
Abstract
Studies on preparation of in situ gel formulations containing diphtheria toxoid as the model active substance and their intranasal administration have been conducted in this study. The objective of mucosal vaccination is to stimulate both systemic and mucosal immune responses. In situ gel formulations were prepared by using, in different ratios, mixtures of Poloxamer 407 and Poloxamer 188 polymers, which gelate in a temperature-dependent manner, and mucoadhesive polymers carbopol 934, hydroxypropyl methyl cellulose, hydroxypropyl cellulose or chitosan. Following pre-formulation studies, F1, F2, F3, F4, F5, F6 and F7 formulations, which gelate at intervals and temperatures in accordance with nasal temperatures, were subjected to more comprehensive studies. For this purpose, organoleptic characteristics of the formulations were identified, their pH and mucoadhesive potencies were measured and rheological behaviors were characterized. Calculated amounts of diphtheria toxoid were added to formulations after optimization of formulations was achieved, and assay and in vitro release studies were carried out. Formulations coded F3 and F7 were considered to be superior to other formulations given the in vitro test results. Therefore, these formulations were tested in guinea pigs to determine immune responses, which they would produce following intranasal and subcutaneous administration. Absorbance values of ELISA tests and antibody neutralization test showed that formulations coded F3 and F7 were unable to stimulate adequate systemic immune response when either of the formulations was administered alone intranasally, whereas F7 resulted in significantly increased neutralizing antibody titers with intranasal administration as a booster dose following subcutaneous administration.
Introduction
The development of vaccines is one of the most important breakthroughs in the immunization at medicine and public health. Immunization with vaccines saves approximately 3 million lives worldwide each year, and it saves millions more from suffering illness and lifelong disability (Bellamy, Citation2005; Matthias et al., Citation2007).
Most of the registered human vaccines are currently available for non-mucosal administration via subcutaneous or intramuscular routes. However, most pathogens gain access to their hosts via the mucosal surfaces (Aggerbeck et al., Citation1997; De Magistris, Citation2006). It is therefore important to provide an immunization by mucosal routes and it may be more effective at inducing protective immunity against mucosal pathogens at their sites of entry (Saatçi & Bozkır, Citation2003).
The application of vaccines on mucosal surfaces provides systemic immune responses as well as the stimulation of mucosal immune responses (Neutra & Kozlowski, Citation2006). Mucosal vaccination can be thought to be alternative to parenteral administration because of stimulating both systemic and mucosal immune system and producing humoral and cellular immune responses (Illum & Davis, Citation2001).
Mucosal vaccines can be administered orally or nasally rather than via injection (O’Hagan, Citation1998). Mucosal routes for local immunization are oral, bronchial, genital, rectal and nasal ways (Sharma et al., Citation2009). Recently nasal vaccination has emerged as an attractive alternative. Most infectious agents enter the body by inhalation and therefore mucosal immune responses function as a first line of defense. The mucosal immune response is rapid and nasal vaccines may also induce protection in distant mucosal sites (Saatçi & Bozkır, Citation2003; Vila et al., Citation2004).
After intranasal immunization, both humoral and cellular immune responses may occur. When the antigens entered into organism, they are taken by sub-mucosal lymphoid cells. Thus, B cells and helper-T cells are activated to create IgA in plasma (Davis, Citation2001). The low molecular weight soluble antigens can penetrate all of the nasal mucosa and then they reach to cervical lymph nodes, which are responsible for the actual systemic immune responses. These soluble antigens can also stimulate humoral immunity (Illum et al., Citation2001).
Nasal mucosa became a region of interest for vaccine administration, because of its large absorption surface, low proteolytic activity and potential of stimulation of both mucosal and systemic immune responses. However, for the vast majority of peptide and protein structure of the active substances in the form of simple solutions administered nasal cavity, less than 1% bioavailability occurred. The major factors affecting the nasal absorption of proteins are: (i) low transfection of antigens through nasal mucosa; (ii) mucociliary clearance mechanism (Soane et al., Citation1999; Amidi et al., Citation2006; Bertram & Bodmeier, Citation2006).
Liquid between 0.02 mL and 0.15 mL volume can be applied to the nasal cavity. Researchers are focused on the use of gelling agents or viscosity enhancers to use this volume effectively. Gelling agents slow down the removal of formulations from the mucous membrane and prolong the duration of the retention (McNeela et al., Citation2001; Park et al., Citation2001; Arora et al., Citation2002).
The goal of vaccination is to provide long-term protection against infection by creating a strong immune response (Aguilar & Rodriguez, Citation2007). Aluminum salts, which are the most commonly used adjuvants for humans, are not effective enough for mucosal vaccination (Jung et al., Citation2001). Generally effective adjuvants when given parenterally have been reported to be toxic or not stable when applied mucosally (Nagler-Anderson, Citation2001). Promising mucosal adjuvants which improve the immune response can be classified under two main groups, as particulate systems and immune stimulating adjuvants (Alpar et al., Citation2005; Devrim et al., Citation2007).
Recently researchers are working on in situ gelling mucosal delivery systems in order to provide retention on application zone and ease of administration (Han et al., Citation2006). These formulations are liquid at room temperature, when they are applied to nose they turn into gel state depending on the temperature. Polymers like poloxamer, tetratonic, ethyl(hydroxy ethyl) cellulose, hydroxypropyl methyl cellulose, and methyl cellulose are used for this purpose (Lin & Sung, Citation2000; Srividya et al., Citation2001; Nanjawade et al., Citation2007).
Poloxamers are non-ionic and ABA-type triblock copolymers that are composed of poly(oxyethylene) and poly(oxypropylene) units (Matschke et al., Citation2002; Kang et al., Citation2007). Most commonly used poloxamers in pharmaceutical industry are Poloxamer 407 (Pluronic F127) and Poloxamer 188 (Pluronic F68). Carbopol (carbomer) is a synthetic high molecular weight polymer of acrylic acid (Han et al., Citation2006).
Talasaz et al. indicate that hydroxypropyl methyl cellulose (HPMC) and hydroxypropyl cellulose (HPC), formed temperature-dependent gelling systems when they were added into aqueous solutions of Poloxamer 407 (Talasaz et al., Citation2008). Furthermore, HPC and HPMC extend the duration of contact of in situ gelling systems with the mucosa due to their viscosity enhancing effects (Bilensoy et al., Citation2006; Zaki et al., Citation2007).
Chitosan is a cationic polysaccharide composed of glucosamine and N-acetyl-glucosamine copolymers. Chitosan presents excellent biological properties such as biodegradability and immunological, antibacterial and mucoadhesive activity (Illum, Citation1998; Davis, Citation2006; Saka & Bozkir, Citation2012). The mucoadhesive properties of chitosan can reduce the clearance rate of drugs from nasal cavity. The interaction of the positively charged amino group of chitosan with the negatively charged sialic acid residues in mucus causes the transient opening of the tight junctions and allows paracellular transport of the compounds (Kotze et al., Citation1998; Davis, Citation2006; Andrews et al., Citation2009).
Diphtheria toxoid was used as a model active substance. Immunologic, but non-toxic diphtheria toxoid is obtained via reaction of diphtheria toxin with formaldehyde. Concentration of formaldehyde, reaction duration, reaction temperature and composition of matrix are the most important conditions.
The purpose of this paper is to evaluate mucosal vaccines in order to prolong the stay in the contact region by in situ gelling systems. In situ gel mucosal vaccines were prepared with Poloxamer 407, Poloxamer 188, Chitosan, Carbopol 934, HPMC and HPC polymers. Moreover, in vitro and in vivo studies were also investigated.
Materials and methods
Materials
Diphtheria toxoid (675 Lf/mL) was kindly provided from Refik Saydam Health Centre, Turkey. Anti-guinea pig IgG peroxidase conjugate, Poloxamer 188 (Pluronic F68, m.wt: 8400) and Poloxamer 407 (Pluronic F127, m.wt: 12500) were purchased from Sigma-Aldrich Chemie GmbH. Hydroxypropyl cellulose (HPC) and Carbopol 934 BF were obtained from Sigma-Aldrich Chemie GmbH and Goodrich Chem. Co., respectively. Hydroxypropyl methyl cellulose (HPMC), low MW chitosan (m.wt. 150 000) and medium MW chitosan (m.wt. 400 000) were purchased from Fluka, Chemie GmbH, Steinheim, Germany. All other chemicals were reagent grade chemicals.
Characterization of diphtheria toxoid
The antigenicity of diphtheria toxoid was 675 Lf/mL as measured by the Ramon flocculation test. The final concentration of diphtheria toxoid was 2000 µg/mL as calculated. The toxoids were stored at 4 °C prior to analysis. Diphtheria toxoid was prepared in PBS (pH 7.4) and acetate buffer (pH 4.5) and peak wavelength (λmax) of diphtheria toxoid solutions of known concentrations were determined.
SDS-PAGE
SDS-PAGE under reducing conditions was performed. Different concentrations of diphtheria toxoid were prepared in PBS (pH 7.4). For reduction of the disulphide bridges, 5 µL sample buffer (1.0 M Tris (pH 6.8), 1% bromophenol blue, 2-mercapto ethanol, 50% glycerol diluted in distilled water) was added into the protein solutions (20 µL) and the resulting mixture was boiled for 10 min at 100 °C. The samples of 20 µL were loaded onto 10% SDS-PAGE gels and electrophoretically separated. Protein bands were visualized by using Coomassie brilliant blue.
Preparation of in situ gels
Thermosensitive and mucoadhesive formulations were prepared using temperature-sensitive Poloxamer and the mucoadhesive polymers (HPC, HPMC, Carbopol 934 and Chitosan). The Pol vehicles used throughout this study were composed of two kinds of polymers, Polaxamer 407 and Polaxamer 188. First, mucoadhesive polymers HPC (0.2%), HPMC (0.2%) and Carbopol 934 (0.2 w/w %) were stirred in the calculated amount of distilled water or phosphate-buffered saline (PBS) at room temperature. Chitosan (medium MW and low MW) was dissolved in 2% acetic acid solution and used as chitosan stock solution. The dispersions were cooled down to 4 °C and Poloxamer 407 and Poloxamer 188 were added slowly with continuous stirring. The final concentrations of each polymer in the Pol vehicles were 18% and 10% for Poloxamer 407 and Poloxamer 188, respectively. The dispersions were then stored in a refrigerator (2 °C–8 °C) until clear solutions were obtained. Diphtheria toxoid was slowly added at 4 °C with continuous stirring before use.
Measurement of gelation temperature by visual inspection
Gelation temperature is defined as the temperature at which the liquid phase makes a transition to gel (Han et al., Citation2006). Briefly, a 10-mL transparent vial containing a magnetic bar and each formulation were placed in a water bath. The vial was heated at a constant rate while stirring (90 rpm). The gelation temperature was measured when the magnetic bar stopped moving due to gelation. Each formulation was tested thrice to control the repeatability of the measurement.
Measurement of gelation duration
Briefly, the formulations were placed on magnetic stirrer (100 rpm) heated to 37 °C. All formulations were removed from water bath at every minute and their flowabilities were checked by turning the vials upside-down. The period from beginning to not flowing has been considered as gelation time.
Viscosity measurement of in situ gels
The viscosity of the formulations was determined with Brookfield RVTDV-II viscometer using spindle T-C and T-F. The measurements were done either in solution at room temperature (25 °C) and in gel at 34 °C. Each formulation was tested thrice to control the repeatability of the measurement.
Evaluation of the mucoadhesive strength
Mucoadhesive forces of the formulations were determined by measuring the tension forces during the separation of the mucin-coated surfaces. We used an instrument which was described by Choi et. Al. with some modifications (Choi et al., Citation1998). Briefly, under the formulations’ applied area, we seated a vial with a weight in its inside on a movable platform and covered its cap (1.1 cm in diameter) with aluminium foil. In addition, a vial cap was covered with aluminium foil as well as lower part, and connected to the right scale pan with the help of a thin wire. Saettone et al. indicate that alginic acid solutions show the closest values of mucin characteristics, so we used alginic acid solution (1% w/w in water) instead of mucin (Saettone et al., Citation1989). Alginic acid solution was spreaded homogeneously both on the vial caps placed at the upper and lower part of the right scalepane. Formulations were also applied onto the vial cap placed at the lower part homogeneously and cooled with cold air. Bottom part of the system was raised slowly by movable platform until the two surfaces touch. By the force of the upper portion, contact was provided between the surfaces in a minute. Then 10 mg/s were added to the left scalepan. We measured the weight (mg) when the two surfaces were separated, at a constant temperature (34 °C, n = 3).
Diphtheria toxoid assay
The concentration of diphtheria toxoid in the formulations were determined by the micro-bichinchoninic acid (micro BCA) protein assay. Each formulation was dissolved in PBS (pH 7.4) or acetate buffer (pH 4.5). The absorption of the samples were read at 560 nm and the amount of diphtheria toxoid in the formulations was calculated from the difference between the amount determined from diphtheria toxoid free formulations and the amount present in the formulations containing diphtheria toxoid. This test was done for each formulation thrice to control the repeatability of the measurement.
In vitro release studies from gel formulations containing diphtheria toxoid
In vitro release of diphtheria toxoid from formulations was evaluated by a membrane-less dissolution model (Barichello et al., Citation1999). Formulations were transferred into eppendorfs and placed in a 37 °C shaking water bath. The formulations gelled within 3 min at 37 °C. Then 1000 μL of the release medium (PBS, pH 7.4), pre-equilibrated at the experimental temperature, was layered over the surface of the gels. In vitro release studies were carried out at 100 rpm. At certain sampling times, supernatant was completely removed and the supernatant withdrawn for the analysis were replaced with equal volumes of fresh dissolution medium at 37 °C. The collected samples were centrifuged at 4000 rpm for 15 min. The amount of diphtheria toxoid present in the dissolution medium was determined by micro BCA protein assay. The absorptions of the samples were read at 560 nm. The absorptions of the samples were read in triplicate for each experiment.
In vivo studies
For in vivo studies, in situ gel formulations containing diphtheria toxoid were administered intranasally (i.n.) or subcutaneously (s.c.) to 45 guinea pigs (n = 5) aged up to 7–8 weeks and weighing about 300–350 g. Diphtheria toxoid in PBS was also given intranasally (i.n.) or subcutaneously (s.c.) for comparison. Animals were maintained on an ad libitum diet throughout the study. The Ethical Committee of Presidency of Refik Saydam Health Centre approved the study and the studies were carried out at Presidency of Refik Saydam Health Centre Biologic Control Division. Formulations were administered either intranasally or subcutaneously as priming dose on day 0 and boosting on days 7 and 21. Both the priming and boosting doses were 4 Lf DT/guinea pig dose for each formulation. Intranasal formulations were applied to unanaesthetized animals and 20 µL (10 µL/nostril) applied into their nares. 200 µL of subcutaneous injections were applied to unanaesthetized animals.
To assess immune responses, blood samples from heart were collected on days 20 and 42. The collected blood samples were stored at room temperature for 1 h, and then blood samples were centrifuged for 10 min at 10 000 rpm. The serum was collected and stored at −20 °C until analysis for IgG titers.
Enzyme-linked immunosorbent assay
Titration of DT-specific Ig G antibody in serum samples collected from vaccinated animals was determined with modified enzyme-linked immunosorbent assay (ELISA) method (European Pharmacopoeia Online, Citation2009). Briefly, 100 µL of diphtheria toxoid solution (diluted to 2 Lf/mL with pH 9.6 carbonate coating buffer) was added to micro-titration plates and allowed to incubate for 16 h at 4 °C. The plates were washed with PBS containing 0.5 g/L Tween 20 for three times and were blocked with 200 µL blockade buffer (washing buffer containing 5.0% skim milk) for 1 h at 37 °C. Washing buffer containing 2.0% skim milk was added to wells, which were washed for three times following incubation. Two-fold dilutions of serum samples were applied to the wells to a final volume of 100 µL. Plates were incubated for 1.5 h at 37 °C and then washed for five times with washing buffer. After washing with washing buffer, plates were incubated with 100 µL of anti-guinea pig IgG peroxidase conjugate diluted 1:5000 with solution containing 2% skim milk/well, for 1.5 h at 37 °C. Unbound conjugate was removed by washing with washing buffer for five times and enzyme activity was determined by adding a 100 µL tablet of o-Phenyldiamine Dihydrochloride (OPD) (Sigma, USA) in 0.05 M citric acid buffer which contains 0.03% sodium perborate/well. Plates were placed in dark for 30 min and then reaction was stopped by adding 100 µL of 1 N H2SO4 solution/well. The absorbance was read at 450 nm with an ELISA reader (Molecular Devices Spectra Max190, USA).
Antibody neutralization test
We determined serum antitoxin titers of vaccinated animals by antibody neutralization test in cell culture, which is more sensitive than ELISA. Vero cells (African green monkey kidney cells) are lysed with diphtheria toxins, and this causes cytopathogenic effect. This method is based on inhibition of cytopathogenic effect by antibodies (European Pharmacopoeia Online, Citation2009).
Cell culture studies were carried out under aseptic conditions at laminar air flow cabinet. Standard serum (1060 units/vial) was used for determination of the endpoint, which is the highest serum dilution that protects the cells from the influence of the diphtheria toxins. We prepared dilutions with vaccinated animals sera and eagle MEM (3% fetal bovine serum). 50 µL of dilution was applied to row A, and 25 µL to row B. Then 25 µL of 9230 CD 50/mL toxins were added to all wells. Plates were immediately shaken and incubated for 1 h at 37 °C for neutralization. After neutralization 100 µL of medium and 50 µL of Vero cells (3 × 105 cells/mL) were added and each well was covered with a transparent film. Plates were incubated at 37 °C for five days in the incubation oven and at the end of the fifth day the calculations were made by counting viable cells with a microscope.
Statistical analysis
Results from in vivo studies were evaluated with the Kruskal–Wallis test and the differences between the groups were determined. Kruskal–Wallis Multiple Range Test (Dunn Test) was then used to determine differences between groups. Time-dependent evaluation of related groups was done with Wilcoxon’s test. p < 0.05 was taken as significant.
Results
Characterization of diphtheria toxoid
The antigenicity of diphtheria toxoid was measured as 675 Lf/mL by the Ramon flocculation test. Peak wavelengths (λmax) of diphtheria toxoid solutions of known concentrations were determined at 560 nm by an ELISA reader. Diphtheria toxiod was analyzed on an acrylamide gel after reduction of the disulphide bridges. Toxoid appeared as a single chain of 66 kDa and in the nicked form as two fragments of 29 kDa (A-fragment) and 45 kDa (B-fragment). Apart from these bands, no additional band was seen.
Gelation temperature and duration
The gelation temperatures have been considered to be suitable if they are in the range of 25 °C to 37 °C. Since the physiological temperature of the nasal cavity is 34 °C (Keck et al., Citation2000), this study aimed at preparing the liquid formulations of Pol 407 and Pol 188 that will gel below 34 °C. The gelation temperatures of formulations were determined by visual inspection.
Preliminary studies showed that gelation temperature of formulations containing 16%, 18% and 20% Pol 407 was found to be 35 °C, 29 °C and 27 °C. Various combinations of Pol 407 and Pol 188 were also examined and the ratio 18/10 Pol 407/Pol 188 (w/w) was chosen for further studies, whereby their mucoadhesive and release properties were improved by the addition of mucoadhesive and PEG polymers. The mucoadhesives used were the neutral polymers hydroxypropyl cellulose (HPC), hydroxypropyl methylcellulose (HPMC), the cationic polymer chitosan (CS, LMW) as well as the anionic polymer carbopol® 934 P (CBP). The gelation temperatures of Pol-based formulations were lowered by mucoadhesive polymers (), but not significantly affected by DT (). The gelation temperatures of Pol-based formulations were lowered from 33 °C to 31 °C in the presence of 0.2% mucoadhesive polymer CBP and 33 °C to 32 °C in the presence of HPC. In the presence of DT, the mixture of Pol 407 and Pol 188 (18/10%) gelled at 32 °C. When DT was added to Pol-based formulations containing CBP or HPC, the gelation temperature was 30 °C and 31 °C, respectively. The time for the complete gelation in vivo might take approximately 5 min following mucosal administration (Park et al., Citation2001). All of the formulations were gelled within 5 min ().
Figure 1. The effect of DT on the gelation temperatures of poloxamer solutions.
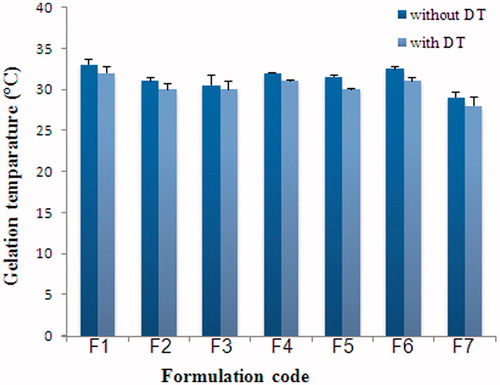
Table 1. Gelation temperatures and duration of poloxamer solutions.
Viscosity of in situ gels
Shear rate viscosity was measured at 25 °C and 34 °C. It was shown that formulation viscosity decreased as shear rate increased, which means that the formulations were pseudoplastic. Presence of the mucoadhesive polymers results in gels becoming more viscous. Viscosities of the formulations ranged as follows: F6 > F5 > F7 > F3 > F2 > F4 > F1. Viscosities of the formulations with chitosan were significantly higher than the other formulations’ viscosity, whereas in the absence of mucoadhesive polymers the formulation showed the lowest viscosity. Rheological properties of gels are largely affected by their polymer content and the thickening behavior of the mucoadhesive polymer used in the formulation.
Mucoadhesive strength
Mucoadhesive strengths of selected formulations are given in . It was clear that mucoadhesive strengths of the formulations showed parallelism with their viscosities.
Table 2. Mucoadhesive strengths of poloxamer solutions.
Diphtheria toxoid assay
The antigen content of formulations was determined between 95.92% and 101.82% using the MicroBCA protein assay. Recovery values of diphtheria toxoid in the formulations were within the 95% confidence limit and the standard errors were small. It was concluded that diphtheria toxoid was dispersed in the formulations homogeneously and the intended dose can be given accurately.
In vitro release studies from gel formulations containing diphtheria toxoid
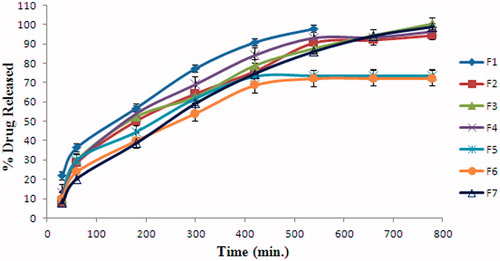
The in vitro release profiles of DT from Pol-based formulations at pH 7.4 PBS are shown in . Non-mucoadhesive gel formulation, F1 showed the fastest release of DT. F1 released its DT content in 540 min. This result was consistent with viscosity values determined before gelation and after gelation. This indicates that, these formulations remain much longer in nasal mucosa and show slow release of DT. This is an important clue for the necessity to use mucoadhesive polymer. Formulations with mucoadhesive polymers (F2, F3, F4, F5, F6 and F7) retarded the diphtheria toxoid release. It was thought that, mucoadhesive polymers enhance the viscosity and cause a delay by compressing the DT diffused water channels in poloxamer micells. Mucoadhesive polymers CBP and HPC retarded the drug release. F3 released its loaded DT in 780 min, whereas F2 released 94% (w/w) of DT in the same duration. Drug release from F4 was the fastest compared to the other formulations with mucoadhesive polymers. This data was compatible with viscosity results. Meanwhile F5 and F6 released only 72%–73% (w/w) of their drug content after 780 min. Therefore, it was decided not to use F5 and F6 for the following studies. Low release of DT may be associated with pH of the dissolution medium or undissolved chitosan in PBS (pH 7.4). F7 formulation which has the highest viscosity values before and after gelation, showed parallel results with release of the DT. However, F7 released its loaded DT in 780 min. It was considered that this difference came from the use of different solutions for the preparation of formulations.
Figure 2. In vitro release profiles of DT from F1, F2, F3, F4, F5, F6 and F7 formulations.
In vivo study results
We considered F3 and F7 formulations to be applied in vivo animal experiments belonging to their physical characteristics, in vitro release characteristics, and mucoadhesive properties. IgG titers of the formulations which generate immunity at guinea pigs, were measured by ELISA. When the 42nd day’s blood samples were analyzed, we observed that immunization was not enough even the animals were immunized intranasally for three times. Conversely, when the F3 and F7 formulations were applied subcutaneously initially and intranasally on day 21, we observed higher IgG responses than the control group consisted of diphtheria toxoid in PBS. While the comparison of the immunity between formulations F3 and F7 was done, higher immune response in animals were obtained in formulation F7.
Immune responses of the formulations at the end of 20th and 42nd days were statistically examined with Kruskal–Wallis Multiple Comparison Test (Dunn test). There is no significant difference (p > 0.05) between three times nasally administered F3, F7 and control group. Therefore, only nasal administration is insufficient to provide systemic immune response against diphtheria. Antitoxin titers of F3 and F7 formulations which were administered subcutaneously initially and intranasally on day 21 are shown in . There is a significant difference between F7 formulation and control group (p < 0.05), but the difference is not significant between F3 formulation and control group (p > 0.05). We compared the antitoxin titers at the end of 42nd day, while F3 and F7 formulation administered both subcutaneous-intranasal combination and subcutaneous only (). Antitoxin titers of F7 formulation which was administered subcutaneously and intranasally are significantly higher than only nasally administered both at the end of 20 and 42 days (p < 0.05). These increases were also investigated while it is come forward by intranasal administration on day 21 or beginning of subcutaneous administration at the beginning. The results indicate that in situ gel formulation of primary vaccination by parenteral route followed by intranasal administration causes both systemic and mucosal strong responses. Unfortunately, F3 formulation did not generate immune response compared to control groups (p > 0.05).
Figure 3. Antibody neutralization titers of formulation at the end of 42 days, which are vaccined subcutaneously at the beginning and initial intranasal administration at 21st day or only subcutaneously at the begining (n:5).
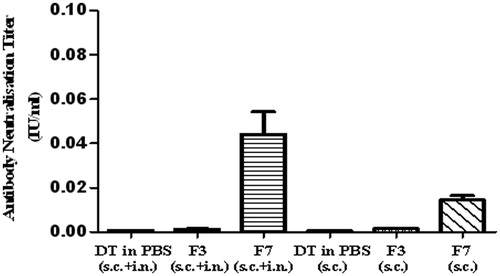
Significant differences of immune responses for each formulation between the beginning and after the 20th and 42nd days were also determined by Wilcoxon’s test. There are no any significant differences for F3 formulation between beginning–20th day, beginning–42nd day and 20th–42nd days (p > 0.05). On the other hand, F7 formulation administered subcutaneously initially followed by intranasal administration as booster, antitoxin titers show significant increases at the end of 42nd day (p < 0.05) and provided partial protection against diphtheria toxoid after 42 days. There is no any significant difference between beginning–20th day (p > 0.05), while significant difference was observed between 20th and 42nd days for F7 formulation (p < 0.05). This is caused by systemic antibodies (IgG) which cannot be formed immediately. While F7 formulation was administered only subcutaneously, there is also significant difference between beginning and interval days (p < 0.05). In this case, although antitoxin titers are low compared to the subcutaneous administration strengthened by initial intranasal administration, acquisition of partial protection of the animals against diphtheria toxoid was maintained at the end of 42nd day.
Discussion
The objective of this study was to develop formulations that contain diphtheria toxoid given intranasally to achieve active immunization against diphtheria, a contagious infection of the upper respiratory tract. In situ gel formulations, which can be administrated as intranasal drops have been developed with the aims of prolonging the residence time of the diphtheria toxoid in the mucosa, improve its therapeutic efficacy and facilitate its administration and to produce both mucosal and systemic immune response. Gelling of the formulations in a manner dependent on the temperature of the nose was achieved by using Poloxamer 407 and Poloxamer 188 polymers and attempts were made to prolong the retention of the formulations in the mucosa by adding the mucoadhesive polymers of Carbopol 934, HPMC, HPC or chitosan. In vitro and in vivo studies were performed for this purpose. Based on the pre-formulation studies, the formulations coded F1, F2, F3, F4 and F5 were selected and their suitability for intranasal administration of diphtheria toxoid was supported by the tests performed. Following in vitro studies, it was decided to determine the immune response of the formulations coded F3 and F7 in vivo, which were found to be superior in rheological and diphtheria toxoid-releasing properties to the remaining formulas. Antitoxin titers in the sera collected from guinea pigs were assayed by the antibody neutralization test using Vero cells. The results of the analyses indicated that the formulations coded F3 and F7 were inadequate to produce systemic immune response when either of the formulas was administered alone intranasally. On the other hand, antitoxin titers of in situ gel formulations given subcutaneously initially and intranasally on day 21 were found to increase significantly with supporting doses. Comparison of the activities of the formulas coded F3 and F7 demonstrated a higher immune response obtained in animals immunized with F7. Both Kruskal–Wallis and Wilcoxon tests showed that combined use of chitosan and Poloxamer gel resulted in an increased systemic immune response with diphtheria toxoid. The mixture of Poloxamer 407 18% and Poloxamer 188 10% acts as a depot for the antigen due to its gelling properties and protects the antigen from break down by proteases until it is diffused from the polymer matrix. There are reports in the literature supporting that chitosan gives high responses in parenteral vaccinations due to its adjuvant properties (Coeshott et al., Citation2004). It has been suggested that the efficacy of intranasal administration of chitosan was associated with its activity of preventing mucociliary clearance due to its better mucoadhesive properties compared to HPMC and improving absorption by opening intracellular links (Kotze et al., Citation1998; Davis, Citation2006; Andrews et al., Citation2009). When in situ gel formulations containing the Poloxamer gel and chitosan (low-molecular weight) mucoadhesive polymer is administered intranasally following subcutaneous dosing, the antitoxin titers they produce indicate that they would provide partial protection according to the ranges defined by the World Health Organization and supporting vaccinations would be necessary at certain intervals.
Conclusion
In this study, we prepared in situ gel formulations of diphtheria toxoids for increasing the therapeutic effect by prolonged mucosal contact and stimulating both systemic and mucosal immune system. Another advantage is that gel formulations can be applied easily by droplet into intranasal way.
We provided protection in the limits of WHO when in situ gel formulations administered intranasally after subcutaneous administration. But we need rapel intranasal doses at intervals. According to these results, our group continue to work on improving the immune response.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Aggerbeck H, Gizurarson S, Heon I, Wantzin J. (1997). Intranasal booster vaccination against diphtheria and tetanus in man. Vaccine 15:307–16
- Aguilar JC, Rodriguez EG. (2007). Vaccine adjuvants revisited. Vaccine 25:3752–62
- Alpar HO, Atuah KN, Bramwell VW, Somavarapu S. (2005). Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv Drug Deliv Rev 57:411–30
- Amidi M, Borchard G, Hennink WE, et al. (2006). Preparation and characterization of protein-loaded N-trimethyl chitosan nanoparticles as nasal delivery system. J Control Rel 111:107–16
- Andrews GP, Jones DS, Laverty TP. (2009). Mucoadhesive polymeric platforms for controlled drug delivery. Eur J Pharmaceut Biopharmaceut 71:505–18
- Arora P, Garg S, Sharma S. (2002). Permeability issues in nasal drug delivery. Drug Discov Today 7:967–75
- Barichello JM, Morishita M, Nagai T, Takayama K. (1999). Absorption of insulin from Pluronic F-127 gels following subcutaneous administration in rats. Int J Pharmaceut 184:189–98
- Bellamy R. (2005). Freedman A. Immun Med 33:17–21
- Bertram U, Bodmeier R. (2006). In situ gelling, bioadhesive nasal inserts for extended drug delivery: ın vitro characterization of a new nasal dosage form. Eur J Pharmaceut Sci 27:62–71
- Bilensoy E, Hincal AA, Rouf MA, et al. (2006). Mucoadhesive, thermosensitive, prolonged-release vaginal gel for clotrimazole: β-cyclodextrin complex. AAPS Pharm Sci Tech 7:E1–7
- Choi HG, Jung JH, Kim CK, et al. (1998). Development of in situ-gelling and mucoadhesive acetaminophen liquid suppository. Int J Pharmaceut 165:33–44
- Coeshott CM, Blonder JM, Rosenthal GJ, et al. (2004). Pluronic® F127-based systemic vaccine delivery systems. Vaccine 22:2396–405
- Davis SS. (2001). Nasal vaccines. Adv Drug Deliv Rev 51:21–42
- Davis SS. (2006). The use of soluble polymers and polymer microparticles to provide improved vaccine responses after parenteral and mucosal delivery. Vaccine 24:7–10
- De Magistris MT. (2006). Mucosal delivery of vaccine antigens and its advantages in pediatrics. Adv Drug Deliv Rev 58:52–67
- Devrim B, Bozkir A, Canefe K. (2007). Preparation and evaluation of surface-modified PLGA microparticles with chitosan for pulmonary delivery of proteins. Adv Chitin Sci 10:349–54
- European Pharmacopoeia Online. (2009). Monographs: Aassay of diphtheria vaccine (adsorbed). 217–22
- Han IK, Cho HJ, Jung WW, et al. (2006). Thermosensitive and mucoadhesive delivery systems of mucosal vaccines. Methods 38:106–11
- Illum L. (1998). Chitosan and its use as a pharmaceutical excipient. Pharmaceut Res 15:1326–31
- Illum L, Davis SS. (2001). Nasal vaccination: a non-invasive vaccine delivery method that holds great promise for the future. Adv Drug Deliv Rev 51:1–3
- Illum L, Davis SS, Fisher AN, et al. (2001). Chitosan as a novel nasal delivery system for vaccines. Adv Drug Deliv Rev 51:81–96
- Jung T, Breitenbach A, Hungerer K, et al. (2001). Tetanus toxoid loaded nanoparticles from sulfobutylated poly(vinyl alcohol)-graft-poly(lactide-co-glycolide): evaluation of antibody response after oral and nasal application in mice. Pharmaceut Res 18:352–60
- Kang ML, Cho CS, Guo DD, et al. (2007). Pluronic® F127 enhances the effect as an adjuvant of chitosan microspheres in the intranasal delivery of Bordetella bronchiseptica antigens containing dermonecrotoxin. Vaccine 25:4602–10
- Keck T, Leiacker R, Riechelmann H, Rettinger G. (2000). Temperature profile in the nasal cavity. Laryngoscope 110:651–4
- Kotze AF, de Boer AG, Junginger HE, et al. (1998). Chitosan for enhanced intestinal permeability: prospects for derivatives soluble in neutral and basic environments. Eur J Pharmaceutl Sci 7:145–51
- Lin HR, Sung KC. (2000). Carbopol/pluronic phase change solutions for ophthalmic drug delivery. J Control Rel 69:379–88
- Matschke C, Fahr A, Isele U, van Hoogevest P. (2002). Sustained-release injectables formed in situ and their potential use for veterinary products. J Control Rel 85:1–15
- Matthias DM, Garrison MM, Nelson C, et al. (2007). Freezing temperatures in the vaccine cold chain: A systematic literature review. Vaccine 25:3980–6
- McNeela EA, Davis SS, Illum L, et al. (2001). A mucosal vaccine against diphtheria: formulation of cross reacting material (CRM197) of diphtheria toxin with chitosan enhances local and systemic antibody and Th2 responses following nasal delivery. Vaccine 19:1188–98
- Nagler-Anderson C. (2001). Man the barrier! Strategic defences in the intestinal mucosa. Nature Rev Immunol 18:59–67
- Nanjawade BK, Manjappa AS, Manvi FV. (2007). In situ-forming hydrogels for sustained ophthalmic drug delivery. J Control Rel 122:119–34
- Neutra MR, Kozlowski PA. (2006). Mucosal vaccines: the promise and the challenge. Nat Rev Immunol 6:148–58
- O’Hagan DT. (1998). Microparticles and polymers for the mucosal delivery of vaccines. Adv Drug Deliv Rev 34:305–20
- Park JS, Kim CK, Kim JM, et al. (2001). In situ gelling and mucoadhesive polymer vehicles for controlled intranasal delivery of plasmid DNA. J Biomed Mater Res 59:144–51
- Saatçi F, Bozkır A. (2003). Aşıların nazal yoldan uygulanışı. J Fac Pharm 32:175–93
- Saettone MF, Burgalassi S, Chetoni P, et al. (1989). Evaluation of muco-adhesive properties and in vivo activity of ophthalmic vehicles based on hyaluronic acid. Int J Pharmaceut 51:203–12
- Saka OM, Bozkir A. (2012). Formulation and in vitro characterization of PEGylated chitosan and polyethylene imine polymers with thrombospondin-I gene bearing pDNA. J Biomed Mater Res – Part B Appl Biomater 100B:984–92
- Sharma S, Benson HAE, Chen Y, Mukkur TKS. (2009). Pharmaceutical aspects of ıntranasal delivery of vaccines using particulate systems. J Pharmaceut Sci 98:812–43
- Soane RJ, Davis SS, Jones NS, et al. (1999). Evaluation of the clearance characteristics of bioadhesive. Int J Pharmaceut 178:55–65
- Srividya B, Amin PD, Cardoza RM. (2001). Sustained ophthalmic delivery of ofloxacin from a pH triggered in situ gelling system. J Control Rel 73:205–11
- Talasaz AHH, Atyabi F, Dinarvand R, et al. (2008). In situ gel forming systems of poloxamer 407 and hydroxypropyl cellulose or hydroxypropyl methyl cellulose mixtures for controlled delivery of vancomycin. J Appl Polym Sci 109:2369–74
- Vila A, Alonso MJ, Behrens I, et al. (2004). Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharmaceut Biopharmaceut 57:123–31
- Zaki NM, Abd Elhadyb SS, Awada GA, Mortadaa ND. (2007). Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharmaceut Sci 32:296–307