
Abstract
Nanotechnology has brought a revolution in the field of science, which has subsequently lead to development of novel dosage forms such as niosomes, liposomes and proniosomes. Proniosomes overcome the demerits involved with niosomal and liposomal drug delivery systems. Proniosomes are liquid crystalline compact niosome hybrids which upon hydration form niosomes. They help in reducing physical stability problems involved with niosomes such as leaking, fusion, aggregation and provide convenience in dosing, distribution, transportation and storage showing improved results than conventional niosomes. This review focuses on different aspects of proniosome such as preparation, characterization, drug release, applications, merits, demerits, present scenario in market and future trends.
Introduction
Novel drug delivery systems have emerged embracing various routes of administration, to attain targeted and controlled drug delivery. Drug encapsulation in the vesicles is one such system which helps to prolong drug duration in systemic circulation and decreases the toxicity by selective uptake. Based on this technique, a number of vesicular drug delivery systems such as liposomes, niosomes and provesicular systems like proliposomes and proniosomes have been developed.
Liposomes are colloidal, vesicular structures that are organized in one or several concentric phospholipidic bilayers with an aqueous core inside which encloses a wide variety of substances and drugs (Charcosset et al., Citation2012). However, liposomes have limited success in terms of oral delivery and suffer from physicochemical stability problems such as sedimentation, aggregation, fusion, phospholipid hydrolysis and/or oxidation.
To resolve this stability issues, proliposome approach has provided a major breakthrough by using dry, free-flowing product, which is more stable during sterilization and storage (Raju et al., Citation2012). Proliposomes are the dry, free-flowing powder formulations containing water soluble carrier particles coated with phospholipids which, upon the addition of water, disperse to form a multi-lamellar liposomal suspension. Despite of these merits, proliposomes preparation involves technical difficulties like usage of vacuum or nitrogen atmosphere during preparation and storage to prevent oxidation of phospholipids (Payne et al., Citation1986; Katare et al., Citation1990).
To overcome the limitations associated with liposomes and proliposomes, niosomes () have gained attention as drug carriers and drug-targeting agents, from the early 1980s (Baillie et al., Citation1985; Schreier & Bouwstra, Citation1994). Niosomes are better alternatives to liposomes as promising drug carriers with greater chemical stability, entrapment efficiency of both hydrophobic and hydrophilic drugs and are less toxic due to their non-ionic nature (Uchegbu & Florence, 1995; Uchegbu & Vyas, 1998). They overcome the demerits associated with liposomes (Muller et al., Citation2002) such as phospholipid purity (Vora et al., Citation1998), difficulty in sterilization and high cost. The large-scale production of niosomes does not require any special conditions, unacceptable solvents or precautions (Carafa et al., Citation2002; Alsarra et al., Citation2005). However, like liposomes, niosomes also have physical stability problems such as leakage, fusion, aggregation and sedimentation (Solanki et al., Citation2007).
Figure 1. Schematic representation of various niosomes and proniosomes.
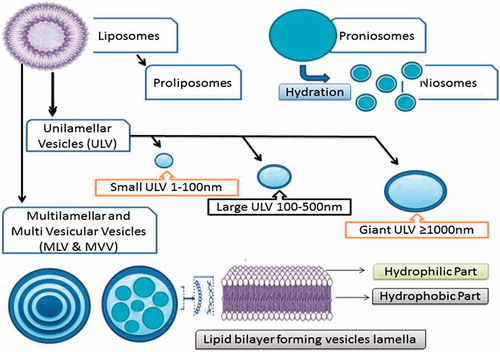
These problems can be evaded by proniosomes. Proniosomes () are dry, free-flowing formulations of surfactant-coated carrier, which can be rehydrated by brief agitation in hot water to form a multi-lamellar niosome suspension suitable for administration by oral or other routes (Hu & Rhodes, Citation1999; Mokhtar et al., Citation2008). Since proniosomes are available as dry powders, they have an additional convenience in transportation, distribution, storage, processing, packaging, providing optimal flexibility, unit dosing as capsule and stable during sterilization. All these advantages make them a promising candidate for industrial production. These versatile delivery systems have potential to be used as a carrier, a wide range of active compounds (Hu & Rhodes, Citation1999; Blazek-Welsh & Rhodes, Citation2001a,Citationb).
Proniosomes preparation methods
The different methods and materials used for the preparation of proniosomes are shown in and .
Figure 2. Materials and methods used.
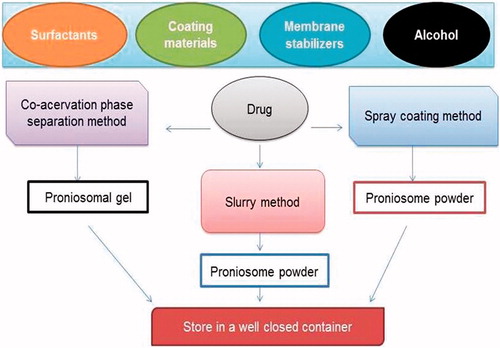
Table 1. Different materials used for the preparation of proniosomes.
Slurry method
This method involves preparation of slurry in a round bottom flask using carrier and surfactant solution. Additional amount of organic solvent can be added if desired to obtain slurry. The slurry is dried by applying vaccum to get free-flowing powder of proniosomes. The powder should be stored at 4 °C in a sealed container. The time required to produce proniosomes is independent on the ratio of carrier material to surfactant solution and appears to be scalable (Perrett et al., Citation1991; Blazek-Welsh & Rhodes, Citation2001a,Citationb; Solanki et al., Citation2007).
Co-acervation phase separation method
This is the widely used method to prepare proniosmal gel. Weighed quantities of drug, lipid and surfactants are taken in a dry wide-mouthed glass beaker followed by the addition of solvent. The ingredients are mixed well and warmed over water bath at 60–70 °C until the surfactant mixture dissolves completely. During the process care must be taken to prevent loss of any solvent due to evaporation. Finally, the aqueous phase is added to the mixture and warmed on water bath. The resultant solution is cooled overnight to obtain proniosomal gel (Vora et al., Citation1998; Gupta et al., Citation2007).
Spray-coating method
In this method, proniosomes are prepared by spraying surfactant in organic solvent onto a carrier/coating material followed by evaporation of solvent. The surfactant forms a thin film on the carrier and subsequent hydration causes formation of multi-lamellar vesicles (Bangham et al., Citation1965; Yoshioka et al., Citation1994).
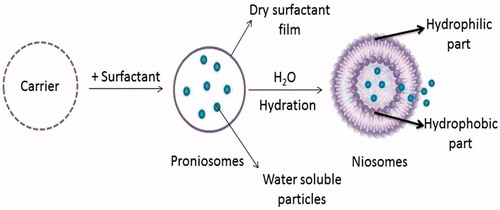
Niosomes formation from proniosomes by hydration
The niosomes can be prepared by hydration of proniosomes, where aqueous phase containing the drug should be added to the proniosomes with brief agitation at a temperature greater than the mean transition phase temperature of the surfactant (Vora et al., Citation1998; Blazek-Welsh & Rhodes, Citation2001a,Citationb). The process is as shown in .
Figure 3. Niosomes formation from provesicular forms by hydration.
Characterization of proniosomes
Proniosomes are characterized for morphology, angle of repose, rate of hydration, aerodynamic behavior, penetration and permeation studies as enlisted in .
Table 2. Various parameters and methods employed for proniosomes characterization.
Particle size analysis (SEM)
Particle size of proniosomes is a very important characteristic. Size distribution and surface morphology (smoothness, roundness and aggregates formation) of particles can be studied by scanning electron microscopy (SEM). The vesicle formation by the particular procedure can be confirmed by optical microscopy. The niosome suspension has to be placed over a glass slide and dried at room temperature, the dry thin film of niosome suspension formed has to be observed for the formation of vesicles.
Measurement of zeta potential
Another characteristic of proniosomes that is of extreme interest is zeta potential. It is a measure of the particle charge, the larger the zeta potential absolute value the larger the amounts of surface charge. Logically, the zeta potential is an index for particle stability. In case of charged particles with increase in zeta potential, the repulsive interactions also increase leading to the more stable particle formation with a more uniform size distribution. A physically stable proniosomal formulation solely stabilized by electrostatic repulsion will have a ± 30 mV of minimum zeta potential and this stability helps in preventing aggregation.
Osmotic shock
This study helps to determine the changes in the vesicle size. The niosomal formulations have to be incubated for 3 h with hypertonic, isotonic, hypotonic solutions and should be observed under optical microscope for vesicle size changes (Malhotra & Jain, Citation1994; Biju et al., Citation2006).
Separation of free (unentrapped) drug
The free drug can be separated from the entrapped drug using techniques as mentioned below.
Dialysis
Using suitable dissolution medium, the aqueous niosomal dispersion has to be dialyzed in dialysis tubing at room temperature. At appropriate time intervals, the samples should be withdrawn from the medium, centrifuged and analyzed for drug content using suitable method UV spectroscopy, HPLC, etc. (Vora et al., Citation1998; Solanki et al., Citation2007).
Gel filtration
Gel filtration is another method used for separation of unentrapped drug from niosomal dispersion using a Sephadex-G-50 column, eluted with suitable mobile phase and analyzed with suitable analytical techniques (Gayatri et al., Citation2000; Vyas & Khar, Citation2002).
Centrifugation
By centrifugation of niosomal suspension (Chandraprakash et al., Citation1990; Vora et al., Citation1998; Hu & Rhodes, Citation1999), the pellets and supernatant are separated. The obtained pellets are washed and then resuspended to obtain a niosomal suspension free from unentrapped drug (Hu & Rhodes, Citation1999; Blazek-Welsh & Rhodes, Citation2001a; Fang et al., Citation2001; Alsarra et al., Citation2005; Gupta et al., Citation2007; Solanki et al., Citation2007; Abd-Elbary et al., Citation2008).
Determination of entrapment (entrapped) efficiency
The entrapment efficiency can be determined by separating the untrapped drug by using any one of the method described above, then the remaining entrapped drug can be determined by complete disruption of the vesicles using propylene glycol: absolute alcohol mixture in 1:1 ratio (Vora et al., Citation1998) or 50% n-propanol followed by the addition of Triton X-100 solution (0.1% v/v) to solubilize vesicles. The resulting clear solution is analyzed for drug content (Chauhan & Luorence, Citation1989; Solanki et al., Citation2007; Ajay et al., Citation2008).
The percentage of entrapped drug is calculated using the following formula (Hao et al., Citation2002; Abd-Elbary et al., Citation2008):
where EE% is the percentage of entrapment efficiency, ED is the concentration of drug entrapped and TD is the concentration of theoretical drug.
In vitro drug release from proniosomes
Skin permeation and in vitro drug release studies for proniosomes can be done by various techniques such as USP Dissolution apparatus Type I (Abd-Elbary et al., Citation2008), Franz diffusion cell (Fang et al., Citation2001; Ruckmani et al., Citation2002; Vyas & Khar, Citation2002; Puglia et al., Citation2004), dialysis tubing (Muller et al., Citation2002), reverse dialysis (Muller et al., Citation2002), cellophane dialyzing membrane (Alsarra et al., Citation2005; Ankur et al., Citation2007), keshary-chien diffusion cell (Vora et al., Citation1998) and spectrapor© molecular porous membrane tubing (Abd-Elbary et al., Citation2008).
In vitro skin permeation studies can be carried out using flank skin, dorsal skin of albino rabbit (Alsarra et al., Citation2005), female albino rat (Sprague-Dawley strain) (Vora et al., Citation1998), Wistar rat skin (7–9 weeks old) (Fang et al., Citation2001). Niosomal vesicles derived from proniosomes can follow any one or more of the drug release mechanisms like drug diffusion from bilayered membrane, desorption from vesicles surface or a combined diffusion and desorption mechanism.
Drug release kinetics
To elucidate mode and mechanism of drug release, the in vitro data have to be transformed and interpreted at graphical interface constructed using various kinetic models (Gibaldi & Perrier, Citation1982) like zero order, first order, Higuchi’s and Peppa’s model.
The zero-order release Equation (1), where the drug release is independent of concentration
where Qt is the amount of drug released in time t, Q0 is the initial amount of the drug in the solution and K0 is the zero-order release constant.
The first-order Equation (2) describes the release from the system where release is concentration dependent
where Qt is the amount of drug released in time t, Q is the initial amount of drug in the solution and K is the first-order release constant.
Higuchi described the release of drug from insoluble matrix as a square root of time
where Qt is the amount of drug released in time t, KH is Higuchi’s dissolution constant.
The following plots can be made: cumulative percentage drug release versus time (zero-order kinetic models); log cumulative of % drug remaining versus time (first-order kinetic model); cumulative percentage drug release versus square root of time (Higuchi’s model).
Peppa’s developed a simple drug release mechanism, semi-empirical model, relating exponentially the drug release to the elapsed time (t)
K is a constant incorporating structural and geometrical characteristic of the drug dosage form, n is the release exponent; ft is Mt/M∞ (fractional release of drug).
If the release exponent n = 0.5 or near, then the mechanism of drug release is fickian diffusion and if n = 1.0 or near, then it is non-fickian diffusion.
Stability studies
The stability studies (Ajay et al., Citation2008) can be performed by storing the samples at different temperatures like freezing temperature (2–8 °C), room temperature (25 ± 0.5 °C) and higher temperature (45 ± 0.5 °C) for a period of 1–3 months. Periodically, drug content and difference in the average vesicle diameter can be observed (Vora et al., Citation1998; Gupta et al., Citation2007; Abd-Elbary et al., Citation2008). According to ICH guidelines, dry proniosome powder meant for re-formulation should be considered for accelerated stability at relative humidity 75%/40 °C as per international climatic conditions and zones. Long-term stability studies have to be conducted based on the climatic zones of the countries. Temperature and relative humidity to be maintained for zones I&II and III&IV are 25 °C/60% RH and 30 °C/65% RH, respectively. The product should be evaluated for appearance, surface characteristics, drug content, color change, pH, particulate matter, assay, preservative content, pyrogenicity and sterility.
Applications
Proniosomes as drug carriers
In vivo behavior of proniosome derived niosomes will be similar to liposomes showing advantages as drug carriers, comprising lower cost and toxicity (Uchegbu et al., Citation1995), easy storage and handling, increased stability. Encapsulation of drug in niosomal formulations reduces the toxicity in various therapies and applications and also prolongs the encapsulated drug circulation time and changes drug distribution in the body (Azmin et al., Citation1985; Baillie et al., Citation1985; Ruckmani et al., Citation2002). Drug delivery through niosomes has been studied using different methods of administration (Blazek-Welsh & Rhodes, Citation2001b), including intravenous (i.v.) (Namdeo & Jain et al., Citation1999), peroral (Rentel et al., Citation1999), transdermal (Yoshioka et al., Citation1994; Blazek-Welsh & Rhodes, Citation2001a; Solanki et al., Citation2007; Ajay et al., Citation2008), inhaled aerosols and intramuscular (i.m.) (Runothayanun et al., Citation1999). Niosomes as drug delivery vesicles, increases absorption of some drugs through cell membranes and cellular uptake via endocytosis (Baillie et al., Citation1985) and so confines the drug in tissues and targeted organs (Namdeo & Jain et al., Citation1999) and also helps to evade the reticulo-endothelial system (Devaraj et al., Citation2002). Various applications of proniosomes are shown in .
Table 3. Applications.
Future trends
Proniosomal formulations are becoming a useful dosage forms for different delivery systems. Still there is a need for discovering the new delivery systems using proniosomes in the field of cosmetics, nutraceuticals, herbal actives and other synthetic formulations. Hence, a wider research should be done to develop scale-up batches for drug and natural preparations.
Conclusion
Proniosomes are promising drug carriers for the future. Proniosomes derived niosomes are better alternative to the liposomal vesicular system due to their greater physical, chemical stability and potentially scalable for commercial viability. Amphiphilic drugs can also be incorporated into the proniosomal delivery system. Proniosomes has attracted a greater deal of attention for delivery of drugs through transdermal route, due to the advantages like non-toxicity, penetration enhancing effect of surfactants and effective modification of drug release. Dry powder form of proniosomes makes them suitable for preparing unit dosage forms such as tablets, capsules and beads. Owing to all these advantages proniosomes are widely investigated as drug carriers. There is lot of scope to investigate new carrier material for preparation of proniosomes and their potential remains to be investigated to full extent.
Declaration of interest
The authors report no conflicts of interest.
References
- Abd-Elbary A, El-laithy MI, Tadros HM. (2008). Sucrose stearate-based proniosome-derived niosomes for the nebulisable delivery of cromolyn sodium. Int J Pharm 357:189–98
- Adnan A, Nilu J, Zeenat I, et al. (2008). Feasibility of proniosomes-based transdermal delivery of frusemide: formulation optimization and pharmacotechnical evaluation. Pharm Dev Technol 13:155–63
- Ajay S, Jolly P, Rajesh P. (2008). Preparation, characterization, optimization, and stability studies of Aceclofenac Proniosomes. Iran J Pharm Res 7:237–46
- Alsarra IA, Bosela AA, Ahmed SM, Mahrous GM. (2005). Proniosomes as a drug carrier for transdermal delivery of ketorolac. Eur J Pharm Biopharm 59:485–90
- Ammara HO, Ghorabb M, El-Nahhasc SA, Higazya IM. (2011). Proniosomes as a carrier system for transdermal delivery of tenoxicam. Int J Pharm 405:142–52
- Ankur G, Sunil KP, Balamurugan M, et al. (2007). Design and development of a proniosomal transdermal drug delivery system for captopril. Trop J Pharm Res 6:687–93
- Azmin MN, Florence AT, Handjani-Vila RM, et al. (1985). The effect of non-ionic surfactant vesicle (niosome) entrapment on the absorption and distribution of methotrexate in mice. J Pharm Pharmacol 37:237–42
- Baillie A, Florence A, Hume L, et al. (1985). Preparation and properties of niosomes-nonionic surfactant vesicles. J Pharm Pharmacol 37:863–8
- Bangham AD, Standish MM, Watkins JC. (1965). Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol 13:238–52
- Biju SS, Talegaonkar S, Misra PR, et al. (2006). Vesicular systems: an overview. Indian J Pharm Sci 68:141–53
- Blazek-Welsh AI, Rhodes DG. (2001a). Maltodextrin-based proniosomes. AAPS PharmSci Tech 3:1–8
- Blazek-Welsh AI, Rhodes DG. (2001b). SEM imaging predicts quality of niosomes from maltodextrin-based proniosomes. Pharm Res 18:656–61
- Brinon L, Geiger S, Alard V, et al. (1999). Peercutaneous absorption of sunscreen from liquid crystalline phases. J Control Release 60:67–76
- Carafa M, Santucci E, Lucania G. (2002). Lidocaine loaded non-ionic surfactant vesicles: characterization and in-vitro permeation studies. Int J Pharm 231:21–32
- Charcosset C, Pham TT, Jaafar-Maalej C, et al. (2012). Liposome and niosome preparation using a membrane contactor for scale-up. Colloids Surf B 94:15–21
- Chandraprakash KS, Udupa N, Umadevi P, et al. (1990). Pharmacokinetic evaluation of surfactant vesicles containing methotrexate in tumor bearing mice. Int J Pharm 61:R1–3
- Chauhan S, Luorence MJ. (1989). The preparation of polyoxyethylene containing non-ionic surfactant vesicles. J Pharm Pharmacol 41:6--11
- Cioca G, James HA, Manuel LT, et al. (1991). Liquid crystal containing cosmetic US 4,999,348 issued 12 Mar 1991
- Devaraj GN, Parakh SR, Devraj R, et al. (2002). Release studies on Niosomes containing fatty alcohols as bilayer stabilizers instead of cholesterol. J Colloid Interface Sci 251:360–5
- El-Laithy HM, Shoukry O, Mahran LG. (2011). Novel sugar esters proniosomes for transdermal delivery of vinpocetine: preclinical and clinical studies. Eur J Pharm Biopharm 77:43–55
- Fang JY, Yu SY, Wu PC, et al. (2001). In-vitro skin permeation of estradiol from various proniosome formulations. Int J Pharm 215:91–9
- Gayatri DS, Venkatesh P, Udupa N. (2000). Niosomal sumatriptan succinate for nasal administration. Int J Pharm Sci 62:479–81
- Gibaldi M, Perrier D. (1982). Pharmacokinetics, 2nd ed. New York: Marcel Dekker
- Gupta A, Prajapati SK, Balamurugan M, et al. (2007). Design and development of a proniosomal transdermal drug delivery system for captopril. Trop J Pharm Res 6:687–93
- Hao Y, Zhao F, Li N, et al. (2002). Studies on a high encapsulation of colchicine by a niosome system. Int J Pharm 244:73–80
- Hiremath PS, Soppimath KS, Betageri GV. (2009). Proliposomes of exemestane for improved oral delivery: formulation and in-vitro evaluation using PAMPA, Caco-2 and rat intestine. Int J Pharm 380:96–104
- Hu C, Rhodes DG. (1999). Proniosomes: a novel drug carrier preparation. Int J Pharm 185:23–35
- Iwai H, Fukasava J, Suzuki TA. (1998). Liquid crystal application in skin care cosmetics. Int J Cosmet Sci 20:87–102
- Katare OP, Vyas SP, Dixit VK. (1990). Effervescent granule based proliposomes of ibuprofen. J Microencapsul 7:455–62
- Lieberman H, Lachman L, Schwartz J. (1990). Pharmaceutical dosage forms: tablets, Vol. 2, 2nd ed. New York: Marcel Decker, 229
- Malhotra M, Jain NK. (1994). Niosomes as drug carriers. Indian Drugs 31:81–6
- Mitsuno Y, Nomaguchi K, Suzuki T. (1988). Lamella type single phase liquid crystal composition and oil-base compositions using the same US 4,767,625 issued Aug 1988
- Mokhtar M, Sammour OA, Hammad MA, et al. (2008). Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm 361:104–11
- Muller RH, Radtke M, Wissing SA. (2002). Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev 54:131–55
- Namdeo A, Jain NK. (1999). Niosomal delivery of 5-fluorouracil. J Microencapsul 16:731–40
- Payne NI, Browning I, Hynes CA. (1986). Characterization of proliposomes. J Pharm Sci 75:330–3
- Perrett S, Golding M, Williams WP. (1991). A simple method for the preparation of liposomes for pharmaceutical applications: characterization of the liposomes. J Pharm Pharmacol 43:154–61
- Puglia C, Trombetta D, Venuti V, et al. (2004). Evaluation of in vivo topical anti-inflammatory activity of indometacin from liposomal vesicles. J Pharm Pharmacol 56:1225–32
- Raju J, Karthik YJ, Ashok V, et al. (2012). Bioavailability enhancement of zaleplon via proliposomes: Role of surface charge. Eur J Pharm Biopharm 80:347–57
- Rentel CO, Bouwstra JA, Naisbett B, et al. (1999). Niosomes as a novel peroral vaccine delivery system. Int J Pharm 186:161–7
- Ruckmani K, Jayakar B, Ghosal SK. (2002). Nonionic surfactant vesicles (niosomes) of cytarabine hydrochloride for effective treatment of leukemias: encapsulation, storage, and in-vitro release. Drug Dev Ind Pharm 26:217–22
- Runothayanun P, Sooksawate T, Florence AT. (1999). Extrusion of niosomes from capillaries: approaches to pulsed delivery device. J Control Release 60:391–7
- Saettone MF, Perini G, Carafa M, et al. (1996). Non-ionic surfactant vesicles as ophthalmic carriers for cyclopentolate. A preliminary evaluation. S.T.P Pharma Sci 6:94–8
- Schreier H, Bouwstra J. (1994). Liposomes and niosomes as topical drug carriers-dermal and transdermal drug-delivery. J Control Release 30:1–15
- Solanki AB, Parikh JR, Parikh RH. (2007). Formulation and optimization of piroxicam proniosomes by 3-factor, 3-level box-behnken design. AAPS Pharm Sci Tech 8:E1–7
- Tamizharasi S, Sunil B, Rathi V, et al. (2009). Formulation and evaluation of maltodextrin based proniosomes loaded with indomethacin. Int J Pharm Tech Res 13:517–23
- Thakur R, Anwer MK, Shams MS, et al. (2009). Proniosomal transdermal therapeutic system of losartan potassium: development and pharmacokinetic evaluation. J Drug Target 17:442–9
- Uchegbu IF, Double JA, Turton JA, et al. (1995). Niosome encapsulation of a doxorubicin polymer conjugates. Pharm Res 12:1019–24
- Uchegbu IF, Florence AT. (1995). Non-ionic surfactant vesicles (niosomes): physical and pharmaceutical chemistry. Adv Colloid Interface Sci 58:1–55
- Uchegbu IF, Vyas SP. (1998). Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int J Pharm 172:33–70
- Varshosa J, Pardhakhty A, Mohsen S, et al. (2005). Sorbitan monopalmitate-based proniosomes for transdermal delivery of chlorpheniramine maleate. Drug Deliv 12:75–82
- Vora B, Khopade AJ, Jain NK. (1998). Proniosome based transdermal delivery of levonorgesterel for effective contraception. J Control Release 54:149–65
- Vyas SP, Khar RK. (2002). Targeted and controlled drug delivery: novel carrier systems. New Delhi, India: CBS Publications
- Vyas SP, Mysore N, Jaittley V, et al. (1998). Discoidal niosome based controlled ocular delivery of timolol maleate. Pharmazie 53:466–9
- Yoshioka T, Sternberg B, Florence AT. (1994). Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60, and 80) and a sorbitan triester (Span 85). Int J Pharm 105:1–6