM) was used to study the shape and surface morphology of the microspheres. Photographs were taken using scanning electron microscope (6390, Jeol JSM, Tokyo, Japan).
Thermal analysis
The thermoanalytical examinations were carried out with a differential scanning calorimeter equipped with a thermal analysis data system (Perkin Elmer DSC7, Waltham, MA). The DSC curves of the pure drug carvedilol, the polymers-ethyl cellulose, hydroxypropyl methyl cellulose and the microsphere formulation were analyzed and compared.
Percentage yield of microsphere formulations
The percentage yield was found out for all the formulations based on the dry weight of the drug and the polymers taken. The percentage yield can be calculated using the following equation:
Percentage drug entrapment efficiency
Twenty-five milligrams of floating microspheres were dissolved in 10 ml ethanol. The samples were then assayed using UV spectrophotometer at 242 nm after suitable dilution for drug content. Percentage drug entrapment efficiency was then calculated using the following equation:
Micromeritic properties
The micromeritic properties of microspheres like bulk density, tapped density, angle of repose, compressibility index and Hausner’s ratio were evaluated to analyze the flow properties of floating microspheres.
In vitro evaluation of floating ability
Floating behavior of carvedilol microspheres was studied using an USP dissolution test apparatus II (Electrolab, Mumbai, India). The microspheres (100 mg) were spreaded on 900 ml of 0.1 mol/l HCl containing the surfactant Tween-80 at a concentration of 0.02%. The medium was agitated at 100 rpm with a paddle, and the temperature was maintained at 37 °C. After 12 h, both the settled and floating portions of microspheres were collected separately. Then the microspheres were dried and weighed. The percentage of floating microspheres was calculated using the following equation:
In vitro drug release study
Carvedilol floating microspheres were evaluated for the in vitro drug release studies in simulated gastric fluid. USP dissolution test apparatus I (Basket type) was used to find out the drug release rate from microspheres. The dissolution test was performed using 900 ml of 0.1 N HCl, at 37 ± 0.5 °C and 100 rpm. Microspheres equivalent to 50 mg carvedilol were accurately weighed and added to the dissolution medium, aliquots (2 ml) were withdrawn at hourly intervals for a period of 12 h. Perfect sink condition was established during the drug dissolution study period by replacing an equivalent volume of dissolution medium. The samples were filtered, and solutions were analyzed at 240 nm using a UV Spectrophotometer (UV 1800, Shimadzu, Kyoto, Japan).
Data analysis
A statistical model incorporating polynomial and interactive terms were used to evaluate the responses:
where β0, the intercept is the arithmetic average of all quantities outcomes of 13 runs, X1 and X2 are the coded levels of the independent variables. The terms X1X2 and
(i = 1,2) are the interaction terms, respectively.
Determination of mechanism of drug release
To understand the mechanism of drug release, in vitro drug release data of the optimized formulation was treated to kinetic models such as zero order, first order, Higuchi model and Peppa’s model. The most appropriate model was selected based on best goodness-of-fit criteria (Korsmeyer et al., Citation1983; Peppas, Citation1985; Costa & Sousa Lobo, Citation2001; Jose et al., Citation2013).
Results and discussion
Pre-optimization studies
In the pre-optimization studies, the solvent ratio (ethanol:dichloromethane ratio) and the concentration of HPMC were optimized. EC was used as the polymer for the preparation of floating microspheres because of its release rate controlling ability, non-toxicity, film forming ability and stability at GI pH.
Different formulations of floating microspheres were prepared by varying the solvent ratio. Drug:polymer concentration was fixed to be 1:3. Studying the shape and surface topography as well as the results of entrapment efficiency and floating ability of the prepared formulations showed that formulation B1 yielded irregular solid microspheres with no considerable buoyancy whereas formulation B3 yielded spherical hollow microspheres with very good floating ability (64.78%) and entrapment efficiency (67.48 ± 0.46%). From the results, formulation B3 with solvent ratio 1:1 was selected for further studies.
In the optimization of HPMC concentration studies, 12 different formulations were prepared by changing the amount of EC and HPMC. The amount of drug added was fixed to be 250 mg. Amount of EC used was found to have a significant impact on drug entrapment efficiency and percentage drug release at 10th hour. For formulation C1 to C3 as the concentration of EC increased; entrapment efficiency was found to increase from 78.18% to 88.93% (), because higher the polymer content, higher is the probability of drug surrounded by the polymer, that act as a barrier for the diffusion of the drug to the external phase, thereby increasing the entrapment efficiency. However, at higher concentrations of EC, drug release was found to be greatly retarded, since ethyl cellulose is a release rate retardant when used in high concentrations. The percentage drug release ranged from 56.06% to 41.12%. Therefore, to increase the drug release rate, HPMC was selected to be used in combination with EC. Formulations C4 to C12 were formulated by combining EC and HPMC. It was observed that as the concentration of HPMC increased drug release increased (from 62.14% to 77.73%), same time drug entrapment efficiency (from 71.54% to 60.71%) was found to be decreased. As the concentration of EC increased percentage floating ability increased but as the concentration of HPMC increased (for the preparations with same EC concentration) floating ability decreased. Concentrations of EC and HPMC showed an adequate drug release along with drug entrapment efficiency in the case of formulation C8. Hence, formulation C8 with entrapment efficiency (81.34 ± 0.57%), floating ability (75.88%) and drug release (68.72 ± 0.19%) was selected for the optimization studies. From the literature review, it was found that stirring speed and EC concentration shows a significant effect on drug entrapment efficiency, drug release and particle size. So stirring speed and EC concentration was selected as the independent variables and 13 formulations were prepared as per 32 full factorial design.
Table 2. Evaluation of carvedilol floating microspheres subjected to pre-optimization of HPMC concentration.
Characterization and evaluation of formulation subjected to optimization
Particle size
The particle size of the formulation was determined, and the mean particle size of the microspheres was found to be increasing with increasing EC concentration (from 184 µm to 330 µm). The viscosity of the medium increased at higher concentrations of EC resulting in increased interfacial tension and decreased shearing efficiency. Effect of different stirring speed (800 rpm to 1600 rpm) on particle size was also studied. And results showed that the particle size decreased as stirring speed increased.
Thermal analysis
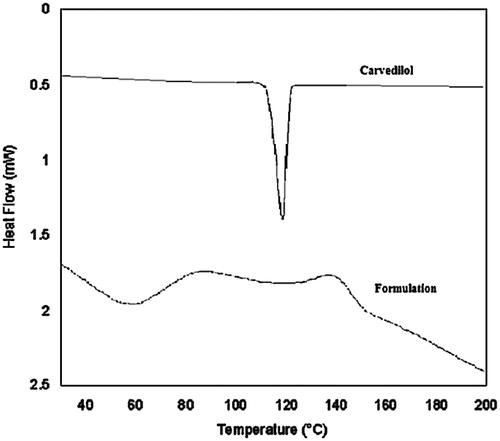
The DSC thermogram of the microsphere formulation is depicted in . DSC is an important technique used to characterize the solubility and physical state of drug in the polymeric matrix. No melting point, typical of carvedilol (118 °C), was detected in the DSC curve of the drug loaded hollow microsphere indicating that the drug was present as molecular dispersion in the polymer matrix.
Figure 1. DSC thermogram of the carvedilol alone and carvedilol formulation.
Shape and surface morphological analysis
The shape and surface morphology of the hollow microspheres were studied and the images showed microspheres with a hollow cavity at the center, smooth surface and spherical shape as in . The microspheres were having a porous internal surface and a smooth and dense outer surface. The microsphere shell also showed some porous structure (). It may be caused by the evaporation of solvent entrapped within the shell of the microspheres.
Figure 2. SEM image showing microsphere with a central hollow cavity (a); SEM image of the surface of a hollow microsphere – enlarged view (b); SEM image of a hollow microsphere wall-enlarged view (c).

Determination of percentage yield
Percentage yield of microspheres varied from 64.43% to 84.43% (). The results showed that upon increasing the concentration of the polymer, the yield of the microspheres increased. When stirring speed increased from 800 to 1200 rpm, the percentage yield increased but decreased with further increment to 1600 rpm. Again even at higher stirring speed (1600 rpm), the percentage yield decreased as it leads to the breakup of microspheres and formation of coagulant masses of microspheres. Hence, the stirring speed should be optimized to obtain the desired percentage yield.
Table 3. Evaluation of carvedilol floating microspheres formulated by 32 full factorial design.
Determination of percentage drug entrapment efficiency
The drug entrapment efficiency of microspheres varied from 60.53% to 88.71% (). Results showed that, as the concentration of EC increased the entrapment efficiency of the drug increased. However, stirring speed was found to have a negative effect on entrapment efficiency.
Micromeritic properties
The angle of repose of formulations of the microspheres ranged from 21.80° to 40.91°. The tapped density values of formulations of the microspheres ranged from 0.2713 to 0.3350 g/cm3. The percentage compressibility index (Carr’s index) was ranged between 11.63% and 25.00%. The Hausner’s ratio was calculated to find out whether the formulations are free flowing or cohesive in nature and the results are given in the . The values of Carr’s index, Hausner’s ratio and the angle of repose indicate excellent flow properties for the formulations except formulation F2, F3, F6 and F9.
In vitro evaluation of floating ability
The floating ability test was carried out and from the results obtained, formulations F7 and F8 have floating ability greater than 80% and formulations F1, F4, F5, F11, F12 and F13 were found to have floating ability between 70% and 80%. The results also showed that, the larger the particle size, the longer the floating time (). However, the in vivo situation may be quite different depending on the phase of gastric motility, but the pH of the stomach in fed and fasted state does not affect the floating ability or drug release as the main polymer used in this study is pH insensitive in nature.
In vitro drug release study
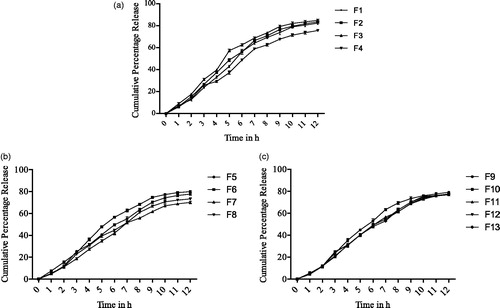
In vitro dissolution studies of carvedilol from floating microspheres were performed in 0.1 N HCl (pH 1.2) for 12 h using the USP dissolution test apparatus. shows the cumulative percentage drug release at 10th hour for various formulations from F1 to F13. It was found that formulations F1, F2 and F3 showed 78.95%–82.05% of drug release at 10th hour and above 80% drug release in 12 h. In order to achieve a sustained drug release, the EC concentration was increased in the formulations. For formulations F4, F5 and F6, the drug release was 71.4%–77.30% within 10 h and 75.6%–79.98% in 12 h. Formulations F7, F8 and F9 showed 66.98%–76.01% of drug release at 10th hour and 70.05%–79.06% drug release in 12 h. Furthermore, from the results, it is also clear that no burst effect was seen, and drug release was significantly sustained (). It was found that as the concentration of EC increased, the percentage cumulative release of drug decreased.
Figure 3. Cumulative in vitro drug release from formulations F1 to F4 (a); cumulative in vitro drug release from formulations F5 to F8 (b); cumulative in vitro drug release from formulations F9 to F13 (c).
32 full factorial design
The responses obtained from the design matrix, like particle size, drug entrapment efficiency, cumulative percentage drug release at 10th hour were statistically evaluated using Design Expert 8.0.3.1 software (Statease, Minneapolis, MN) and is given in . The model developed can be characterized by using the polynomial equation representing the respective response data, which is as follows:
where Y1 is the particle size, Y2 is the drug entrapment efficiency, Y3 is the CPRD at 10th hour, A is the concentration of ethyl cellulose in mg and B is the stirring speed in rpm.
showed nearly linear decreasing patterns for the particle size values as the stirring speed increased. But at higher levels of the polymers, the response surface showed a slightly linear shape. Maximum particle size is observed at high levels of ethyl cellulose concentration.
Figure 4. Response surface plot for the response, particle size (a); entrapment efficiency (b); and cumulative percentage drug release at 10th hour (c).
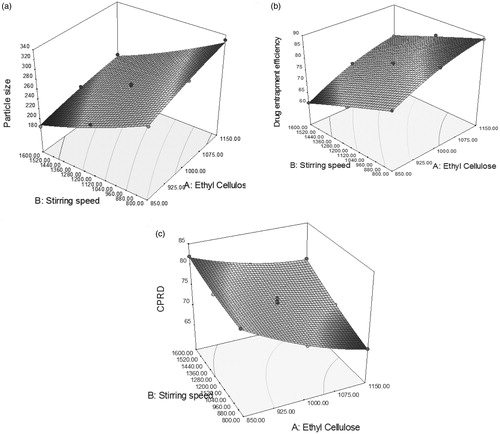
depicts a quite linear increasing pattern in the drug entrapment efficiency values with increased value of ethyl cellulose concentration and nearly linear decreasing trend with stirring speed. However, the influence of stirring speed is more significant than that of the ethyl cellulose concentration. Hence, the lower levels of stirring speed have to be complemented with higher levels of EC to maintain drug entrapment at a constant level.
reveals a decrease in the cumulative percentage release of drug (CPRD) values at 10th hour with the increase in the concentration of ethyl cellulose. A linear increasing pattern is seen in the values of CPRD (10 h) with the increase in stirring speed. It also showed that the variation in CPRD (10 h) is a complex function of the concentration of EC and the effect of stirring speed is less significant.
Formulation F8 showed comparatively smaller particle size than other formulations. Though formulations F2, F3, F6, F9 have smaller particle size than F8, these formulations have low drug entrapment efficiency and their drug release rate at 10th hour were too high so that a sustained drug release was not attainable in those formulations.
Percentage floating ability of formulation F8 was higher when compared with other formulations. Although formulation F7 has highest floating ability, its particle size was very high; which is not suitable.
Although the formulations F3, F6, F9 (stirring speed 1600 rpm) resulted in smaller microspheres compared to other formulations, their yield was low due to the formation of coagulant masses of microspheres during their formation due to very high stirring speed. As a result floating ability of the microspheres also decreased. lists the actual and predicted values of all the three responses. The observed values are within the limits.
Table 4. Comparative levels of actual and predicted responses of carvedilol floating microspheres.
Formulation F8 showed a controlled release of drug and only 70.66% of drug was released at 10th hour. So a sustained drug release is possible with F8. Formulations F1, F2, F3 showed very high drug release (above 75%) at 10th hour, which is not suitable. So from the optimization results, formulation F8 was selected as the optimized formulation.
Determination of mechanism of drug release
In order to understand the mechanism of drug release, in vitro drug release data of the optimized formulation F8 were treated to kinetic models such as zero order, first order, Higuchi’s model and Peppa’s model. The interpretation of data was based on the value of the resulting regression coefficient R2 value which was 0.955 for zero order, 0.825 for first order, 0.991 for Higuchi’s model and 0.969 for Peppa’s model. The in vitro drug release showed the highest regression coefficient values for Higuchi’s model, so diffusion was found to be the predominant mechanism of drug release. When analyzed according to Peppa’s model, the release exponent was greater than 1.0. Therefore, the diffusional release was found to follow Super Case II transport.
The results of the 32 full factorial design revealed that the concentration of EC and stirring speed significantly affected the dependent variables such as drug entrapment efficiency, drug release at 10th hour and particle size of the microspheres. The polynomial equation–based optimization model was generated and evaluated. In vitro data obtained for the floating microspheres of carvedilol showed good floating ability and sustained drug release. The microspheres of the optimum batch (F8) exhibited 83.26% drug entrapment efficiency, mean particle size of 261 µm and 70.66% drug release at 10th hour. Further, diffusion was found to be the main release mechanism. Thus, the prepared floating microspheres may prove to be the potential candidate for multiple-unit drug delivery devices adaptable to any intra gastric condition.
Conclusion
Gastro retentive drug delivery system based on non-effervescent approach was successfully designed and developed. In this study, floating microspheres of carvedilol were prepared and optimized. The microspheres were discrete, spherical with a central hollow cavity and showed sustained drug release patterns in simulated GI fluids. The drug entrapment efficiency, drug release and particle size of the microspheres were dependent on the concentration of ethyl cellulose and stirring speed. The formulated microspheres floated in the simulated gastric fluid for over a period of 10 h. Thus floating microspheres of carvedilol with good buoyancy and modified drug release were obtained.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the article.
References
- Awasthi R, Kulkarni G, Pawar V, Garg G. (2012). Optimization studies on gastroretentive floating system using response surface methodology. AAPS PharmSciTech 13:85–93
- Bhadouriya P, Kumar M, Pathak K. (2012). Floating microspheres – to prolong the gastric retention time in stomach. Curr Drug Deliv 9:315–24
- Chakraborty S, Shukla D, Mishra B, Singh S. (2010). Clinical updates on carvedilol: a first choice beta-blocker in the treatment of cardiovascular diseases. Expet Opin Drug Metabol Toxicol 6:237–50
- Costa P, Sousa Lobo JM. (2001). Modeling and comparison of dissolution profiles. Eur J Pharmaceut Sci 13:123–33
- Dave BS, Amin AF, Patel MM. (2004). Gastroretentive drug delivery system of ranitidine hydrochloride: formulation and in vitro evaluation. AAPS PharmSciTech 5:77--82
- El-Kamel AH, Sokar MS, Al Gamal SS, Naggar VF. (2001). Preparation and evaluation of ketoprofen floating oral delivery system. Int J Pharm 220:13–21
- Goyal P, Gill S, Gupta UD, et al. (2011). Development and characterization of rifampicin loaded floating microspheres. Artif Cell Blood Substit Biotechnol 39:330–4
- Gurnany E, Manwani R, Singhai P, et al. (2012). Gastro-retentive floating multiparticulate drug delivery system: a review. Curr Drug Deliv [CDD-EPUB-20120521–5]
- Hoffman A, Stepensky D, Lavy E, et al. (2004). Pharmacokinetic and pharmacodynamic aspects of gastroretentive dosage forms. Int J Pharm 277:141–53
- Jain SK, Agrawal GP, Jain NK. (2008). Floating microspheres as drug delivery system: newer approaches. Curr Drug Deliv 5:220–3
- Jose S, Fangueiro JF, Smitha J, et al. (2013). Predictive modeling of insulin release profile from cross-linked chitosan microspheres. Eur J Med Chem 60:249–53
- Korsmeyer RW, Gurny R, Doelker E, et al. (1983). Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm 15:25–35
- Li S, Lin S, Daggy, BP, et al. (2003). Effect of HPMC and Carbopol on the release and floating properties of gastric floating drug delivery system using factorial design. Int J Pharm 253:13–22
- Nagarwal RC, Srinatha A, Pandit JK. (2009). In situ forming formulation: development, evaluation, and optimization using 3(3) factorial design. AAPS PharmSciTech 10:977–84
- Peppas NA. (1985). Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv 60:110–11
- Ramachandran S, Shaheedha SM, Thirumurugan G, Dhanaraju MD. (2010). Floating controlled drug delivery system of famotidine loaded hollow microspheres (microballoons) in the stomach. Curr Drug Deliv 7:93–7
- Roy P, Shahiwala A. (2009). Multiparticulate formulation approach to pulsatile drug delivery: current perspectives. J Contr Release 134:74–80
- Sathish D, Himabindu S, Kumar YS, et al. (2011). Floating drug delivery systems for prolonging gastric residence time: a review. Curr Drug Deliv 8:494–510
- Singh BN, Kim KH. (2000). Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release 63:235–59
- Streubel A, Siepmann J, Bodmeier R. (2006). Drug delivery to the upper small intestine window using gastroretentive technologies. Curr Opin Pharmacol 6:501–8
- Tinny T, Chacko AJ, Jose S. (2013). Formulation development and statistical optimization of chronotherapeutic tablets of indometacin. Drug Dev Ind Pharm 39:1357--63
- Yedurkar P, Dhiman MK, Petkar K, Sawant K. (2012). Mucoadhesive bilayer buccal tablet of carvedilol-loaded chitosan microspheres: in vitro, pharmacokinetic and pharmacodynamic investigations. J Microencapsul 29:126–37