Abstract
The purpose of this paper is to prepare a less-irritating and lipid-based clarithromycin complex (LCC) by the route of intravenous administration qualified with high drug-loading and fine particle size. The LCC was prepared by the injection of organic solvent phase (containing clarithromycin, sodium cholesterol sulfate and phospholipid at a molar ratio of 1:1:1.5) into the Tris buffer solution (0.05 M, pH 7.2) at 40 °C, and then was concentrated by ultrafiltration to remove the organic solvent. The technique of lyophilization was applied to obtain the LCC lyophilized products. Evaluation of the injectable LCC was performed by particle sizes analysis, transmission electron microscope, entrapment efficiency, stability and irritation tests. The LCC possessed a log-normal size distribution with an average size of 75.4 nm. The drug entrapment efficiency was above 97.0%, which was influenced by the amount of phospholipid, pH value of the media and ionic strength of aqueous suspension. The results of stability and irritation tests proved that LCC had more stability and less irritation. This unique LCC formulation may be applied to the human by the route of injection with less or no irritation. LCC could be an appropriate candidate for intravenous preparation and has a great potential for clinical and industrial-scale production.
Introduction
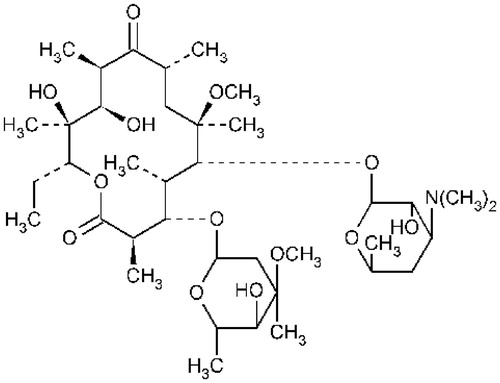
Clarithromycin (CL) is a semi-synthetic macrolide antibiotic with identical structure to erythromycin except that the methoxyl group has been substituted by the hydroxyl group (), which makes CL more stable than erythromycin in acidic condition (Kohno et al., Citation1989; Nakagawa et al., Citation1992), and become a better alternative over erythromycin. Clarithromycin also shows higher bioavailability, better tissue distribution and more antimicrobial activity against a wide range of gram-positive and gram-negative organisms, such as Staphylococcus aureus. Streptococcus pyogenes. Moraxella catarrhalis and Streptococcus pneumonia (Langtry & Brogden, Citation1997). It is also highly active against pathogens caused atypical pneumonia.
Figure 1. The chemical structure of clarithromycin.
Clarithromycin is currently administered in oral dosage forms, such as tablets, capsules and suspensions. Pharmacokinetics study indicates that the absolute bioavailability of oral administration is just 50% (Chu et al., Citation1992). Therefore, the market would be in great need of an alternative intravenous injection preparation of CL to improve its bioavailability and efficiency in clinic. But the irritation caused by the injectable CL lactobionate preparation has limited the intravenous administration to a certain extent (Zimmermann et al., Citation2001; Zimmerm et al., Citation2002). For researchers it is necessary to research and develop CL formulations with less or no irritation by the route of intravenous administration. It was reported that the interaction between drug molecules and local tissues might be the main reason for irritations following intravenous administration (Brown, Citation1970; Hecker et al., Citation1984). Therefore, the selection of a desired drug carrier to shield the drug molecule portions which might cause irritation would be the more effective method to reduce the irritation (Jiang et al., Citation2012; Zhao et al., Citation2013).
A number of CL formulations for intravenous administration had been reported. Lovell had developed a less irritating emulsion formulation for CL, but the formulation was not stable enough (Lovell et al., Citation1994). Tang had reported an intravenous CL emulsion containing vitamin E or loading with a CL–phospholipid complex, which had successfully reconstituted the matter of stability (Lu et al., Citation2008, Citation2009). But the drug concentration in CL emulsion was relatively low (2.5 mg/ml or 5.0 mg/ml) compared with the CL lipid-based complex prepared in the study (15 mg/ml), which may need to increase the dosage to obtain efficiency in clinical therapy. Cannon had studied a bile salt formulation for CL, which could reduce the irritation approximately by 50%, but the preparation might cause a rapid release of the drug following an intravenous injection (Cannon et al., Citation1995). What is more, the microspheres or the nanoparticles for CL had been prepared (Qin, Citation2007; Mohammadi, Citation2011), not only the reduction of irritation and/or the content of drug-loading were not satisfactory, but also the particle size was not suitable for intravenous injection. There was also a patent about the CL liposome, but it similarly exhibited the less stability and low drug-loading content (Flood et al., Citation1998).
Since the reported CL formulations for intravenous administration could not meet the requirements of less irritation, good stability and high drug-loading content more or less, the development of a new CL delivery system by the intravenous route would be indeed necessary.
Methods
Materials
Freeze-dried CL lactobionate (250 mg/vial) and sodium cholesterol sulfate (SCS) was supplied by Regenex Biotechnology Co. Ltd (Guangzhou, China). Clarithromycin was supplied by Huayi Pharmaceutical Co. Ltd (Zhejiang, China). Phospholipid (PC 72%) was obtained from Toshisun Enterprise Co. Ltd. (Shanghai, China). Soybean oil was obtained from Tieling BeiYa Pharmaceutical Co. Ltd. (Tieling, China). Medium chain triglycerides and oleic acid were purchased from Lipoid Company (Ludwigshafen, Germany). Lactose was obtained from Huamao Pharmaceutical Co. Ltd. (Shanghai, China). Hydrochloric acid solution and tri(hydroxymethyl) aminomethane were obtained from Tianjin Fuchen Chemicals Reagent Factory (Tianjin, China). All chemicals and reagents were of chromatographic or analytical grade.
Preparation of lipid-based clarithromycin complex
Clarithromycin (5 g) and SCS at a suitable ratio were dissolved in 100 ml dimethyl sulfoxide (DMSO) and heated by a water bath at 40 °C. Phospholipid solution was prepared by dissolving phospholipid into 100 ml ethanol at the same temperature. The organic phase was obtained by a mixture of CL DMSO solution and phospholipid solution, and then the organic phase solution was injected into 2000 ml Tris buffer solution (0.05 M, pH 7.2) at a certain speed stirring with the magnetic stirrer to form a transparent lipid-based clarithromycin complex (LCC) colloidal suspension at 40 °C. The LCC colloidal suspension was concentrated by ultrafiltration (molecular weight cut-off 100 kDa) several times to remove the organic solvent and then lyophilized (Four-Ring Science Instrument Plant Co. Ltd, Beijing, China) in the presence of 10% lactose (g/ml). The pre-freezing time and drying duration were 4 hours and 24 hours, respectively. During the pre-freezing period quick-frozen technique was applied because of the products having good appearances and fine particle sizes.
Characterization of LCC
Determination of particle size and particle size distribution
A vial of lyophilized LCC containing 250 mg CL was suspended into 50 ml double-distilled water to make a colloidal suspension. A sample of 2 ml was taken and determined by Mastersizer 1000HSA (Malvern Instruments, Malvern, UK) to obtain the average particle size and particle size distribution of the LCC.
Morphological property of LCC
The lyophilized LCC formulations were reconstituted in water to acquire the transparent colloidal suspension. Then the LCC colloidal suspensions were added onto copper grid and then stained with 2% phosphotungstic acid. Transmission electron microscope (TEM) (Hitachi H-600, Minato-Ku, Japan) was used to observe morphological characteristics of LCC.
Determination of drug entrapment efficiency
Lyophilized LCC containing 250 mg CL was reconstituted and diluted to 250 ml with double-distilled water. A sample of 0.2 ml was placed in an ultrafiltration cup (PALL Ltd. Co., Port Washington, NY), which is composed of a filter membrane (molecular weight cut-off 5 kDa). The sample was centrifuged at the speed of 5000 rpm for 5 min to separate the free drug and lipid complex by the membrane filtration. The concentrations of both free drug in the filtrate (B) and total drug in the solution without ultrafiltration (A) were analyzed by a reverse-phase HPLC-Coularry ECD (ESA HPLC Systems, Chelmsford, MA) method at 860 mV and Hypersil C18 column (4.6 mm × 250 mm, 5 μm). The mobile phase consisted of a mixture of 0.067 mol ċ L−1 potassium dihydrogen phosphate (pH was adjusted to 4.0 by HCl) and acetonitrile (60:40, v/v) at a flow rate of 1.0 ml/min. The detection was carried out at 210 nm. The standard curve of CL between the range of 0.1 and 8 µg/ml was linear with r2 ≥ 0.99. The efficiency of drug loading was calculated from the following equation, entrapment efficiency = (1 − B.A) × 100%, where A was the concentration of total drug in the solution without ultrafiltration and B was the concentration of free drug.
Drug release in vitro
Release test of CL complex were performed and evaluated according to the dialysis method. 20 ml of suspension of CL complex were performed and evaluated according to the dialysis method. 20 ml of suspension (appropriate 200 mg of CL) were placed into a dialysis bag and then put into 500 ml volume of release medium at 37 ± 0.5 °C with a rotate speed of 50 r/min. During the test, 20 μl samples were withdrawn at scheduled periods and then an equal volume of temperature equilibrated blank media was added. The samples were injected into HPLC the percentage of accumulated release was calculated.
Osmotic swelling test
LCC formulations composed of SCS, CL and phospholipid at a molar ratio of 1:1:1.5 and 1:1:2.5 were prepared separately. These two LCC dispersions were concentrated by ultrafiltration with a hollow fiber ultrafiltration membrane device (Motimo, Tianjin, China) and diluted with double-distilled water separately to assure that the lactose concentration of the final dispersion is not more than 1%. The particle size in different time after dilution was measured by Mastersizer 1000HSA (Malvern, UK).
Stability test
Stability studies of LCC in 5% dextrose
In order to evaluate the stability of LCC, a vial of lyophilized LCC containing 250 mg CL was reconstituted in 250 ml 5% dextrose (D5W). Then the particle size and drug entrapment efficiency were determined at different time.
Accelerated test
In the test, three batches of lyophilized LCC were put into transparent ampoules and then sealed with rubber stopper and aluminum cap. These investigated samples were placed in constant temperature humidity chamber at 40 °C ± 2 °C with RH 75% ± 5% for six months and were taken to study appearance, color, redispersibility, free drug and drug content at predetermined time.
Irritation tests in vivo
Sprague–Dawley rats and Albino New Zealand rabbits were obtained from Guangzhou University of Chinese Medicine Animal Center (Guangzhou, China). All animal studies were conducted in accordance with the Principles of Laboratory Animal Care and approved by the Animal Ethical Committee of Guangzhou University of Chinese Medicine.
Paw lick test in rats
In the test, a group of 10 rats (Sprague–Dawley, 180–220 g each) received a subcutaneous injection of 0.1 ml LCC suspension (5 mg/ml) each one (Celozzi et al., Citation1980; Comereski et al., Citation1986; Gupta et al., Citation1994). Thereafter, the number of time the animals licked their paws as well as the total time for which the paws were licked were monitored over a period of 15 min. Once again, 0.1 ml CLS and D5W served as positive and negative controls, respectively. The concentration of the CLS was the same as that of the LCC.
Rabbit ear vein test
Groups of three rabbits were given an infusion of 5 mg/ml LCC, CLS and D5W into their marginal ear veins. The rate of infusion was maintained 1 ml/min at a dose of 40 mg/kg daily for four days. Following infusion, visual observations of the vascular reaction were made every day. Twenty-four hours after the last administration, three rabbits from each group were killed and a piece of vascular tissue at the site of injection was removed for histopathological examination.
Results and discussions
Formulation investigations of LCC
Sodium cholesterol sulfate as the main excipient of LCC
In preliminary experiments, we found that SCS and CL could form a stable lipid composition in aqueous medium at neutral pH. The binding of the cholesterol sulfate and CL might cover the molecule portions of CL which causes irritation. In view of this, SCS was selected as the main excipient of LCC. It was possible that the partial positively charged CL and the negatively charged cholesterol sulfate could hang together in aqueous phase and form a stably colloidal particle dispersion system.
The molar ratio of SCS to CL in LCC
The molar ratio of SCS to CL was found to have a marked influence on the physical stability of LCC. When the molar ratio of SCS to CL is less than 1:1, it was found that only metastable or supersaturated solutions could be prepared which precipitated the drug within several hours or days. As the molar ratio of SCS to CL was 1:1, a relatively stable colloidal suspension was prepared without precipitation within five days. However, the particle size was increasing during storage in the form of solution at room temperature. With the molar ratio of SCS to CL increasing from 1:1 to 2:1, the lipid composition showed more and more physical stability and only few free drugs were detected in the medium (). Unfortunately, as the molar ratio increased, the lipid composition had a trend of hemolysis, which is dangerous for intravenous administration. So the molar ratio of SCS to CL of 1:1 was determined due to its less hemolysis and more stability.
Table 1. Influences of the molar ratio of SCS to CL on the size distribution and free drug content.
Phospholipid as the adjuncts in LCC
Several pharmaceutical adjuncts such as phospholipid, soybean oil, medium chain triglycerides and oleic acid were selected to control the particle size change during storage and to make the formulation more stable. Previous studies showed that the addition of soybean and medium chain triglycerides increased the particle size of LCC and formed a milky white suspension, and the addition of oleic acid had little influence on the particle size of LCC but accelerated drug precipitation. On the other hand, the addition of phospholipids made the formulation more stable with little particle size change even during storage in solution at room temperature. The reason why phospholipids made the formulation more stable needs further investigation. Our experiments indicated that the optimal molar ratio of SCS to CL to phospholipids is 1:1:1.5, which could form an optically clear suspension in Tris buffer solution (pH 7.2) with particle size distribution of 52 ∼ 105 nm.
Characterization of LCC
Particle size and particle size distribution of LCC
Size and size distribution are of great importance for nanoparticles drug delivery systems. Different particle sizes may result in different drug release in vitro and different drug diffusion in vivo. The LCC possessed a log-normal size distribution (). The intensity-averaged diameter of the LCC was 75.4 nm and the PDI was 0.318 nm.
Figure 2. Size distribution of clarithromycin lipid-based complex.
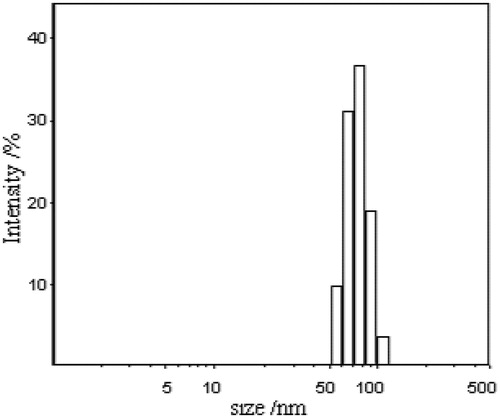
The particle size was influenced by the ionic strength of aqueous solutions and the concentration of phospholipids in the preparations. The results showed that the higher the ionic strength of the aqueous phase produced the larger the diameter for the prepared LCC. When the ionic strength increased to 0.35 M, the increase of LCC particle size became more rapidly. As mentioned above, the binding of CL and sodium cholesterol was due to their opposite charges. However, the higher ionic strength might decrease the binding affinity resulting in the increase of particle size.
Morphological studies of LCC
Images of the LCC prepared at the molar ratios of SCS to phospholipid of 1:1.5 and 1:2.5 were acquired by TEM () to explain the micro-structure of nanoparticles. The micrographs showed that all particles in were of sphere structure. Maybe the phospholipid bilayer structure could be demonstrated by . But it was not enough to conclude that these two types of particles had different structures only based on the TEM micrographs.
Figure 3. Microscope photograph (×10 000) of CLC (A) at The molar ration of sodium cholesterol sulfate and phospholipid of 1:1.5; (B) at the molar ration of sodium cholesterol sulfate and phospholipid of 1:2.5.
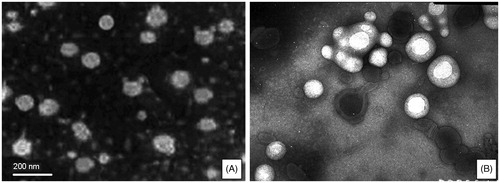
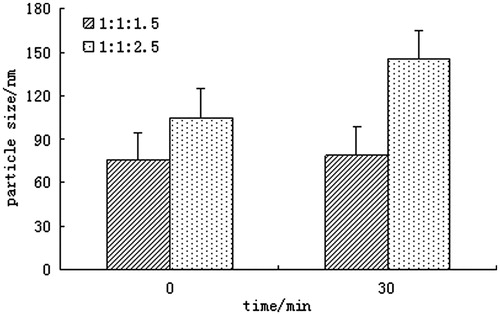
To further determine whether these two particles had the same structure, osmotic swelling test was used to prove whether there was phospholipids’ bilayer when the molar ratio of SCS to phospholipid was increased to 1:2.5. As we all know, liposomes are self-assembling vesicles, which are composed of phospholipid bilayer membranes surrounding an inner aqueous core. One characteristic of liposomes is that isotonic liposomes swell in a hypotonic medium (Grazia & Gerald, Citation1968). In view of this unique feature, two formulations of LCC composed of SCS, CL and phospholipid in molar ratio of 1:1:1.5 and 1:1:2.5 were tested, respectively, in double-distilled water at different time.
It could be seen from that there was no appreciable swelling at the molar ratio of CL/SCS/PC 1:1:1.5. However, an obvious swelling was detected and the mean particle size increased by approximately 40% at the molar ratio of CL/SCS/PC 1:1:2.5, which indicated the forming of phospholipid bilayer. Consequently, the micro-structure of LCC might have an essential change from lipid complex to liposome at the molar ratio of SCS to phospholipid of 1:2.5.
Figure 4. Particle size of CLC after osmotic swelling test at different time.
Entrapment efficiency of LCC
In our experiment the drug entrapment efficiency was above 97% and remarkably influenced by the molar ratio of phospholipid to SCS and the pH value of aqueous phase. Increase of the molar ratio of phospholipid to SCS would decrease the affinity of CL and SCS leading to more drug leakage from LCC and lower drug entrapment efficiency. This could be explained by the change of the particle structure. When the molar ratio of phospholipid to SCS was above 2:1, the lipid composition changed to liposome in which the drug was loaded into the phospholipid bilayer.
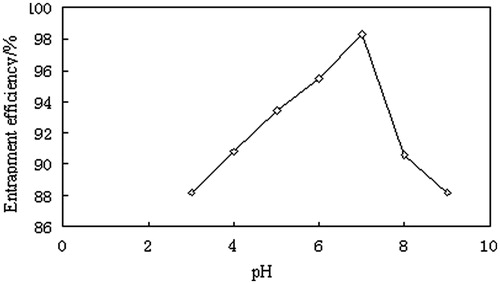
In our study, at the pH range of 3∼7.5, the drug solubility can play a more important role on the entrapment of LCC than the affinity of CL and SCS. While, at the pH range of 7.5∼9, the affinity of CL and SCS was more important. This might be why the entrapment of LCC increased firstly and then decreased as the pH value varying from 3 to 9 shown in . The Tris buffer solution (0.05 M, pH 7.2) selected as the aqueous phase can obtain higher entrapment efficiency and more stable LCC.
Figure 5. Effect of aqueous pH on entrapment efficiency of clarithromycin.
Drug release
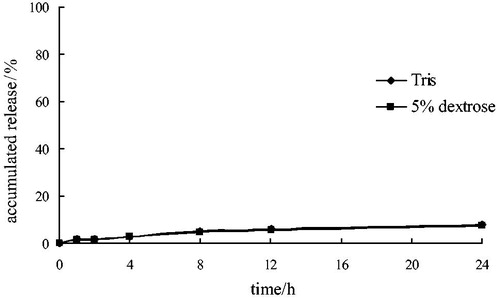
indicated that the release of CL lipid complex in the media of pH 7.2 Tris buffer and 5% dextrose was considerably slow. The accumulated release amount of CL was less than 2% and 10% at 1 hour and 24 hours, respectively. While the length of medication use in clinic was about 1 hour, the release of CL lipid complex can meet the clinical requirements.
Figure 6. The accumulated release of clarithromycin from lipid-based complex in different media.
Stability studies of LCC
The lyophilized LCC prepared in this study was designed to reconstitute and dilute in 5% dextrose solution before intravenous infusion in clinical application, so the stability of LCC in 5% dextrose solution was studied. We studied the variation of particle size distribution and drug entrapment efficiency in 5% dextrose after dilution for 6 hours. The results are shown in . No obvious changes of the particle size and drug entrapment efficiency were observed after dilution over 6 hours. It demonstrated that the formulation had a relatively good stability in 5% dextrose.
Table 2. Particle size and entrapment efficiency of LCC at different time in 5% dextrose.
In the accelerated test, LCC samples collected at different periods occurred as white powder and showed good redispersibility in D5W. Both drug content and free drug content in lyophilized LCC meet the requirements described in . And during the six months the particle size of LCC had no obvious change just increasing from 75.4 nm to 81.4 nm.
Table 3. Stabilities of CL lipid-based complex in the accelerated tests (40 °C ± 2 °C, RH 75% ± 5%, n = 3).
Irritation tests
Rat paw lick test is a model that assesses the reaction in rats due to pain or irritation caused by injection. The principle of rat paw lick test is that the more painful or irritating, the more animal licks its paws. In addition, the times of the animal licks their paws also increased with pain and/or irritation. When CLS was injected, all of the animals licked their paws immediately. The average number of times was 28 and the total licking time was 155 s. As LCC (SCS/CL/PC = 1:1:2.5) suspension was injected, eight out of 10 rats licked their paws. The average number of times was 15 and the total licking time was 84.5 s. However, when LCC (SCS/CL/PC = 1:1:1.5) was injected, the average number of times each rat licked its paws was about 7 and the average total licking time was 32.2 s.
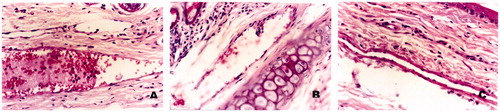
The rabbit vein irritation test was carried out to measure the irritation caused by LCC following intravenous injection and the observations were compared with LCC and D5W. The area around the rabbit ear vein in the group receiving D5W remained normal over the study period. In the group of LCC, the ears of all three rabbits were flushed with blood and obvious discoloration following infusion. With 5 mg/ml LCC, one rabbit exhibited slight discoloration of the vein during dosing while other two animals remained normal. The histopathological slides of the rabbit ear-rim auricular vein are shown in . From the macroscopic observations, vascular engorgement and dropsy were seen at the injection site, simultaneously angiectasia and erythrocyte aggregation were investigated at or away from the site of administration in the CLS group. However, these phenomena were not apparent in the group of LCC and D5W. Based on these investigations, LCC formulation produced less irritation than CLS.
Figure 7. The histopathologic slide photos (×40) of rabbit ear-rim auricular vein following the administration of CLS (A), CLC (B) and D5W (C).
Conclusions
Colloid is one of the dispersion states of materials. No matter what kind of material it is, if it was dispersed in another material within the size range of 1∼100 nm it can be called colloids, which is a relatively stable disperse system. Many hydrophilic polymer compounds following hydration reaction with water can dispersed into water and formed transparent and hydrophilic colloidal solutions which can be used in drug delivery systems. In this study, we developed an alternative lipid-based CL carrier by the route of intravenous administration which has high drug-loading (the final drug concentration in the system was 15 mg/ml) and less irritation. The drug delivery system consisting of SCS, CL and phospholipids (1:1:1.5) has fine and uniform particle size. The transparent colloidal suspension of CL was concentrated and lyophilized by membrane filtration and freeze-drying method to obtain the final lyophilized LCC. The procedure can be accomplished efficiently and is adapt to industrial-scale production. The lyophilized LCC dissolved in 5% dextrose solution before intravenous infusion can provide more stability and safety during the course of storage, transportation and application.
Declaration of interest
Authors have no declarations of interest to report.
References
- Brown GA. (1970). Infusion thrombophlebitis. Br J Clin Pract 24:197–200
- Cannon JB, Adeyinka Williams N, Papp KJ. (1995). Reduction of pain on intravenous infusion with bile salt formulations for a macrolide antibiotic. Int J Pharm 114:65–74
- Celozzi E, Lotti VJ, Stapley EO, Miller AK. (1980). An animal model for assessing pain-on-injection of antibiotics. J Pharmacol Methods 4:285–9
- Chu SY, Deaton R, Cavanaugh J. (1992). Absolute bioavailability of clarithromycin after oral administration in humans. Antimicrob Agents Chemother Agents Chemother 36:1147–50
- Comereski CR, Williams PD, Bregman CL, Hottendorf GH. (1986). Pain on injection and muscle irritation: a comparison of animal models for assessing parenteral antibiotics. Fundam Appl Toxicol 6:335–8
- Flood KM, Liu R, Peck KD, Zheng J. (1998). Pain reducing parenteral liposome formulation. CA Patent 2279259
- Gupta PK, Patel JP, Hahn KR. (1994). Evaluation of pain and irritation following local administration of parenteral formulations using the rat paw lick model. J Pharm Sci Technol 48:159–66
- Grazia S, Gerald W. (1968). Phospholipid spherules (liposomes) as a model for biological membranes. J Lipid Res 9:310–18
- Hecker JF, Fisk GC, Lewis GBH. (1984). Phlebitis and extravasation (“tissuing”) with intravenous infusions. Med J Aust 140:658–60
- Jiang Y, Li F, Luan Y, et al. (2012). Formation of drug/surfactant catanionic vesicles and their application in sustained drug release. Int J Pharm 436:806–14
- Kohno Y, Yoshida H, Suwa T, Suga T. (1989). Comparative pharmacokinetics of clarithromycin (TE-031), a new macrolide antibiotic, and erythromycin in rats. Antimicrob Agents Chemother 33:751–6
- Langtry HD, Brogden RN. (1997). Clarithromycin – a review of its efficacy in the treatment of respiratory tract infections in immunocompetent patients. Drug 53:973–1004
- Lovell MW, Johnson HW, Hui HW, et al. (1994). Less-painful emulsion formulations for intravenous administration of clarithromycin. Int J Pharm 109:45–57
- Lu Y, Wang YJ, Tang X. (2008). Formulation and thermal sterile stability of a less painful intravenous clarithromycin emulsion containing vitamin E. Int J Pharm 346:47–56
- Lu Y, Zhang Y, Yang ZY. (2009). Formulation of an intravenous emulsion loaded with a clarithromycin-phospholipid complex and its pharmacokinetics in rats. Int J Pharm 366:160–9
- Mohammadi G. (2011). Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf B Biointerfaces 88:39–44
- Nakagawa Y, Itai S, Yoshida T, Nagai T. (1992). Physicochemical properties and stability in the acidic solution of a new macrolide antibiotic, clarithromycin, in comparison with erythromycin. Chem Pharrn Bull 40:725–8
- Qin LH, Leng W. (2007). Formulation and evaluation of less-painful clarithromycin lipid microspheres. Arch Pharm Res 30:1336–43
- Zhao L, Liu J, Zhang L, et al. (2013). Self-assembly properties, aggregation behavior and prospective application for sustained drug delivery of a drug-participating catanionic system. Int J Pharm 452:108–15
- Zimmerm C, Gea E, Roig J, et al. (2002). Comparative tolerability of intravenous erythromycin and clarithromycin in hospitalised patients with community-acquired pneumonia results of a double-blind, randomised, prospective study. Clin Drug Invest 22:393–8
- Zimmermann T, Laufen H, Riedel KD, et al. (2001). Comparative tolerability of intravenous azithromycin, clarithromycin and erythromycin in healthy volunteers results of a double-blind, double-dummy, four-way crossover study. Clin Drug Invest 21:527–36