Abstract
Objective: Monocholesterylsuccinate (CHS)-modified paclitaxel-loaded discoidal reconstituted high density lipoproteins (cP-d-rHDL) as novel biomimetic nanocarriers that were developed for tumor targeting delivery to avoid unexpected drug leakage from discoidal reconstituted high density lipoproteins (d-rHDL) during remodeling process associated with lecithin-cholesterol acyltransferase (LCAT).
Methods: Their in vitro characterizations and biomimetic properties, simultaneously tumor distribution and pharmacodynamics in tumor bearing mice were elaborately investigated.
Results. In vitro characterization results showed that cP-d-rHDL had nano-size diameter, high negative zeta potential and high entrapment efficiency (EE). Furthermore, morphology study indicated that cP-d-rHDL did not remodel in the presence of LCAT, compared with that of paclitaxel-loaded d-rHDL (P-d-rHDL, not modified). And cellular uptake, together with cytotoxicity toward tumor cells of cP-d-rHDL was not affected after interaction with LCAT. Tumor distribution and pharmacodynamics tests revealed that cP-d-rHDL possessed specific targeting property and anti-tumor efficacy.
Conclusion: cP-d-rHDL served to restrain remodeling process and drug leakage, at the same time reinforce the targeting effect, and could act as a potential drug delivery system for cancer therapy.
Introduction
High density lipoproteins (HDL) are one species of natural bioactive particles, existing in two different architectures: discoidal HDL (nascent HDL) and spherical HDL (mature HDL). Discoidal HDL are composed of apolipoproteins (mainly apoA-I), phospholipids bilayer and free cholesterol. While spherical HDL consist of a hydrophobic core of triglycerides and cholesterol esters surrounded by an outer monolayer of phospholipids, free cholesterol and apolipoproteins. Catalyzed by lecithin-cholesterol acyltransferase (LCAT), discoidal HDL could transform into spherical HDL (Eirini et al., Citation2010).
HDL have long been considered as potential drug transporting vehicles since they are well biodegradable and biocompatible, and could extend residence time in the circulation (Rensen et al., Citation2001; Murakami, Citation2012). Moreover, they are suited to accommodate lipophilic drugs, including anti-cancer drugs (Mooberry et al., Citation2010). It has been demonstrated that the expression of HDL receptors, i.e. scavenge receptor class BI (SR-BI), are enhanced in cancer cells, thus favoring specific high-affinity uptake of drugs (Lacko et al., Citation2002; Marina et al., Citation2012). A series of native or reconstituted HDL (rHDL) encapsulating therapeutic or diagnostic agents have been reported (McConathy et al., Citation2008; Kim et al., Citation2009; Jae et al., Citation2012). Additionally, bilayer-structured discoidal rHDL (d-rHDL) are capable of fusing with cell membranes. And Liadaki et al. (Citation2000) have reported that d-rHDL exhibited higher affinity to SR-BI receptor than spherical rHDL. In a word, d-rHDL seem to be more advantageous as carriers of anti-cancer drugs.
We have previously prepared biocompatible d-rHDL that are able to stably incorporate paclitaxel (PTX), a lipophilic natural product from Taxus baccata. Biomimetic property examinations showed that P-d-rHDL maintained remodeling behaviors similar to their native counterparts when incubated with LCAT. However, the drug release rate of P-d-rHDL became faster when LCAT was added, and cellular drug uptake and cytotoxicity were attenuated. This might be ascribed to PTX leakage from P-d-rHDL, which was provoked by the structural conversion in the remodeling process before cellular internalization (Jia et al., Citation2012). Therefore, it is urgent to prevent the remodeling behaviors of the drug system induced by LCAT. ApoA-I, lecithin and cholesterol are known to play essential roles in the remodeling process of d-rHDL. After apoA-I activation, LCAT catalyzes the transfer of the sn-2 acyl group from lecithin to the hydroxyl group of cholesterol, finally generating cholesteryl esters (Alessandro et al., Citation2012). Then cholesteryl esters are sequestered into the center of the discoidal particles to form a hydrophobic core and ultimately these particles were gradually converted into spherical ones (Rader, Citation2006). And modifications for the three elements above might influence the remodeling behaviors induced by LCAT (Roosbeek et al., Citation2001; Nobecourt et al., Citation2007). Whereas, it is difficult to effectively characterize the modification result for the heterogeneity of phospholipids in d-rHDL. Consequently, modifying cholesterol and apoA-I would be more reasonable. Based on the above principle, the cholesterol was esterified with butanedioic anhydride to synthesize monocholesterylsuccinate (CHS) so as to eliminate substrate of LCAT, as described previously by Zhang et al. (Citation2012); moreover, as CHS had free carboxyl group and fine amphipathicity, when they were covalently attached to apoA-I, they could improve the amphipathic properties of apoA-I and thus enable easier and more stable binding of apoA-I to lipid cores with an increased binding efficiency ().
Figure 1. Different actions between P-rHDL or cP-rHDL with LCAT.
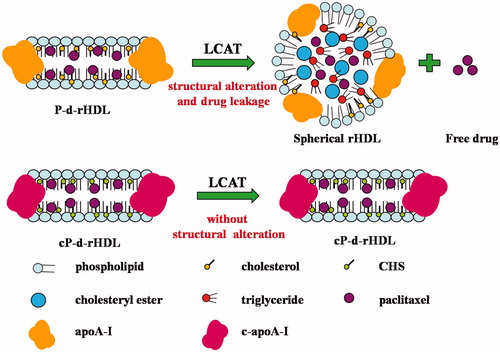
In the present work, novel mimic cP-d-rHDL were constructed with PTX, phosphatidylcholine, CHS (instead of cholesterol) and CHS-modified apoA-I (c-apoA-I) by thin-film dispersion method. The cP-d-rHDL were characterized by particle size, zeta potential and morphology. Phagocytic uptake and cytotoxicity of cP-d-rHDL with and without LCAT were also evaluated. To further estimate the effectiveness of modification for d-rHDL, tumor distribution, adverse side effects and anti-tumor activity of various PTX formulations, including cP-d-rHDL, P-d-rHDL, CHS-modified paclitaxel-loaded liposomes (cP-liposome), and commercial PTX (Taxol) were examined systematically in tumor bearing mice. The ultimate purpose of this study was to provide a new method for improving in vivo stability of d-rHDL and promoting the application of d-rHDL as drug delivery systems for anti-cancer agents.
Materials and methods
Materials
Paclitaxel was obtained from Yew Pharmaceutical Company Ltd. (Jiangsu, China). Lipoid S 100 was purchased from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol was the product of Sigma-Aldrich (Saint Louis, MO). ApoA–I was isolated from precipitate IV (plasma fractionation) (Kistler & Nitschmann, Citation1962; Wang et al., Citation2010). CHS, c-apoA-I were synthesized in our laboratory. LCAT was isolated from human plasma in our laboratory. Human breast cancer cells (MCF-7) were supplied by the cell bank of Chinese Academy of Science. Tumor bearing mice were supplied by KeyGEN Biotech Co. Ltd. (Nanjing, China). All other chemicals of analytical or HPLC grade were purchased from Chemical Reagent Company, Ltd. (Nanjing, China).
Preparation of cP-d-rHDL
The cores of cP-d-rHDL were prepared by thin-film dispersion method (Jia et al., Citation2012). Briefly, soy phosphatidylcholine (SPC), CHS and PTX (29.37: 1: 1.08, molar ratio) were dissolved in absolute ethanol and dried under reduced pressure. Then 0.01 M phosphate-buffered saline (PBS, pH 7.4, containing 0.138 M NaCl and 0.0027 M KCl) was added to hydrate the dry lipid film at room temperature by thoroughly vortexing, followed by an ultrasonication (200 W, 300 s) on ice with an Ultra-Homogenizer (JY92II, Ningbo, China). The suspensions were extruded through 0.45 μm and then 0.22 μm filter several times to homogenize the particles. The CHS-modified paclitaxel-loaded liposomes (cP-liposome) were obtained.
Then c-apoA-I was synthesized as reported previously (Zhang et al., Citation2012). Briefly, CHS was firstly reactivated by 1-Ethyl-3-(3-dimethylamino)propylcarbodiimide hydrochloride (EDC ċ HCl) and N-hydroxysuccinimide (NHS) to obtain CHS-NHS active esters (CHE-NHSE). Second, the active ester was added into apoA-I solution, after agitation overnight at 4 °C, the mixtures were dialyzed, centrifuged to remove the extra active esters. Finally, c-apoA-I was acquired.
After that, cP-liposomes were incubated with c-apoA-I to form cP-d-rHDL. Briefly, 2 mL of cP-liposome were incubated with PBS solution containing 30 mg c-apoA-I and sodium cholate (final sodium cholate to SPC molar ratio 1: 1.6) under magnetic stirring at 4 °C. After incubation for 10 h, the mixtures were subjected to dialysis against PBS for 48 h to remove sodium cholate.
Size, zeta potential and drug entrapment efficiency
The mean diameters and surface potentials of cP-liposome and cP-d-rHDL were measured by dynamic light scattering (DLS, Zetasizer 3000 HAS, Malvern Instruments, Malvern, UK), respectively. Samples were diluted with aqueous phase appropriately before determination.
Drug entrapment efficiency (EE) was measured with mini-column centrifugation method (Jain et al., Citation2008; Li et al., Citation2008). The concentrations of PTX incorporated in cP-liposome/cP-d-rHDL (C) and the total drug in cP-liposome/cP-d-rHDL dispersions (C0) were analyzed by HPLC, respectively. Briefly, 20 μL of each sample was subjected into the system. The mobile phase was methanol: water (75:25, v/v), and flow rate was maintained at 1.0 mL/min. HPLC system comprises a pump (LC-10AT VP), a UV detector (Shimadzu, Kyoto, Japan) set at 227 nm, and a Synergi Hydro-RP C18 (5 μm, 150 mm × 4.6 mm) column applied at 30 °C.
EE was calculated by the following equation:
Biomimetic properties
Morphology
Morphological examinations of P-d-rHDL and cP-d-rHDL with or without LCAT incubation were performed under transmission electron microscopy (TEM, H-7650, Hitachi High Technologies Corporation, Tokyo, Japan).
Influence of LCAT on cytotoxicity and cellular uptake in vitro
Cell culture
The culture of human breast cancer cells (MCF-7) was described previously by Jia et al. (Citation2012). The cells were cultured in DMEM medium supplemented with 10% FBS (v/v), penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C under 5% CO2.
Cytotoxicity evaluation
The cytotoxicities of P-d-rHDL and cP-d-rHDL with or without LCAT were determined through the MCF-7 cells viability using MTT assays (Xu et al., Citation2010). The cells were seeded in 96-well plates at a density of 105 cells/well in their media for 24 h. Then the medium was washed with PBS (pH 7.4) twice, and then incubated with samples diluted in DMEM at 37 °C for 6 h. 100 μL of MTT (1 mg/mL) was added at given time intervals, and after incubation for 4 h, the medium was replaced with 200 μL DMSO so as to dissolve the formazan crystals. The samples were read at 570 nm in a Microplate Reader. All experiments were performed in triplicate. Cell viability was expressed as the formula below:
where ODtest stands for the absorbance intensity of cells treated with various preparations, and ODcontrol stands for the absorbance intensity of cells incubated only with the medium.
Cellular uptake study
MCF-7 cells were seeded in 24-well plates at a density of 105 cells/well and incubated in complete culture medium for 24 h. Then the cells were washed with PBS twice and incubated with P-d-rHDL and cP-d-rHDL with or without LCAT (drug concentration at 5.2 μg/mL) at 37 °C for 0.5, 1, 2, 4, 6 h. At the end of incubation, the non-phagocytosed drug was removed and the cells were washed twice with ice-cold PBS. Subsequently, the cells were subjected to five cycles of freezing (−40 °C) and thawing (37 °C) to split before dissolved in 400 μL methanol. Then, samples were centrifuged at 12 000 rpm for 10 min. The protein content of each cell lysate was determined by BCA protein assay. The amount of PTX was measured by HPLC as described above. And the intracellular drug percentages were calculated:
where C represents the concentration of phagocytosed drug at different time, C0 represents the initial drug concentration, while M means the unit weight (mg) of cellular protein at different time, M0 means the initial unit weight (mg) of cellular protein.
Tumor distribution of PTX formulations
For tumor targeting studies, ICR male mice (20 ± 2 g) bearing breast tumor were used. All care and handling of animals were carried out according to the Guidelines evaluated and approved by the Ethics Committee of China Pharmaceutical University (Nanjing, China). In order to explore the tumor targeting effects of PTX formulations, 48 tumor bearing mice were randomly divided into four groups, Taxol group, cP-liposome (P-L) group, P-d-rHDL group and cP-d-rHDL group (12 mice each), respectively.
Each mouse in four groups was given a single 10 mg/kg dose of PTX by the tail vein injection. At 0.5, 2, 6 and 12 h after injection, three mice in one group were sacrificed. Tumors were excised, thoroughly washed with ice-cold saline, dried with filter paper and weighed.
Tumor tissues were mixed with saline and homogenized. The resultant tissue homogenate (200 μL) was added to 10 μL of 1 mg/mL diazepam solution as internal standard. After vortexing for 3 min, 200 μL of PBS was added, and another 1 min vortex. Finally, these samples were vortexed for 3 min and centrifuged at 3000 rpm for 10 min after mixing with ethyl acetate. The supernatant (900 μL) was transferred into a 10 mL of tube and dried under nitrogen. Then residues were reconstituted with 100 μL of methanol, followed by centrifugation at 12 000 rpm for 10 min. Aliquots (20 μL) of supernatant were collected and injected into the HPLC system. The HPLC method was described previously in the current article.
In vivo anti-tumor efficacy of PTX formulations
Since 24 h after inoculation, tumor bearing mice were randomized into five groups (six mice per group): group 1 for saline (control group), group 2 for Taxol, group 3 for P-L, group 4 for P-d-rHDL and group 5 for cP-d-rHDL, respectively. Group 2 to 5 were used as treatment group. All mice in treatment group received four dosages on day 0, day 2, day 4 and day 6 through the tail vein injection at a dose of 5 mg/kg as PTX and the mice were checked for survival every day.
Tumor inhibition evaluation
The tumor was measured every other day in two dimensions, and its volume was calculated as (Maha et al., Citation2008):
where a and b denote the shortest and longest diameter of the tumor in mm, respectively.
Tumor volume–time curve was obtained from days 0 to 8 after administration.
Animals in all groups were killed by cervical dislocation on day 8 and their tumors were immediately harvested, washed by saline, weighed and the inhibition rate was computed as follows (Yan et al., 2011):
where Wc is the tumor weight of control group and Wt is the tumor weight of treatment group.
Safety studies
To evaluate the toxicity of PTX treatment, as indicated by loss of body weight, each mouse was weighed every alternate day. And body weight versus time curve was established.
Statistical analysis
All results were expressed as the mean ± SD. Both ANOVA and Student’s two-sample t-test were utilized for statistical analysis. p < 0.05 was considered significant.
Results
Size, zeta potential and drug entrapment efficiency
As shown in , the mean diameters of cP-d-rHDL were smaller than those of cP-liposome, merely about 54.5 ± 0.32 nm; after incubation with apoA-I, surface charges of cP-d-rHDL increased from−17.96 ± 0.43 mV to −26.05 ± 1.31 mV. Whereas, the EE did not change notably before and after incubation with apoA-I.
Table 1. Average diameter, zeta potential value and entrapment efficiency of cP-liposome and cP-d-rHDL (mean ± SD, n = 3).
Biomimetic properties
Morphology
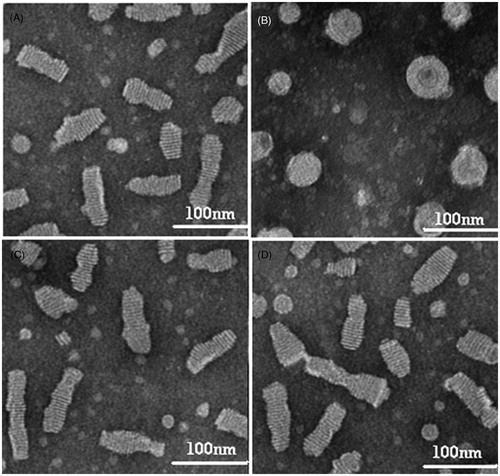
As seen in , unlike P-d-rHDL, discoidal cP-d-rHDL did not transform into spherical ones when interacted with LCAT.
Figure 2. Microphotographs of different preparations using transmission electron microscope. (A) P-d-rHDL in the absence of LCAT; (B) P-d-rHDL incubation with LCAT; (C) cP-d-rHDL in the absence of LCAT; (D) cP-d-rHDL incubation with LCAT.
Influence of LCAT on cytotoxicity and cellular uptake in vitro
Cytotoxicity evaluation
MTT assay was conducted to compare the cytotoxicity of P-d-rHDL and cP-d-rHDL, and to estimate the actions of LCAT on cP-d-rHDL. The 50% cellular growth inhibitions (IC50) of various preparations against MCF-7 cells were calculated from the experiment and listed in . As depicted in the table, cP-d-rHDL presented higher toxic effect on MCF-7 cells than P-d-rHDL. The IC50 value of cP-d-rHDL was 1.38-fold lower than that of P-d-rHDL (p < 0.01). Furthermore, there was distinct influence of LCAT on the viability of cells for P-d-rHDL and cP-d-rHDL (p < 0.001). Specifically, the IC50 value of P-d-rHDL increased greatly after LCAT addition (p < 0.001), but the value of cP-d-rHDL in the presence of LCAT was hardly affected (p > 0.05).
Table 2. Cytotoxicity of P-d-rHDL and cP-d-rHDL with or without LCAT against MCF-7 cells (mean ± SD, n = 3).
Cellular uptake study
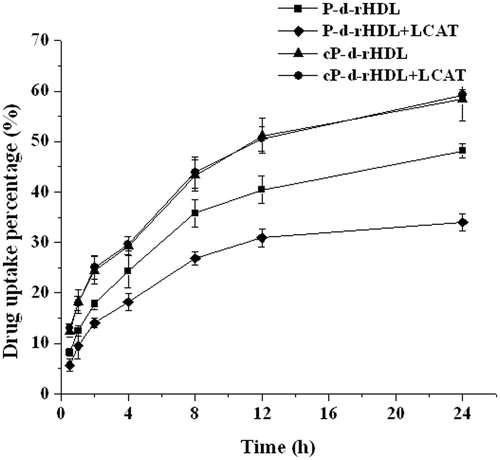
The internalization of P-d-rHDL and cP-d-rHDL with or without LCAT by MCF-7 cells was examined. exhibited that the cellular uptake percentage of cP-d-rHDL was higher than that of P-d-rHDL at each time point. And when LCAT was added, the uptake amount of P-d-rHDL was reduced markedly, while cP-d-rHDL remained unaltered.
Figure 3. Profiles of uptake percentages versus time of different preparations (mean ± S.D., n = 3). (▪) P-d-rHDL; (♦) P-d-rHDLs incubation with LCAT; (▴) cP-d-rHDL; (•) cP-d-rHDL incubation with LCAT.
Tumor distribution of PTX formulations
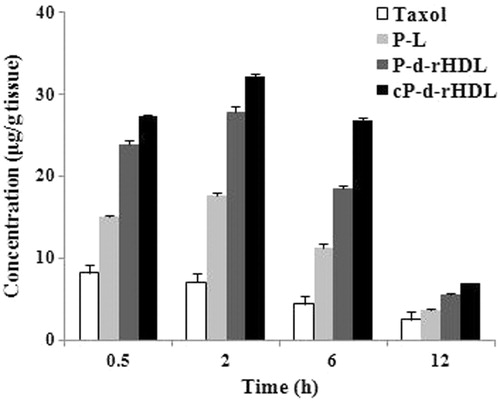
Drug distribution in tumor tissues at different time was recorded after administration of PTX formulations (). revealed that drug concentrations in tumors of four groups at each time point declined as below: cP-d-rHDL > P-d-rHDL > P-L > Taxol, the drug concentrations in Taxol and P-L groups were significantly lower than those in P-d-rHDL and cP-d-rHDL groups. Moreover, the tumor distribution of PTX in cP-d-rHDL was obviously higher than that of P-d-rHDL, illustrating that CHS-modified rHDL carriers had the best tumor targeting property.
Figure 4. Tumor distribution of PTX after intravenous administration of Taxol, P-L, P-d-rHDL and cP-d-rHDL in tumor bearing mice (n = 3).
In vivo anti-tumor efficacy of PTX formulations
Tumor inhibition evaluation
Tumor volume
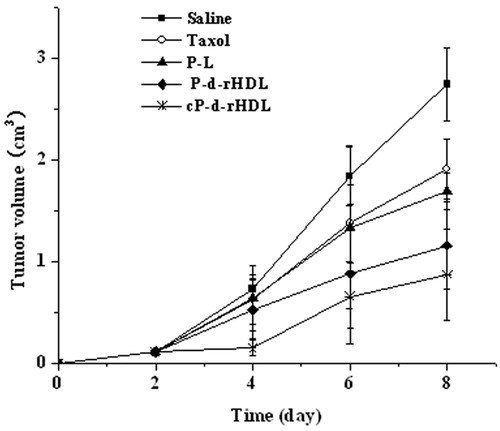
The tumor growing curves of different groups were acquired as shown in . The tumor growing rates of mice increased as the following sequence: cP-d-rHDL < P-d-rHDL < P-L < Taxol < Saline. And the tumor volume in the cP-d-rHDL group was remarkably smaller than those of other groups. In the saline control group, the tumor volume–time curve almost presented a straight line; nevertheless, tumors grew slowly for the mice which received cP-d-rHDL therapy.
Figure 5. The tumor growing curves for different treatment groups (n = 6).
Tumor weight
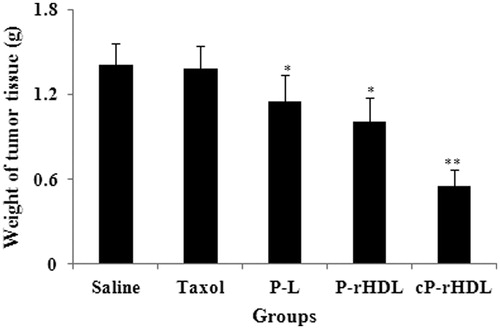
The average tumor weight at the end of treatment was 1.41 ± 0.25, 1.38 ± 0.18, 1.15 ± 0.39, 1.01 ± 0.37 and 0.55 ± 0.16 g for Saline, Taxol, P-L, P-d-rHDL, cP-d-rHDL groups, respectively (). In contrast to saline group, Taxol group revealed no significance (p > 0.05), but P-L, P-d-rHDL and cP-d-rHDL exhibited remarkable difference in tumor weight (p < 0.05, p < 0.05, p < 0.01). And inhibition rates of Taxol, P-L, P-d-rHDL and cP-d-rHDL group were 1.78%, 17.86%, 28.11% and 61.21%, respectively.
Figure 6. The tumor weight of mice after treatments with different formulations (n = 6). Significant differences: *p < 0.05, **p < 0.01, compared with saline group.
Safety studies
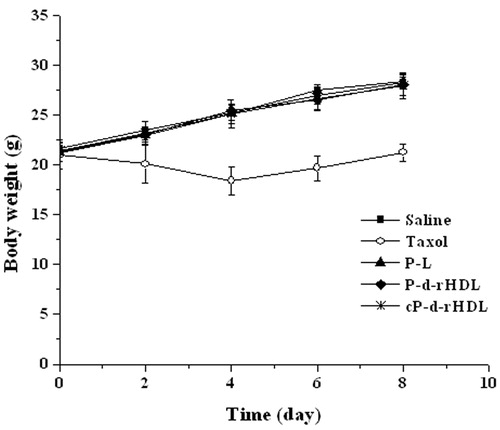
The changes in body weight over time for all groups are summarized in . Apparently, except for Taxol group, body weight in other three PTX groups was comparable to that of the control group, which meant that in vivo toxicity of P-L, P-d-rHDL and cP-d-rHDL was much lower than that of Taxol.
Figure 7. The body weight of mice after treatments with different formulations (n = 6).
Discussions
Several attributes of d-rHDL make them attractive candidates for delivering anti-cancer agents to tumors, primarily owing to their unique structures, biocompatible components and the potential drug uptake via receptor-mediated endocytosis. This report is one of the first to pay attention to the stability of d-rHDL in their metabolic process. Through chemical modification, the reactivity of d-rHDL with LCAT was suppressed, consequently stabilizing the drug carrier.
In the current research, PTX was chosen as the model drug and novel cP-d-rHDL were successfully prepared. The physicochemical properties, biological activities, in vivo performances of cP-d-rHDL such as tumor distribution and pharmacodynamics were amply assessed.
The diameters of cP-d-rHDL were smaller than those of cP-liposome, possibly due to the interaction of apoA-I and phospholipids which transformed vesicles into nanoscale rHDL (Tufteland et al., Citation2008; Jia et al., Citation2012). An increase in surface charge of cP-d-rHDL reflected that negative charged apoA-I in the medium had integrated with the lipids (Gessner et al., Citation2002; Zhang et al., Citation2011). It was well known that small particle size and anionic nanocarriers were able to escape from blood elimination. Meanwhile, as endogenous vehicles, rHDL could efficiently hide from the reticulo-endothelia systems (RES). Thus the three advantages of cP-d-rHDL would facilitate them to circulate longer in vivo and produce enhanced permeability and retention (EPR) effect (Wei et al., Citation2011; Yuta et al., Citation2011). And LCAT did not have an impact on cP-d-rHDL structure after incubation, meaning that remodeling process of cP-d-rHDL was inhibited, which had been identified by TEM microphotographs. Earlier studies by our team showed that the addition of LCAT significantly accelerated the rate of drug release from P-d-rHDL, while it had no significant impact on cP-d-rHDL, indicating that the latter did not experience a remodeling process like the former. Moreover, the drug was released slower from cP-d-rHDL than that from P-d-rHDL in the absence or presence of LCAT, suggesting the modification of carriers had a positive effect on the stability of d-rHDLs (Zhang et al., Citation2012).
Human breast cancer cell line (MCF-7) was selected for cytotoxicity and phagocytosis studies because cellular uptake of PTX from d-rHDL is facilitated by SR-BI which was over-expressed on MCF-7 (Ding et al., Citation2012; Sabnis & Lacko, Citation2012). cP-d-rHDL possessed higher toxic actions than P-d-rHDL, and cytotoxicity of the former was not affected by LCAT. Drug intracellular tests of P-d-rHDL and cP-d-rHDL with or without LCAT were also carried out (). demonstrated that uptake percentage of cP-d-rHDL was higher than that of P-d-rHDL, probably resulting from the higher binding efficiency of apoA-I with cP-d-rHDL, which favored mediation of drug uptake by apoA-I (Mineo & Philip, Citation2012). Cellular uptake of P-d-rHDL decreased under the action of LCAT, indicating that drug might leak from the system during remodeling process before phagocytosis; on the contrary, cP-d-rHDL were not influenced by LCAT. It could be concluded from these results that cP-d-rHDL were stable when interacted with LCAT.
Our previous results from pharmacokinetic experiments in rats of different PTX carriers proved that cP-d-rHDL had longer MRT, larger AUC, lesser body clearance and apparent distribution volume than Taxol, P-L and P-d-rHDL. Thus, cP-d-rHDL could prolong the circulation time and improve the bioavailability of drug (Zhang et al., Citation2012).
Tumor distribution and anti-tumor activities of Taxol, P-L, P-d-rHDL and cP-d-rHDL were explored in tumor bearing mice. It was manifested that P-d-rHDL and cP-d-rHDL evidently displayed a largely increased local concentration of PTX in the tumor. And tumor targeting assay illustrated that targeting property of P-d-rHDL was weaker than that of cP-d-rHDL. Moreover, cP-d-rHDL were observed to be the most prominent in limiting tumor growth. This outcome may be explained as following: first, smaller nanocarrier size and longer half-life in vivo of cP-d-rHDL assisted in their accumulation in the tumors through EPR effect (passive targeting), which were consistent with previous reports (Jie et al., Citation2012; Lee et al., Citation2012). Second, SR-BI has been found to be broadly expressed in breast cancer cells, so drug from d-rHDL could be endocytosed into tumors via SR-BI-mediated mechanism (Cao et al., Citation2004; Zhang et al., Citation2013). Third, more apoA-I were conjugated to the lipid bilayer of cP-d-rHDL, thus contributing to striking enhancement in therapeutic efficacy (positive targeting). These two targeting routes led to the more effective tumor distribution of cP-d-rHDL than the other three treatment groups (Taxol, P-L and P-d-rHDL groups). Besides, drug leakage from cP-d-rHDL during the remodeling behaviors induced by LCAT was restrained, which increased drug delivery to tumors (Jia et al., Citation2012). In addition, the alterations in body weight over time after administration in tumor bearing mice were employed as one of the markers of preparation safety. Just as testified by the safety estimation, there was no obvious body weight loss in mice treated with cP-d-rHDL, compared to that of saline group. This implied that cP-d-rHDL may have less severe side effect and could be exploited as promising drug delivery vehicles for cancer medication.
Conclusions
In this article, in vitro physicochemical characterizations, biological properties as well as in vivo performances of cP-d-rHDL covering morphology, cytotoxicity, cellular uptake, tumor distribution and anti-tumor efficacy were systematically investigated aiming to explore the feasibility of improving d-rHDL tumor targeting features by carrier modification. Specifically, the microstructure, endocytosis and toxic effect of cP-d-rHDL were not influenced under the action of LCAT. In comparison with P-d-rHDL, cP-d-rHDL had reinforced cellular uptake and toxicity; owned optimal tumor targeting properties and anti-tumor efficacy. In summary, our results supported the idea that through modifying the vehicles could not only inhibit remodeling behaviors and drug leakage induced by LCAT, but also exert preferable actions of d-rHDL in drug delivery and cancer treatment.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This study is financially supported by the National Science Foundation Grant of China (No. 81273466), the Specialized Fund for Basic Scientific Research of Higher Education from China Central Authorities (No. ZJ11253), the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20120096120005) and Young Foundation from the Health Department of Fujian Province, China (No. 2013-2-85).
References
- Alessandro C, Luisa C, Bernardetta M, et al. (2012). LCAT cholesterol esterification is associated with the increase of ApoE/ApoA-I ratio during atherosclerosis progression in rabbit. J Physiol Biochem 68:541–53
- Cao WM, Murao K, Imachi H, et al. (2004). A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res 64:1515–21
- Ding Y, Wang W, Feng MQ, et al. (2012). A biomimetic nanovector-mediated targeted cholesterol-conjugated siRNA delivery for tumor gene therapy. Biomaterials 33:8893–905
- Eirini MT, Maria SB, Elisavet HF, et al. (2010). HDL biogenesis and functions: role of HDL quality and quantity in atherosclerosis. Atherosclerosis 208:3–9
- Gessner A, Lieske A, Paulke B, Muller R. (2002). Influence of surface charge density on protein adsorption on polymeric nanoparticles: analysis by two-dimensional electrophoresis. Eur J Pharm Biopharm 54:165–70
- Jae YS, Yoosoo Y, Paul H, et al. (2012). pH-responsive high-density lipoprotein-like nanoparticles to release paclitaxel at acidic pH in cancer chemotherapy. Int J Nanomedicine 7:2805–16
- Jain SK, Chaurasiya A, Gupta Y, et al. (2008). Development and characterization of 5-FU bearing ferritin appended solid lipid nanoparticles for tumour targeting. J Microencapsul 25:289–97
- Jia JT, Xiao Y, Liu JP, et al. (2012). Preparation, characterizations, and in vitro metabolic processes of paclitaxel-loaded discoidal recombinant high-density lipoproteins. J Pharm Sci 101:2900–8
- Jie L, Li ZX, Jeffrey IZ, Fuyuhiko T. (2012). In vivo tumor suppression efficacy of mesoporous silica nanoparticles-based drug delivery system: enhanced efficacy by folate modification. Nanomedicine: N.B.M 8:12–20
- Kim SI, Shin D, Lee H, et al. (2009). Targeted delivery of siRNA against hepatitis C virus by apolipoprotein A-I-bound cationic liposomes. J Hepatol 50:479–88
- Kistler P, Nitschmann H. (1962). Large scale production of human plasma fractions. Vox Sang 7:414–24
- Lacko AG, Nair M, Paranjape S, et al. (2002). High density lipoprotein complexes as delivery vehicles for anticancer drugs. Anticancer Res 22:2045–9
- Lee PY, Zhang RZ, Li V, et al. (2012). Enhancement of anticancer efficacy using modified lipophilic nanoparticle drug encapsulation. Int J Nanomedicine 7:731–7
- Li C, Cui J, Wang C, et al. (2008). Encapsulation of mitoxantrone into pegylated SUVs enhances its antineoplastic efficacy. Eur J Pharm Biopharm 70:657–65
- Liadaki KN, Liu T, Xu S, et al. (2000). Binding of high density lipoprotein (HDL) and discoidal reconstituted HDL to the HDL receptor scavenger receptor class B type I. J Biol Chem 275:21262–71
- Maha S, Olga BG, Elizabeth B, et al. (2008). Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging? J Control Release 130:107–14
- Marina GD, Kannan M, Sushant T, et al. (2012). Templated high density lipoprotein nanoparticles as potential therapies and for molecular delivery. Adv Drug Deliv Rev 65:649--62
- McConathy WJ, Nair MP, Paranjape S, et al. (2008). Evaluation of synthetic/reconstituted high-density lipoproteins as delivery vehicles for paclitaxel. Anticancer Drugs 19:183–8
- Mineo C, Philip WS. (2012). Functions of scavenger receptor class B, type I in atherosclerosis. Curr Opin Lipidol 23:487–98
- Mooberry LK, Nair M, Paranjape S, et al. (2010). Receptor mediated uptake of paclitaxel from a synthetic high density lipoprotein nanocarrier. J Drug Target 18:53–8
- Murakami T. (2012). Phospholipid nanodisc engineering for drug delivery systems. Biotechnol J 7:762–7
- Nobecourt E, Davies M, Brown B, et al. (2007). The impact of glycation on apolipoprotein AI structure and its ability to activate lecithin: cholesterol acyltransferase. Diabetologia 50:643–53
- Rader DJ. (2006). Molecular regulation of HDL metabolism and function: Implications for novel therapies. J Clin Investig 116:3090–100
- Rensen PCN, de Vrueh RLA, Kuiper J, et al. (2001). Recombinant lipoproteins: lipoprotein-like lipid particles for drug targeting. Adv Drug Deliv Rev 47:251–76
- Roosbeek S, Vanloo B, Duverger N, et al. (2001). Three arginine residues in apolipoprotein AI are critical for activation of lecithin: cholesterol acyltransferase. J Lipid Res 42:31–40
- Sabnis N, Lacko A. (2012). Drug delivery via lipoprotein-based carriers: answering the challenges in systemic therapeutics. Ther Deliv 3:599–608
- Tufteland M, Ren G, Ryan R. (2008). Nanodisks derived from amphotericin B lipid complex. J Pharm Sci 97:4425–32
- Wang L, Chen WZ, Wu MP. (2010). Apolipoprotein A-I inhibits chemotaxis, adhesion, activation of THP-1 cells and improves the HDL inflammatory index. Cytokine 49:194–200
- Wei Z, Yuan S, Yan Z, et al. (2011). The potential of Pluronic polymeric micelles encapsulated with paclitaxel for the treatment of melanoma using subcutaneous and pulmonary metastatic mice models. Biomaterials 32:5934–44
- Xu Y, Jin XF, Ping QN, et al. (2010). A novel lipoprotein-mimic nanocarrier composed of the modified protein and lipid for tumor cell targeting delivery. J Control Release 146:299–308
- Yan H, Yong H, Xiang F, et al. (2011). Anti-tumor activity of biodegradable polymer-paclitaxel conjugate micelles on lewis lung cancer mice models. J Biomater Sci Polymer Ed 22:1131--46
- Yuta Y, Yusuke K, Kenichi O, et al. (2011). PEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacy. Int J Pharm 412:132–41
- Zhang MY, Jia JT, Liu JP, et al. (2012). A novel modified paclitaxel-loaded discoidal recombinant high-density lipoproteins: preparation, characterizations and in vivo evaluation. Asian J Pharm Sci 7:364–73
- Zhang WL, He HL, Liu JP, et al. (2013). Pharmacokinetics and atherosclerotic lesions targeting effects of tanshinone IIA discoidal and spherical biomimetic high density lipoproteins. Biomaterials 34:306–19
- Zhang WL, Xiao Y, Liu JP, et al. (2011). Structure and remodeling behavior of drug-loaded high density lipoproteins and their atherosclerotic plaque targeting mechanismin foam cell model. Int J Pharm 419:314–21