
Abstract
Arginine-stabilized, docetaxel-loaded polymeric micelles (AR-DTX-PM) were prepared to enhance the physical stability of micelles and control the degradation of docetaxel (DTX). Amphiphilic diblock copolymers, methoxy-(Polyethylene Glycol)-block-Poly (D, L-lactide) (mPEG-PDLLA) were synthesized and used for the formulation of lyophilized DTX-PM powders. The micelles were found to have diameters of 20–30 nm with narrow polydispersity, and the entrapment efficiency was 90–100%. The accumulative release of AR-DTX-PM was higher than that of glucose-dispersed DTX-PM (Glu-DTX-PM). The results of both physical and chemical stability studies showed that the concentration of arginine required for optimum stability was 2.0 mg/ml. Preliminary investigation of the mechanisms of stabilization by arginine suggested that it is due to the electrostatic interaction as well as hydrogen bonds between DTX and arginine. The acute toxicity studies demonstrated that AR-DTX-PM was better tolerated in beagle dogs than DTX injection. However, the pharmacokinetic studies revealed no significant difference in Cmax and AUC of AR-DTX-PM compared to DTX injection. When AR-DTX-PM was administrated at a dose of 30 mg/kg, the antitumor effect was stronger than that of commercial DTX injection at 10 mg/kg, and the increase of administration dose did not cause higher toxicity. The in vivo imaging test showed that the residence time of AR-DTX-PM at tumor sites was longer than its commercial formulation. In a word, it is expected that AR-DTX-PM can reduce systemic toxicity while retaining antitumor efficacy in cancer patients.
Introduction
Docetaxel (DTX), a semi-synthetic analogue of paclitaxel, is a second-generation taxane derived from the needles of the European yew tree, Taxus baccata. It is known to be more active than paclitaxel and exhibits 1.9 times greater affinity for microtubules (Gaucher et al., Citation2010). It also has been shown to exhibit significant activity against a large spectrum of solid tumors. However, it has an extremely poor aqueous solubility (6–7 μg/ml). Therefore, it requires the use of nonionic surfactants for its commercial formulation. However, nonionic surfactants such as Tween80 are known to exhibit serious side effects such as hypersensitivity reactions, fluid retention and febrile neutropenia (Baker et al., Citation2009). Recently, great efforts have been devoted to develop alternative Tween80-free systems for DTX. The systems that have been reported include liposomes (Immordino et al., Citation2003), micelles (Tong et al., Citation2012), nanoparticles (Dong & Feng, Citation2004), microemulsion (Yin et al., Citation2009) and drug-polymer conjugates (Zhang et al., Citation2005). Among these formulations, polymeric micelles seem to be the most ideal carrier for the delivery of poorly water-soluble drugs.
Polymeric micelles comprise a typical core-shell structure with a drug-loading hydrophobic core and flexible hydrophilic shell, and they are small in size, which are preferable to bypass both renal clearance and entrapment by the mononuclear phagocyte system (MPS), resulting in high accumulation in hypervascular tumor tissues by the enhanced permeability and retention effect (EPR) (Miyata et al., Citation2011). Polymeric micelles are usually formed through the self-assembly of linear amphiphilic block copolymers, among which polyether-polyester diblock copolymers are the most widely used for micelle preparation. Polyether-polyester block copolymers consist of hydrophobic (polyester) and hydrophilic (polyether) segments. Poly (D, L-lactide) (PDLLA) (Zareie et al., Citation1996), poly (lactide-glycolide) (PLGA) (Yoo & Park, Citation2001), poly (β-benzyl-L-aspartate) (PBLA) (La et al., 1996) and poly (epsilon-carprolactone) (PCL) (Shenoy & Amiji, Citation2005) are often chosen as the hydrophobic segments for their biodegradability and biocompatibility, while poly (ethylene glycol) (PEG) (Kataoka et al., Citation2000), poly (N-vinyl pyrrolidone) (PVP) (Benahmed et al., Citation2001), poly (N-isopropyl acrylamide) (PNIPAM) (Hales et al., Citation2004) and poly (hydroxypropyl methacrylamide) (PHPMA) (Krimmer et al., Citation2011) have been reported to be the shell-forming block, among which PEG is the most frequently used hydrophilic block because of its non-toxicity and low immunogenicity (Miyata et al., Citation2011).
In this study, mPEG-PDLLA was used to prepare the DTX-PM. However, according to our previous study, mPEG-PDLLA–based DTX-PM are not stable enough to entrap the drug in the micelle core, the dynamic stabilizing time of DTX-PM was much poorer than that of paclitaxel polymeric micelles (PTX-PM). Therefore, there is a need to explore a suitable technology to produce more stable DTX-PM for clinical use. Interestingly, we have recently found that arginine could enhance the dynamic stability of DTX-PM to a large extent; however, DTX degraded dramatically when exposed to arginine because of the strong alkaline conditions. It also has been reported that DTX degrades easily under acidic or basic conditions (Kumar et al., Citation2007). Therefore, the micelle-stabilizing mechanism of arginine was investigated while maintaining a balance between the physical stability of micelles and the chemical degradation of DTX in the presence of arginine.
Materials and methods
Materials
mPEG2000 and Stannous octoate were purchased from Sigma-Aldrich (Sanfrancisco, USA) and DL-lactide was obtained from Daigang biotechnology Co., Ltd (Shandong, China). Polystyrene monodisperse standards were obtained from Polymer Laboratories (London, England). Docetaxel (purity>99.9%) was purchased from Taihao Pharmaceutical Co., Ltd (Chongqing, China), and DTX injection was purchased from Taiji Pharmaceutical Co., Ltd (20 mg: 0.5 ml, Chongqing, China. DTX vehicle: 11.9% alcohol). DiR was obtained from Fanbo Biochemicals Co., Ltd (Beijing, China), and arginine was purchased from Tian’an Pharmaceuticals Co., Ltd (Tianjin, China). Acetonitrile of HPLC grade was from Tedia (USA). Cancer cell line MCF-7 was purchased from American Type Culture Collection while A549 and H460 were from Basic Medical Cell Center of Peking Union Medical College (Beijing, China). Cell culture media, Roswell Park Memorial Institute-1640 (RPMI-1640), Minimal Essential Medium (MEM) and fetal bovine serum (FBS) were purchased from GIBCO (New York, USA). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories (Tokyo, Japan). Lewis tumor-bearing mice were purchased from KeyGen Biotechnology Co., Ltd (Nanjing, China). All other reagents were of analytical grade.
Synthesis of mPEG-PDLLA
Diblock copolymer of mPEG-PDLLA was synthesized from monomers of methoxy polyethylene glycol 2000 (mPEG2000) and DL-lactide by ring-opening polymerization in the presence of stannous octoate using a monomer ratio of 1:1 as reported previously (Zhang et al., Citation1996; Liggins & Burt, Citation2002).
Characterization of the polymer
Infrared spectroscopy was used to confirm the structure of mPEG-PDLLA. To prepare KBr tablets, the polymer sample was grinded with KBr and then compressed into a transparent tablet. The structure, number average molecular weight and the mPEG content of the copolymer were determined by 1H-NMR in CDCl3 at 300 Hz with TMS as internal reference at 25 °C. The molecular weight distribution was determined by gel permeation chromatography (GPC). The molecular weight was characterized by relative retention time to polystyrene monodisperse standards (molecular weight: 1930, 3070, 5120, 6930, 10680, 12830). Shodex column (KF-800, 4.6 × 300 mm) and Shima RID-10A differential refractometric detector were used. The column temperature was maintained at 40 °C. The mobile phase was THF with a flow rate of 1 ml/min. The injection volume was 20 μl.
Preparation of lyophilized DTX-PM powders
DTX-PM was prepared by rotary evaporation method. Briefly, 100 mg DTX and 500 mg mPEG-PDLLA were dissolved in 15 ml methanol. The organic solvents were removed by rotary evaporation under high vacuum at 30 °C to form a thin film of drug/polymer mixture. Drug-loaded micelles were formed by resuspending the film in 10 ml water at 30 °C for five minutes. The resulting solution was filtered through 0.22 μm membrane filter and then dispensed into five vials (20 mg DTX per vial). Finally, the solution was lyophilized to form white loose acicular powders and stored at 4 °C until use.
Characterization of micelles
Particle size and morphology
Particle mean size and size distribution were determined by dynamic light scattering (DLS) using a ZetaPlus Particle sizer (Brookhaven Instrument Corporation, USA) at a wavelength of 657 nm and 25 °C. The freeze-dried powder was dispersed and diluted to the appropriate concentration with deionized water/0.9% NaCl/5% glucose/arginine to avoid multi-scattering phenomena and was placed into a quartz cuvette. The intensity of the scattered light was detected at a scattering angle of 90 °. The reported experimental results were expressed as mean ± SD of three independent experiments.
The morphology of micelles was studied with Atomic Force Microscope (AFM, diNanoScope, Veeco Instruments Inc.). A drop of micelle solution with the polymer concentration of 10 μg/ml was placed on a clean cover-glass and air-dried before measurement.
Determination of drug encapsulation efficiency
The analysis of DTX was determined by RP-HPLC method on a system equipped with a LC-20AT pump, a SIL-20A autosampler and a SPD-20A UV detector (Shimadzu, Kyoto, Japan) operated at 230 nm. A reversed-phase column (Symmetry 3.5 μm C18, 4.6 mm × 150 mm, Waters) was equilibrated with a mobile phase of acetonitrile:water 55:45 (v/v) flowing at a rate of 1.0 ml/min at 35 °C. Sample solution was injected at a volume of 20 μl, the DTX retention time was around 5 min. The concentrations of DTX were determined by comparing the peak areas with the standard curve. The dispersed micelles were centrifuged at 10 000 rpm at room temperature for 5 min to remove the unloaded DTX. The drug encapsulation efficiency (E.E %) and drug-loading content (D.L %) of DTX-PM were calculated using the following formula:
In vitro drug release profile
The lyophilized DTX-PM powders containing 20 mg DTX were dispersed with arginine (2.0 mg/ml) and 5% glucose separately to give DTX concentration of 10.0 mg/ml, and then diluted 10 times with the release medium (PBS 7.4 containing 0.5% Tween80 and 5% bovine serum) to give DTX concentration of 1 mg/ml. Two hundred microliter of sample (DTX concentration 1 mg/ml) was separately introduced into the dialysis bags and the end-sealed dialysis bags (molecular weight cutoff 3.5 kD, Nanjing ZhuYan Biotechnology, Co. Ltd) were immersed into 9 ml release medium. The system was shaked at 75 rpm for 48 h at 37 °C. At scheduled time intervals, 1 ml aliquots were withdrawn and replaced with the same volume of fresh medium. The sink condition was maintained by the addition of Tween80 and frequent replacement of fresh buffer. The experiments were carried out in triplicates. The samples were added into 2.0 ml acetonitrile, vortexed for 5 min, centrifuged for 10 min at 10 000 rpm, and then the supernatant was filtered through 0.22 μm organic filter and the amount of DTX in the filtrate was determined by HPLC as described in section “Determination of drug encapsulation efficiency”.
Micelle stability
The lyophilized DTX-PM powder was evaluated for their physical stability in arginine solutions of different concentrations at 25 °C with 5% glucose as the control. The lyophilized DTX-PM powder was first dispersed in arginine solution to give DTX concentration of 10 mg/ml and then the solution was diluted 10 times with 5% glucose to give DTX concentration of 1 mg/ml. Time-dependent changes in diameters were monitored by DLS.
The chemical stability was evaluated by measuring the degradation of DTX using HPLC method as described in section “Determination of drug encapsulation efficiency”. The lyophilized DTX-PM powders were first dispersed in arginine solution to give DTX concentration of 10 mg/ml, and then the solution was diluted 10 times with either 0.9% NaCl or 5% glucose. Time-dependent changes in the related substance were studied.
Stabilizing mechanism of arginine in DTX-PM
The effect of arginine on CMC of the polymer
Fluorescence technique based on the selective partition of pyrene in hydrophobic phase against aqueous phase was used to determine the critical micelle concentration (CMC) of mPEG-PDLLA as described in previous studies (Asadi et al., Citation2011). Emission wavelength was set at 390 nm and excitation wavelength ranged from 300 to 360 nm. To study the effect of arginine on CMC of the polymer, the lyophilized blank micelle powder was dispersed in 2 ml arginine solution at concentrations of 2, 4 and 8 mg/ml, and diluted subsequently with deionized water.
Active group for the stabilizing mechanism
The effect of arginine on the physical stability of blank micelles was further investigated. Paclitaxel-loaded polymeric micelles (PTX-PM) and dexoypodophyllotoxin-loaded polymeric micelles (DPT-PM) were investigated to see if there is a specific interaction between arginine and the drug. Lysine, histidine, ammonia, sodium hydroxide, glycocyamine and the mixed solution of glycocyamine and sodium hydroxide/ammonia/ethylenediamine (pH 10.34, pH similar to 5 mg/ml arginine solution) were used to substitute arginine and find the active group which plays a critical role in the physical stability of micelles. Five percent glucose was used as control.
Acute toxicity in beagle dogs
The in vivo acute toxicity studies were conducted in healthy beagle dogs (male/female, 6–8 months old, 6–8 kg), which were purchased from Mars Biological Technology Co., Ltd (Beijing, China). The lyophilized DTX-PM powder was prepared according to the method described in section “Characterization of micelles”. The DTX-PM powder was first dispersed with arginine (2 mg/ml) to give DTX concentration of 10 mg/ml, and then the solution was diluted with 5% glucose to give DTX concentration of 0.15, 0.30 and 0.60 mg/ml, respectively. The DTX injection was prepared according to the operating manual, and diluted to 0.30 mg/ml with 5% glucose. For the control group, 100 mg lyophilized blank micelles were dispersed in 2 ml arginine and diluted with 5% glucose to obtain 3 mg/ml micelle solution. The administration capacity was 5 ml/kg. All the solutions were infused intravenously using an electric pump at a speed of 0.15 ml/kg/min. The beagle dogs were randomly divided into five groups (n = 4, male/female), and these groups were separately infused with blank micelle solution (control), 1.5 mg/kg DTX injection, 0.75 mg/kg DTX-PM, 1.5 mg/kg DTX-PM and 3 mg/kg DTX-PM single dose on the first day (Day 0). The reactions of beagle dogs were assessed during the first 14 days after administration, and the abnormal clinical signs were recorded in details. The body weight of beagle dogs were acquired on Day 0, Day 3, Day 7, Day 10 and Day 14, and body weight variation was calculated. On Day 0, 1, 4, 7 and 14, blood samples were collected from the forelimb/hindlimb subcutaneous veins and examined using the Bayer Advia2120 automated hematology analyzer.
Pharmacokinetics in beagle dogs
Beagle dogs (male/female) weighing 5–8 kg at about 9–10months old were purchased from Mars Biological Technology Co., Ltd (Beijing, China). Twelve beagle dogs were divided into two groups, one group was intravenously infused with 1.5 mg/kg DTX injection single dose, and the other group was intravenously infused with 1.5 mg/kg DTX-PM single dose, and the dispersing method was the same as that described in section 2.5.4. The infusion speed was 0.15 ml/kg/min and the infusion capacity was 5 ml/kg. Blood samples were collected from the forelimb/hindlimb subcutaneous veins at 0 (before administration), 0.55, 0.75, 1.05, 1.55, 2.55, 4.55, 6.55, 8.55 and 24.55 h. At each time point, 1 ml of blood was collected in a heparinized centrifuge tube, centrifuged at 4500 rpm for 5 min at 4 °C, and stored at −70 °C until further analysis by LC/MS/MS. One hundred microliter of plasma was added into a test tube and mixed with 10 µl of the internal standard (2 µg/ml paclitaxel solution). Subsequently, 0.5 ml tert-butyl ether was added and the mixture was vortexed for 2 min. Then, the mixture was centrifuged at 4000 rpm for 10 min. The organic phase was withdrawn and dried under a nitrogen stream at 40 °C. The dried residue was dissolved with the mixed solvent of methanol and water (1:1, v/v). Ten microliter of this sample was injected into the LC/MS/MS system. The separation was performed on a Ultimate XB-C18 column (2.1 mm × 50 mm, 3 μm) using a mobile phase consisting of 0.5% acetic acid in distilled water and 0.5% acetic acid in methanol at a flow rate of 0.5 ml/min. Mass spectra were recorded with positive electrospray ionization (ESI+) and analysis was performed using multiple reaction monitoring (MRM) with a specific transition at 808.4 → 226.1 for docetaxel and 854.6 → 286.2 for the internal standard.
Pharmacokinetic parameters were obtained using the WinNonlin (V6.2). Statistical analysis of the samples was performed using an independent sample t-test, and p values <0.05 were considered statistically significant. All data are reported as the means ± SD.
In vivo antitumor efficacy
The antitumor efficacy studies were conducted in female mice bearing S180 cells. The mice were subcutaneously injected at the right axillary space. Treatments were started when the tumor volume reached 80–130 mm3 and this day was designated as “Day 0”. The mice were randomly assigned to six treatment groups with seven mice in each group. Each group of mice was treated every three days with: (a) 5% glucose injection, (b) DTX injection (10 mg/kg, dispersed in 13% ethanol), (c) AR-DTX-PM (10 mg/kg, dispersed in 2.0 mg/ml arginine), (d) AR-DTX-PM (30 mg/kg, dispersed in 2.0 mg/ml arginine), (e) Glu-DTX-PM (10 mg/kg, dispersed in 5% glucose), (f) Glu-DTX-PM (30 mg/kg, dispersed in 5% glucose).
Tumors were measured every other day with calipers during the period of study. The tumor volume was calculated by the formula: V = (W2 × L)/2, where W is the tumor measurement at the widest point and L is the tumor dimension at the longest point. The relative tumor volume could be defined as follows: RTV = Vt/V0, where Vt is the tumor volume on Day t post administration, and V0 is the tumor volume on Day 0. In addition, the body weights and death rate of mice were recorded every other day.
In vivo and ex vivo imaging
To visualize the real-time biodistribution of DTX-PM in S180 tumor-bearing mice (the tumor volume at the time of administration was 100–200 mm3), a hydrophobic near infrared (NIR) fluorescent dye DiR (excitation at 745 nm and emission at 800 nm) was loaded into the commercial injection and into the micelles. DiR injection was prepared by rotary evaporation method. Briefly, 1 mg DiR and 2.5 ml Tween80 were dissolved in ethanol. Then, the organic solvents were removed by rotary evaporation under high vacuum at 30 °C, the resulting solution was dispensed into five vials (0.2 mg DiR per vial). Finally, the solution was lyophilized to form the white loose acicular powders. The lyophilized powders were stored in a refrigerator until use. The preparation of DiR-PM was the same as that of DTX-PM except that 1 mg DiR was dissolved into methanol instead of 100 mg DTX before rotary evaporation. Before lyophilization, the micelles were centrifuged at 10 000 rpm at room temperature for 5 min to remove the free DiR (Cho et al., Citation2012).
The DiR injection was first dispersed with 13% ethanol and then diluted to the appropriate concentration with 5% glucose before administration. DiR-PM powders were first dispersed with arginine (2 mg/ml, AR-DiR-PM) and then diluted to the appropriate concentration with 5% glucose before injection. S180 tumor-bearing mice were injected via tail vein with 400 μl of DiR injection and AR-DiR-PM (the concentration of DiR was 0.01 mg/ml). Whole body fluorescent images were recorded at 1, 2, 4 and 8 hours after administration. Fluorescent images were captured using Maestro in vivo imaging system (CRI, Inc., Woburn).
Results and discussion
Characterization of mPEG-PDLLA copolymer
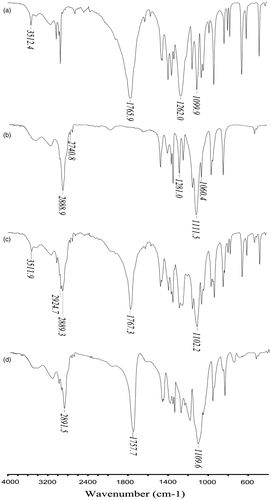
shows the IR spectrum of the monomer DL-lactide (a) and mPEG2000 (b), the physical mixture of the monomers (c) and the diblock copolymer mPEG-PDLLA (d). The characteristic absorption band of DL-lactide () was at 1765.9 cm–1 (stretching band of C=O), while the signals at 1262.0 cm–1 and 1099.0 cm–1 belonged to the stretching band of C–O–C. The sharp absorption peak at about 3510 cm–1 was due to the hydroxyl group of lactic acid, a degradation product of lactide. In the spectra of mPEG2000 (), absorption peaks were at about 2888 cm–1 and 2740 cm–1 (antisymmetric and symmetric stretching band of C-H), 1281 cm–1 and 1060 cm–1 (antisymmetric and symmetric stretching band of C-O-C). The copolymer mPEG-PDLLA () showed a sharp and intense absorption band at 1760 cm–1 due to DL-lactide, a broader absorption band at 2900 cm–1 due to the overlapping C–H stretching band of both the monomers (Kim et al., Citation1998). The absorption peak of the physical mixture () at about 3510 cm–1 (stretching band of –OH group consistent with DL-lactide) was absent from the spectrum of mPEG-PDLLA. The above results confirmed the successful synthesis of the copolymer mPEG-PDLLA.
Figure 1. IR spectrum of D, L-lactide (a), MPEG2000 (b), the physical mixture of lactide and MPEG2000 (c) and the diblock copolymer MPEG-PDLLA (d).
The synthesized polymer was also characterized by 1H NMR in CDCl3 (, ). The peaks at 3.40 ppm and 3.65 ppm corresponded to the methyl and methylene protons of mPEG segment respectively, while the methyl and methylene protons of PDLLA segment were at 1.50 ppm and 5.20 ppm, respectively. The methylene protons next to the PDLLA segment and the methyne protons at the end of the PDLLA segment coincide at 4.35 ppm. These results demonstrate the successful synthesis of mPEG-PDLLA copolymer (Kim et al., Citation1998).
Figure 2. 1H NMR spectrum of mPEG-PDLLA (50:50).
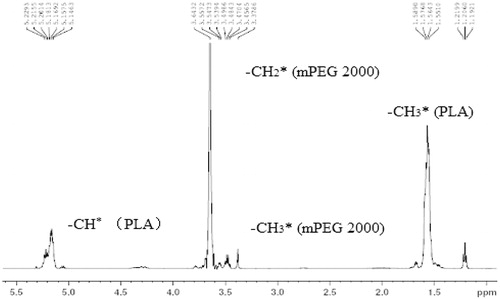
Table 1. Molecular weight and molecular weight distribution of mPEG-PDLLA.
From the ratio of the peak areas at 5.20 and 3.65 ppm, the number average molecular weight of the synthesized copolymers were determined and the weight content of mPEG could be calculated. shows the molecular weight distribution calculated by GPC. The number average molecular weight Mn obtained from 1H-NMR was very close to the theoretical number 4000, the mPEG content was around 50%, and the Mw/Mn determined by GPC (, ) was less than 1.5, indicating the monodispersity of the polymer.
Figure 3. Gel permeation chromatograms of mPEG-PDLLA.
Particle size and morphology
The mean diameters and polydispersity indexes (PDI) of the water-dispersed and arginine-dispersed DTX-PM were measured (), and the results suggest that the micellar size was narrowly distributed and there was no significant difference in both diameter and PDI between the two groups. shows two distributions of the micelles, one was 20–30 nm and the other was 50–60 nm.
Figure 4. Size distribution of arginine-dispersed DTX-PM measured by DLS (a) and Atom Force Microscopy (AFM) of the DTX-PM (b).
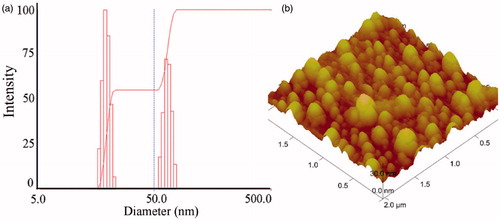
Table 2. Characteristics of dispersed DTX-PM in deionized water or arginine (2.0 mg/ml).
Furthermore, the atom force microscopy (AFM) of the micelles () showed that the micelle particles were spherical in shape with a size ranging from 30 to 60 nm, which was consistent with the measurements made by the DLS analyzer.
Drug encapsulation efficiency
shows that the drug encapsulation efficiency of the redissolved micelles was more than 90% while the drug-loading content was about 16.00%, and there was no significant difference between the two groups, indicating that arginine has no influence on the encapsulation of the drug.
In vitro drug release profile
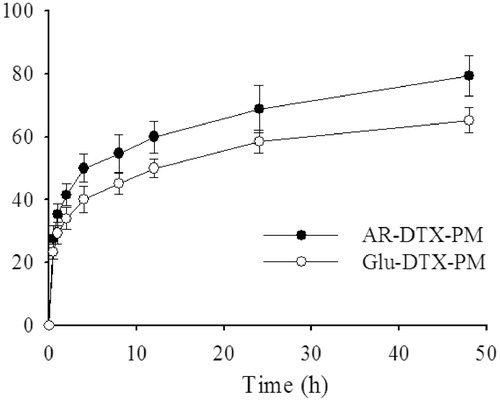
shows that DTX was released quickly from the micelles within the first 6 hours; subsequently, the release was slow and constant. In addition, AR-DTX-PM had a higher cumulative release than Glu-DTX-PM, which could be attributed to the increased dynamic stability of drug-loaded micelles by arginine. As is reported (Liu et al., Citation2011), the release of DTX from commercial DTX injection Duopafei® was fast and approximately 100% of drug was released after 24 h incubation, indicating that DTX released much slower from DTX-PM than from Duopafei®. Therefore, DTX-PM might reduce the frequency of administration, which is good for the clinical application.
Figure 5. In vitro release profile of DTX from AR-DTX-PM and Glu-DTX-PM in PBS7.4 (0.5% of Tween80 and 5% bovine serum) at 37 °C (n = 3).
Micelle stability
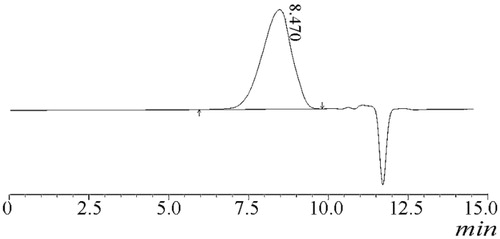
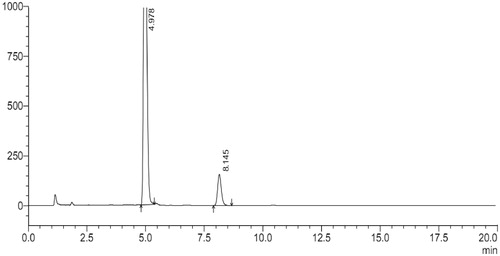
shows that the diameter of Glu-DTX-PM increased significantly within 30 min and the diameter stability time prolonged with the increased arginine concentration. When the arginine concentration is above 2 mg/ml, micelles in 5% glucose were physically stable within the first two hours which is preferable for the intravenous injection. However, DTX seriously degraded in arginine solution where the pH condition was strongly alkaline. Peak area normalization method was used to study the degradation of DTX in arginine. As shown in , the main peak at 4.9 min is for DTX, whereas the peak at 8.1 min belonging to 7-epi DTX was formed by the epimerization of seven hydroxyl groups (Vasu Dev et al., Citation2006; Kumar et al., Citation2007), which has the highest percentage of the degraded substances. Consequently, 7-epi DTX was considered as the main impurity to be controlled. Therefore, it was important to monitor the single impurity content (represented by the area percent of peak 8.1 min) and total impurity content (represented by the total area percent of peaks except solvent and DTX) before injection.
Figure 6. The change of diameter of arginine-dispersed DTX-PM and 5% glucose-dispersed DTX-PM diluted with 5% glucose.
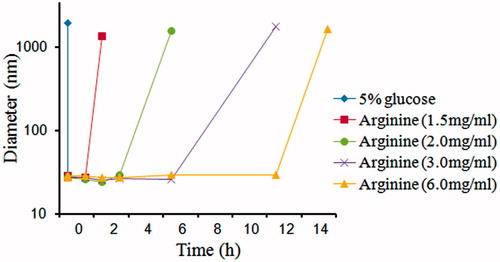
Figure 7. HPLC chromatogram of alkaline degradation of DTX.
The pH of each sample solution was tested (), the values indicated that the degradation of DTX was highly related to the alkalinity of the dispersion and the micelle dispersion diluted with 5% glucose had a lower pH, so 5% glucose solution is the preferred isotonic solvent instead of 0.9% NaCl. shows that both the single and total impurities increased with the arginine concentration. When the arginine concentration was below 3 mg/ml, total impurity content could be controlled less than 5%.
Table 3. PH value of dispersed DTX-PM in arginine of different concentrations and diluted with either 5% glucose or 0.9% NaCl*.
Table 4. Single impurity content (S) and total impurity content (T) of 5% glucose dilution of DTX-PM dispersed in arginine (AR) with a series concentration of 1.5, 3.0, 6.0 mg/ml, respectively.*.
Both physical and chemical stability contemplated, 2 mg/ml was considered to be the best concentration of arginine, and 5% glucose solution was chosen as the isotonic solvent. Lyophilized DTX-PM powder was dispersed with arginine solution (2 mg/ml) to make the drug concentration 10 mg/ml and then diluted with 5% glucose to give DTX concentration of 1 mg/ml for the intravenous injection in clinic.
Stabilizing mechanism
The effect of arginine on CMC of the polymer
and and show that arginine had no effect on CMC of the polymer, which indicated that the interaction of arginine was not with the polymer, and it could be with DTX in the hydrophobic micelle core.
Figure 8. Pyrene excitation spectra of mPEG-PDLLA aqueous solutions (emission wavelength at 390 nm).
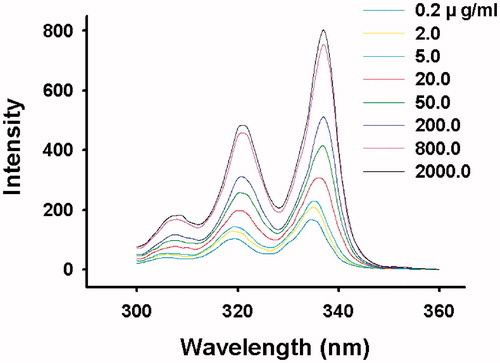
Figure 9. Intensity ratio of I337/I334 versus logC for mPEG-PDLLA (50/50) in (a) water, (b) 2 mgċml–1 arginine solution, (c) 4 mgċml–1 arginine solution and (d) 8 mgċml–1 arginine solution.
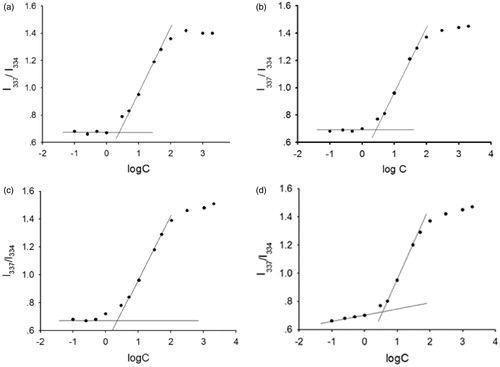
Table 5. CMC value of mPEG-PDLLA in arginine solution of different concentration (n = 5, ).
).
Active group for stabilizing mechanism
Interestingly, it was found that arginine could also enhance the physical stability of PTX-PM, but had no effect on blank micelles or DPT-PM. The arginine-substituted substances including lysine, histidine, ammonia, sodium hydroxide and glycocyamine did not give any promising result. But the physical stabilizing time of DTX-PM powders dispersed with the mixed solution of glycocyamine and sodium hydroxide was increased to a minor extent, and as shown in , the mixed solution of glycocyamine and ammonia or ethylenediamine had a similar stabilizing effect as arginine.
Table 6. The stabilizing time of DTX-PM dispersed with different solutions.
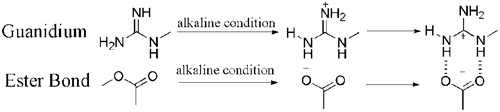
It is noteworthy that the distinct guanidinium group of arginine (pKa = 12.48) is always positively charged even in basic solutions and the typical ester bonds of the taxanes could be hydrolyzed into the negatively charged carboxylate anion under alkaline conditions. Guanidinium salts have been popular as a carboxylate recognition motif, providing the hydrogen bond donor to interact with the carboxylate oxygens, and in the case of a guanidinium cation, providing a complementary electrostatic interaction with the carboxylate anion (Schmuck & Graupner, Citation2005; Cho et al., Citation2012). And it is supposed that the positively charged guanidinium cation interacted with the negatively charged carboxylate anion to form large amounts of hydrogen bonds (), thus strengthening the intermolecular forces of micelle inner core and preventing the micelle structure from disassembling and, thereby, enhancing the stability of the micelle solution. Besides, as shown in , those containing both guanidinium and amino group had a better stabilizing effect than that containing guanidinium only, which indicates that the combined action of guanidinium and amino group played an important role in the micelle stability. Hereby, it is inferred that the electrostatic interaction of guanidinium cation and carboxylate anion drew the two molecules (arginine and DTX/paclitaxel) closer, which facilitated the hydrogen bond formation between arginine –NH– and the taxane oxygen atom, and the amino group of arginine acting as the hydrogen bond donor increased the number of hydrogen bond to a large extent (), making the intermolecular forces of the micelle inner core stronger. In conclusion, both the guanidinium and amino group of arginine were of great importance to the micelle stability, and the strong alkaline condition of arginine facilitated the hydrolysis of the ester bond of the taxanes into carboxylate anion. The stabilizing mechanism was related closely to the electrostatic interaction between the positively charged guanidinium cation and the negatively charged carboxylate anion as well as the hydrogen bond formation between guanidinium/amino –NH– and carboxylate oxygen.
Figure 10. The electrostatic interaction and hydrogen bond formation between arginine and the taxanes.
Acute toxicity
Clinical Observation
No abnormal clinical signs were noted in the control group and 0.75 mg/kg DTX-PM group during the whole observation period. For the DTX injection group, clinical signs like eyelid swelling/redness, conjunctival hyperemia, salivation/hypersalivation, hematochezia, swelling (face/ears/perioral), flushing (perioral/ears), itching were observed within the 30 min post infusion, and loose/watery stools, soft feces were noted during the next 13 days. Only soft feces were found on Day 5 for the 1.5 mg/kg DTX-PM group. Hematochezia, vomiting, loose/watery stools and soft feces were noted for the 3 mg/kg DTX-PM group from Day 3 to Day 9. In general, no toxic signs related to the polymer vehicle of DTX-PM were noted, and DTX-PM was much more tolerated than DTX injection.
Body weight variation
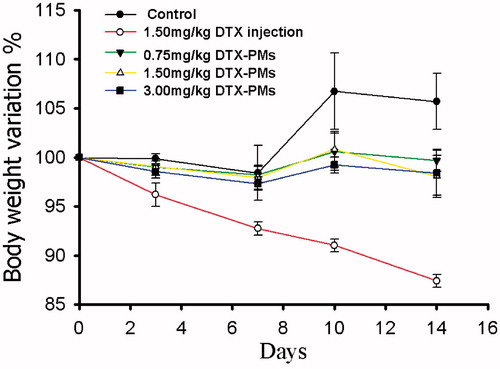
The in vivo toxicity of DTX-PM and DTX injection was evaluated in healthy beagle dogs by monitoring the body weight change (). The control group showed a small increase in body weight while there is almost no change in the DTX-PM groups. Weight loss was found in the group treated with DTX injection, and the maximum weight loss was up to 13%. It demonstrated that DTX-PM formulation was better tolerated in beagle dogs than DTX injection.
Figure 11. Variation of body weight in beagle dogs time curves. The results are shown as the means ± SD (n = 4).
Blood tests
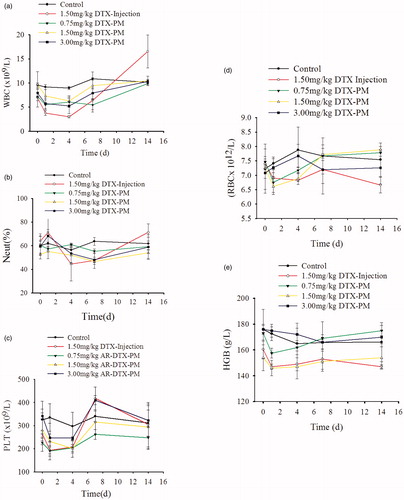
The myelosuppression effects were evaluated by routine peripheral blood examination (). shows that for the control group, 3 mg/kg, 1.50 mg/kg and 0.75 mg/kg DTX-PM group white blood cell (WBC) counts fluctuated within the normal range throughout the observation period. For the DTX injection group, there was an obvious downward trend in the first four days post injection, after that, a significant rebound appeared and the WBC count was above the normal level. shows that there was no obvious fluctuation for neutrophil (Neut)% in the control group, 3 mg/kg, 1.50 mg/kg and 0.75 mg/kg DTX-PM group. However, for the DTX injection group, Neut% declined drastically during the 1 to 4 days post injection, and it went back to normal after that. shows that platelet (PLT) counts for the control group fluctuated within the normal level, but for the DTX injection and 3 mg/kg, 1.50 mg/kg, 0.75 mg/kg DTX-PM group, the PLT counts decreased significantly within the first day post administration, and remained unchanged during the 2 to 4 days post administration, and then it rebounded significantly during the 4 to 7 days, after that, it went back to the normal gradually. The decline and rebound margin of the DTX injection group and the 3 mg/kg DTX-PM group were the biggest, 1.50 mg/kg DTX-PM group secondly and 0.75 mg/kg DTX-PM thirdly. The RBC (red blood cell) and HGB (hemoglobin) counts of all groups were within the normal level (, ). In general, the bone marrow suppression effect for the DTX-PM group was reduced significantly compared to the DTX injection group.
Figure 12. The variation of white blood cells (WBC, a), neutrophil % (Neut, b), platelets (PLT, c), red blood cells (RBC, d) and hemoglobin (HEG, e) during the observation (n = 4).
Pharmacokinetics in beagle dogs
The DTX plasma concentration-time curves for the two formulations are shown in . The pharmacokinetic parameters were calculated by noncompartmental model and the results were listed in . Both DTX-PM and DTX injection displayed the similar distribution trends at the same dose of 1.5 mg/kg. The parameters t1/2, tmax, MRT showed no significant difference between the two formulations. However, the plasma Cmax, AUC(0–24h) of DTX-PM were 0.5 times lower than those of DTX injection, and Vd, Cl of DTX-PM were both two times higher than those of DTX injection. In fact, arginine in micelles could increase the positive charge of micelles, which would facilitate nonspecific attachment of nanoparticles to negatively charged plasma membranes via electrostatic interactions (He et al., Citation2010). Therefore, the interaction with blood components induce rapid uptake by the reticuloendothelial system and rapid elimination from the blood circulation.
Figure 13. The plasma concentration time curves of DTX from DTX-PM and DTX injection. The results are shown as the means ± SD (n = 6).
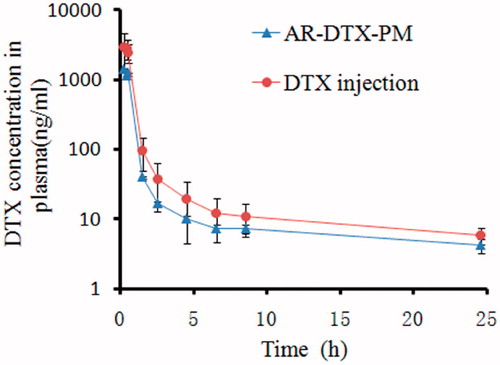
Table 7. The pharmacokinetic parameters of DTX for DTX injection and DTX-PM.
In vivo antitumor efficacy
From the relative tumor volume-time curve in , the following conclusions can be drawn: (1) Mice in all the groups that were administered DTX (DTX injection or DTX-PM) had smaller tumors than the mice in the control group. (2) The AR-DTX-PM was found to be more efficient at inhibiting tumor growth than Glu-DTX-PM at the same dose, and this could be attributed to the increased dynamic stability of DTX-PM by arginine. (3) When the administration dose was increased to 30 mg/kg, the antitumor efficacy of AR-DTX-PM was found to be stronger than DTX injection at the dose of 10 mg/kg. (4) The DTX-PM showed increasing efficacy with increasing dosages of DTX.
Figure 14. The antitumor efficacy against mice bearing S180. (a) The relative tumor volume-time curve; (b) the survival rate change; (b) the body weight change. The results are shown as means ± SD (n = 7).
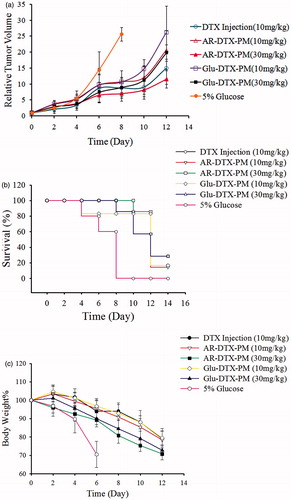
As shown in , the death rate of 5% glucose group was 60%, while that of Glu-DTX-PM is 80%, and there was no death in other groups. On Day10, all the mice in the group administered 5% glucose died, and there was no death in AR-DTX-PM (30 mg/kg) group, while the death rate of other groups was all 80%. On Day12, the death rate of AR-DTX-PM (10 mg/kg) and Glu-DTX-PM (30 mg/kg) groups declined to 60%, while those of DTX injection, AR-DTX-PM (30 mg/kg) and Glu-DTX-PM (10 mg/kg) remained 80%. On Day14, more than half of the mice died in all groups. It is concluded that the death rate changes of AR-DTX-PM (30 mg/kg) group and DTX injection (10 mg/kg) group were similar, that is, the high dose of AR-DTX-PM did not kill the tumor-bearing mice in advance, indicating that the AR-DTX-PM was much better tolerated compared to DTX injection.
depicts the variation of the body weight change of the mice, and it is used as an index of systemic toxicity. The results showed that both DTX injection and DTX-PM group could suppress the body weight loss of mice, and it is dependent with the administration dose. And the percentage of the weight loss could be controlled within 30% in all groups.
In conclusion, when AR-DTX-PM was administrated at a dose of 30 mg/kg, the antitumor effect was stronger than that of commercial DTX injection at 10 mg/kg, and the increase of administration dose did not cause higher toxicity.
In vivo imaging
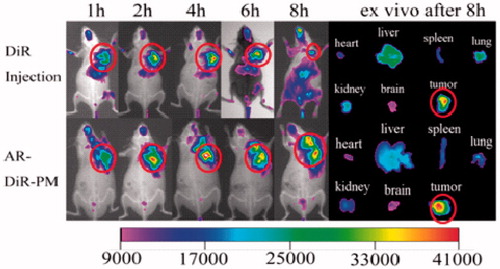
As shown in , it is concluded that DiR injection and AR-DiR-PM had a good targeting effect to tumor site, and AR-DiR-PM had a longer residence time in the tumor than DiR injection. 8 h after injection, DiR injection was gradually distributed to other organs, while AR-DiR-PM was still localized at the tumor site. This could be attributed to the enhanced permeability and retention (EPR) effect in tumors (Li et al., Citation2010).
Figure 15. In vivo infrared (NIR) optical imaging of DiR injection and AR-DTX-PM at different time point on S180 allograft tumor in SPF mice, and ex vivo images of dissected tissues after 8 h.
Conclusion
It has been demonstrated in this study that arginine had an outstanding stabilizing effect on DTX-PM in light of the micelle diameter, and the mechanism was investigated. The acute toxicity, pharmacokinetics, in vivo antitumor effect and distribution were evaluated focusing on the comparison with its commercial DTX injection. The results showed that, DTX-PM was better tolerated than DTX injection, and AR-DTX-PM exhibited stronger antitumor efficacy at the dose of 30 mg/kg than DTX injection at 10 mg/kg. Pharmacokinetic study indicated that the cellular uptake rate of AR-DTX-PM was faster than DTX injection. The in vivo imaging showed that AR-DiR-PM had a longer residence time at tumor site than DiR injection. Therefore, it is expected that AR-DTX-PM can reduce systemic toxicity while retaining antitumor efficacy when the administration dose was increased.
Acknowledgements
We acknowledged Vitus Onjia and Ammar Quahab for their kind help in revising English of this article.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work was financially supported by the major science and technology project, Key New Drug Development (2011ZX09501-001-06), the National Natural Science Foundation of China Project (Grant no. 81072588, 81201182) and the Fundamental Research Funds for the Central Universities (No. JKPZ2013006).
Related Research Data
References
- Asadi H, Rostamizadeh K, Salari D, et al. (2011). Preparation and characterization of tri-block poly(lactide)–poly(ethylene glycol)–poly(lactide) nanogels for controlled release of naltrexone. Int J Pharm 416:356–64
- Baker J, Ajani J, Scotté F, et al. (2009). Docetaxel-related side effects and their management. Eur J Oncol Nurs 13:49–59
- Benahmed A, Ranger M, Leroux J. (2001). Novel polymeric micelles based on the amphiphilic diblock copolymer poly(N-vinyl-2-pyrrolidone)-block-poly(D,L-lactide). Pharm Res 18:323–8
- Cho H, Indig GL, Weichert J, et al. (2012). In vivo cancer imaging by poly(ethylene glycol)-b-poly(ε-caprolactone) micelles containing a near-infrared probe. Nanomed Nanotechnol 8:228–36
- Dong Y, Feng SS. (2004). Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials 25:2843–9
- Gaucher G, Marchessault RH, Leroux JC. (2010). Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. J Control Release 143:2–12
- Hales M, Barner-Kowollik C, Davis TP, et al. (2004). Shell-cross-linked vesicles synthesized from block copolymers of poly(d,l-lactide) and poly(N-isopropyl acrylamide) as thermoresponsive nanocontainers. Langmuir 20:10809–17
- He C, Hu Y, Yin L, et al. (2010). Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31:3657–66
- Immordino ML, Brusa P, Arpicco S, et al. (2003). Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release 91:417–29
- Kataoka K, Matsumoto T, Yokoyama M, et al. (2000). Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-L-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J Control release 64:143–53
- Kim SY, Shin IG, Lee YM. (1998). Preparation and characterization of biodegradable nanospheres composed of methoxy poly(ethylene glycol) and dl-lactide block copolymer as novel drug carriers. J Control Release 56:197–208
- Krimmer SG, Pan H, Liu J, et al. (2011). Synthesis and characterization of poly(ɛ-caprolactone)-block-poly[N- (2-hydroxypropyl)methacrylamide] micelles for drug delivery. Macromol Biosci 11:1041–51
- Kumar D, Tomar RS, Deolia SK, et al. (2007). Isolation and characterization of degradation impurities in docetaxel drug substance and its formulation. J Pharmaceut Biomed 43:1228–35
- La SB, Okano T, Kataoka K. (1996). Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly(ethylene oxide)–poly(β-benzyl L-aspartate) block copolymer micelles. J Pharm Sci 85:85–90
- Li Y, Xiao K, Luo J, et al. (2010). A novel size-tunable nanocarrier system for targeted anticancer drug delivery. J Control Release 144:314–23
- Liggins RT, Burt HM. (2002). Polyether–polyester diblock copolymers for the preparation of paclitaxel loaded polymeric micelle formulations. Adv Drug Deliver Rev 54:191–202
- Liu D, Liu Z, Wang L, et al. (2011). Nanostructured lipid carriers as novel carrier for parenteral delivery of docetaxel. Colloids Surf B Biointerfaces 85:262–9
- Miyata K, Christie RJ, Kataoka K. (2011). Polymeric micelles for nano-scale drug delivery. React Funct Polym 71:227–34
- Schmuck C, Graupner S. (2005). Amino acid binding in water by a new guanidiniocarbonyl pyrrole dication: the effect of the experimental conditions on complex stability and stoichiometry. Tetrahedron Lett 46:1295–8
- Shenoy DB, Amiji MM. (2005). Poly(ethylene oxide)-modified poly(epsilon-caprolactone) nanoparticles for targeted delivery of tamoxifen in breast cancer. Int J Pharm 293:261–70
- Tong SW, Xiang B, Dong DW, et al. (2012). Enhanced antitumor efficacy and decreased toxicity by self-associated docetaxel in phospholipid-based micelles. Int J Pharm 434:413–19
- Vasu Dev R, Moses Babu J, Vyas K, et al. (2006). Isolation and characterization of impurities in docetaxel. J Pharmaceut Biomed 40:614–22
- Yin YM, Cui FD, Mu CF, et al. (2009). Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release 140:86–94
- Yoo HS, Park TG. (2001). Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA–PEG block copolymer. J Control Release 70:63–70
- Zareie HM, Kaitian X, Pişkin E. (1996). STM images of PDLLA-PEG copolymer micelles. Colloid Surface A 112:19–24
- Zhang X, Jackson JK, Burt HM. (1996). Development of amphiphilic diblock copolymers as micellar carriers of taxol. Int J Pharm 132:195–206
- Zhang X, Li Y, Chen X, et al. (2005). Synthesis and characterization of the paclitaxel/MPEG-PLA block copolymer conjugate. Biomaterials 26:2121–8