
Abstract
To evaluate the effects of poly(ethylene glycol) (PEG) grafting density on the tumor targeting efficacy of nanoparticles (NPs) with ligand modification, various amounts of PEG were conjugated to linoleic acid and poly(β-malic acid) double grafted chitosan (LMC) NPs bearing similar substitution degree of folate (FA). Increased particle size, decreased surface charge, reduced contact angle, retarded drug release and suppressed protein adsorption of LMC NPs were detected after surface modification. Compared to LMC NPs, FA-modified LMC NPs (FA-LMC NPs) remarkably enhanced tumor specificity. For PEG-modified FA-LMC NPs, increased drug accumulation in tumor tissues and reduced cellular uptake were observed with the increase of PEG grafting density. In regard to in vivo antitumor efficacy, FA-LMC NPs with moderate PEG grafting density (8.9%) significantly outperformed FA-LMC NP. Therefore, PEG modification with moderate grafting density could be a promising approach to coordinating with the tumor targeting efficacy of ligand-modified NPs.
Introduction
Nanoparticulate systems for targeted drug delivery have been extensively investigated in the treatment of tumor to improve the therapeutic efficacy and reduce the side effects of chemotherapeutic agents (Ruenraroengsak et al., Citation2010; Fang et al., Citation2011). Hydrophilic polymers and targeting ligands are commonly modified to the surface of nanoparticles (NPs), defined as surface modification, to endow the NPs with enhanced passive and active targeting capacities, respectively (Sheng et al., Citation2009; Shen et al., Citation2011). Hydrophilic polymer modification of NPs may increase their accumulation in tumor tissues through avoiding the macrophage uptake (Sheng et al., Citation2009; Fang et al., Citation2011), whereas decrease their uptake by tumor cells (Ruenraroengsak et al., Citation2010; Wang & Thanou, Citation2010). Comparatively, despite no enhancement in tumor accumulation, conjugation of targeting ligands may remarkably facilitate the internalization of NPs into tumor cells after their arrival in tumor tissues (Ruenraroengsak et al., Citation2010). Therefore, many efforts have been focused on coordinating the hydrophilic polymer modification with ligand modification of NPs to enhance their tumor accumulation and subsequent cellular uptake, thereby leading to improved antitumor efficacy of targeted drug nanocarriers (Danhier et al., Citation2009; Ruenraroengsak et al., Citation2010; Wang & Thanou, Citation2010).
For ligand-modified NPs, hydrophilic poly(ethylene glycol) (PEG) has mostly served as the spacer between ligands and NPs with the view of avoiding the phagocytosis of reticuloendothelial systems (RES), while preserving enough exposure of ligands to tumor cell membrane (Ruenraroengsak et al., Citation2010; Maruyama, Citation2011). However, the presence of ligands on the PEG surface may render the PEG layer of NPs obsolete, resulting in their increased protein adsorption, massive capture by the RES and rapid clearance from circulation (Chen et al., Citation2008; Gu et al., Citation2008; Koide et al., Citation2008). In addition, ligands folding inside the PEG globule may not be able to interact with the receptors on the cell membrane of targeting cells, leading to limited therapeutic efficacy of NPs (Sawant et al., Citation2008). To overcome these drawbacks of PEG as the spacer, both PEG and ligands were assumed to be directly conjugated to the surface of NPs. It is known that the higher grafting density and molecular weight of PEG can contribute to the reduced phagocytic uptake and improved passive targeting capacity of NPs without ligand modification (Fang et al., Citation2006; Essa et al., Citation2011). However, considering the shielding effects of PEG on the uptake by tumor cells, the optimal PEG grafting density should be investigated for ligand-modified NPs to fully coordinate the passive targeting of PEG with active targeting of ligands (Owens & Peppas, Citation2006; Chen et al., Citation2008). To our knowledge, the effects of PEG grafting density on the tumor targeting efficacy of ligand directly modified NPs are seldom reported.
In our previous study, the amphiphilic linoleic acid and poly (β-malic acid) double-modified chitosan (LMC) NPs were demonstrated to be able to load paclitaxel (PTX) into the hydrophobic core and show desired antitumor efficacy (Zhao et al., Citation2009). In addition, the lateral carboxyl groups of LMC NPs provided abundant sites for surface modification (Zhao et al., Citation2009). Therefore, in the present investigation, folate (FA) modified LMC NPs (FA-LMC NPs) with various grafting amounts of PEG were prepared to investigate the effects of PEG grafting density on the tumor targeting efficacy of NPs with ligand modification. The NPs were characterized in terms of particle size, surface charge, contact angle, protein adsorption, drug loading and release profile. In vitro cellular uptake was conducted on tumor cells, normal cells and macrophages. In vivo tumor inhibition and PTX accumulation in tumor tissues were monitored in H-22 tumor bearing mice.
Materials and methods
Materials
Chitosan (deacetylation degree of 85% and molecular weight of 105 kDa) was purchased from Golden-shell Biochemical Co., Ltd. (Zhejiang, China). Bovine serum albumin (BSA), FA, rhodamine B (RhB), and 4-dimethylaminopyridine (DMAP) were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). N-Hydroxysuccinimide (NHS) and N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl) were purchased from Yuanju Bio-Tech Co., Ltd. (Shanghai, China). PTX was from Hisun Pharmaceutical Co., Ltd. (Zhejiang, China). Methoxy (polyethylene glycol) (mPEG, molecular weight of 2 kDa) was purchased from Sigma Co., Ltd. (St. Louis, MO). All other chemicals were of analytical grade.
Cell lines and animals
Murine leukemic monocyte macrophages (Raw 264.7) were obtained from the American Type Culture Collection (Rockville, MD). Human liver cells (L02) and human hepatoma cells (SMMC-7721) were provided by Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s-modified Eagle medium containing 10% fetal calf serum.
Male Kunming mice (18–22 g) were obtained from the Animal Care Center of Fudan University. The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee, Fudan University, China.
Surface modification of LMC NPs
LMC NPs (3.0 of linoleic acid groups and 70.0 of poly (β-malic acid) groups per 100 anhydroglucose units of chitosan) were synthesized as described by Zhao et al. FA-LMC NPs was accomplished via EDC·HCl mediated Schiff base reaction at room temperature. FA (89 mg) was added into 50 mL of aqueous solution containing NHS (0.8 mg/mL), DMAP (0.2 mg/mL) and EDC·HCl (0.8 mg/mL). The resultant mixture was stirred for 4 h to form FA succinate, then added into 80 mL of LMC NPs (10 mg/mL) and kept stirring for another 12 h. Thus, FA-LMC NPs were obtained. For PEG modification, 16 g of mPEG and 16 mL of acetic anhydride were added to 80 mL of DMSO and stirred for 30 h at 40 °C. The resultant solution was added to 300 mL of ice-cold ether to precipitate the mPEG aldehyde. The mPEG aldehyde (0.8, 1.6 and 4.9 g) and 50 mL of LMC NPs (10 mg/mL) were mixed and kept stirring for 24 h at room temperature. NaBH4 (0.05, 0.1 and 0.2 g) was added into the mixture to deoxidize the C=N bond between PEG and LMC NPs. Thus, PEG-modified LMC NPs were obtained. The conjugation of FA to PEG-modified LMC NPs was achieved using the same method as FA-LMC NPs. PEG1,FA-LMC NPs, PEG2,FA-LMC NPs and PEG3,FA-LMC NPs referred to FA-LMC NPs with low, moderate and high PEG grafting density, respectively. All modified NPs were precipitated at pH 6.0 and washed with water for purification.
Characterization of NPs
1H NMR measurements were performed on an AVANCE DMX 500 spectrometer (Bruker, Germany). All NPs were dissolved in mixed solvent of DMSO-d6 and trifluoroacetic acid-d. The internal reference was tetramethylsilane. The particle size and surface charge of the NPs were determined with Zetasizer Nano (Malvern, UK). Contact angle measurements were performed using JC 2000A Goniometer (Zhongchen Digital Technology Co., Ltd., Shanghai, China).
Drug loading and encapsulation efficiency
Paclitaxel (3 mg) was dissolved in 60 µL of ethanol, followed by the addition of 3 mL of the NPs (10 mg/mL). The mixture was sonicated at 100 W for 15 min in an ice bath. PTX-loaded NPs were collected by silica column (50–100 mesh) as described previously (Zhao et al., Citation2009). PTX content in eluted solution was determined by high performance liquid chromatography (HPLC). The mobile phase was acetonitrile/water (46/54, v/v) and the flow rate was 1.0 mL/min. The detection wavelength was set at 227 nm. The column was a Hypersil C18 column (150 mm × 4.6 mm, Yilite, China). Drug loading capacity (LC) and encapsulation efficiency (EE) were calculated as described by Zhao et al.
In vitro drug release
In vitro release of PTX from the NPs was investigated using dialysis method. Briefly, 5 mL of PTX-loaded NPs suspension was placed in a dialysis bag (molecular weight cut-off of 14 kDa) and dialyzed against 500 mL of phosphate buffered saline (PBS, pH 7.4) containing 0.1% (w/v) Tween 80 at 37 °C and 100 rpm. At predetermined time intervals, an aliquot of 1 mL was withdrawn and centrifugated at 13 000 rpm for 15 min. The amount of released PTX in the supernatant was determined by HPLC, and the accumulative released amount was calculated.
Protein adsorption
Bovine serum albumin was used to evaluate the protein adsorption of the NPs. The NPs were suspended in PBS (pH 7.4) at a concentration of 10 mg/mL and incubated with BSA solution (0.8 mg/mL) at 37 °C under constant agitation. After 2 h, the mixture was centrifugated at 13 000 rpm for 30 min. The amount of BSA in the supernatant was measured by Lowry method. Results were expressed as the amount of adsorbed protein (mg) per milligram of the NPs.
Cellular uptake
RhB labeled NPs were synthesized to evaluate the cellular uptake. The NPs were covalently labeled with RhB via amide bonding between the residual primary amino groups on the NPs and the carboxyl groups on RhB mediated by EDC·HCl/NHS. The cells were seeded in 24-well plates at a density of 100 000 cell/well and incubated at 37 °C for 24 h. RhB labeled NPs were added at a concentration of 200 µg/mL. Following incubation for 2 h, the culture medium was removed. The cells were washed with PBS (pH 7.4) and lysed with Triton X-100 (1%, v/v) for 30 min. The fluorescence in the cell lysate was quantified for the NPs and the amount of cellular protein was measured by Lowry method. Results were expressed as the amount of internalized NPs (µg) per milligram of cellular protein. To evaluate the effect of free FA on the uptake, FA at a final concentration of 135 μg/mL was added 0.5 h prior to NP application and incubated with the cells throughout the 2-h uptake process.
In vivo antitumor efficacy
Tumor-bearing mice were obtained by subcutaneous inoculation of 0.2 mL of hepatoma H-22 mouse ascites into the axillary region of mice. When the tumor volume reached ∼100 mm3, tumor-bearing mice were randomly assigned to seven groups: saline (control); PTX injection (PTX in ethanol/Cremophor EL/0.9% NaCl (0.5/0.5/9, v/v/v); PTX-loaded LMC NPs; PTX-loaded FA-LMC NPs; PTX-loaded PEG1,FA-LMC NPs; PTX-loaded PEG2,FA-LMC NPs; PTX-loaded PEG3,FA-LMC NPs; PTX dosage was set as 3 mg/kg body weight. Drug administration was conducted three times through intravenous injection with 2 days spaced between each time, and the first administration day was recorded as 0 day. The length of the longest axis (L, mm) and the vertical axis (S, mm) of tumors were measured every other day and the tumor volume (V, mm3) was calculated as follows, V = L × S2/2. The mice were sacrificed on the 10th day after the last administration, and the tumors were excised and weighed. The tumor inhibition ratio (TIR) was calculated according to the following equation:
where WT and WC referred to the tumor weight of the test and control groups, respectively.
PTX accumulation in tumor tissues
Mice bearing H-22 tumor xenograft (∼100 mm3) were injected intravenously with PTX injection and various PTX-loaded NPs as described before. At 2 - and 24-h post-injection, mice were sacrificed and tumors were excised and homogenized. Protein precipitation with zinc sulfate and methanol and extraction with ethyl acetate were used for sample preparation, and diazepam served as the internal standard. The amount of PTX and diazepam were determined with HPLC. The mobile phase was acetonitrile/water (53/47, v/v) and the flow rate was 1.0 mL/min. The detection wavelength was set at 227 nm. The analytic column was a Diamonsil C18 column (150 mm × 4.6 mm, Dikma, China). Results were expressed as the amount of PTX (µg) per milligram of tumor tissues.
Statistical analysis
All results were expressed as mean ± standard deviation. Statistical analysis was evaluated with single factor analysis of variance (ANOVA) followed by Tukey’s post hoc test and statistical significance was considered at p < 0.05.
Results and discussion
Characterization of NPs
As illustrated in , PEG, FA-LMC NPs was synthesized through sequential conjugation of mPEG aldehyde and FA onto the amino groups exposed on the surface of LMC NPs. In the 1H NMR spectrum of FA (), the peaks at 6.8, 7.6 and 8.6 ppm were assigned to the aromatic C-NH, 1-benzene CH and 2-pyrazine CH, respectively. 1H NMR spectrum of FA-LMC NPs showed proton peaks for FA. PEG-modified FA-LMC NPs demonstrated respective proton peaks for FA (6.8, 7.6 and 8.6 ppm) and PEG (CH2 at 3.6 ppm). Such results confirmed that the FA and PEG were conjugated to LMC NPs. The area of proton peak for PEG was enhanced from PEG1,FA-LMC NPs to PEG3,FA-LMC NPs, suggesting the increased PEG grafting density. The degree of substitution (DS) of FA and PEG in the NPs was calculated by comparing the peak area of the corresponding protons (6.8 ppm for FA and 3.6 ppm for PEG) to that of acetyl protons (1.8 ppm) of chitosan. The DS of FA in FA-LMC NPs and PEG-modified FA-LMC NPs was ∼7.5%. When the DS of PEG was calculated, the integral areas of methylene protons on chitosan at 3.6 ppm had been subtracted. Thus, the DS of PEG in PEG1,FA-LMC NPs, PEG2,FA-LMC NPs and PEG3,FA-LMC NPs was 3.7%, 8.9% and 15.1%, respectively.
Figure 1. (a) Schematic illustration summarizing the chemical synthesis of FA and PEG grafting onto LMC NPs. (b) 1H NMR spectra of NPs in a mixed solvent of DMSO-d6 and trifluoroacetic acid-d. Peaks marked by asterisk *, plus + and arrow → are assigned to FA (the aromatic C-NH, 1-benzene CH and 2-pyrazine CH), PEG (CH2) and chitosan (acetyl protons), respectively.
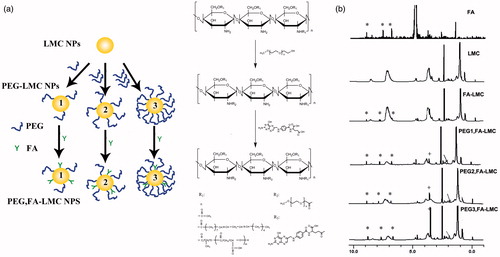
The physicochemical properties of the NPs were illustrated in . Particle sizes of all the NPs displayed unimodal distribution (polydispersity index < 0.3) with a size range of 200–300 nm. The particle size of FA-LMC NPs was similar to that of LMC NPs, suggesting the negligible contribution of FA modification to the alteration of particle size. After PEG modification, no significant augmentation was noted in the particle size of PEG1,FA-LMC NPs, whereas the particle sizes of PEG2,FA-LMC NPs and PEG3,FA-LMC NPs were significantly increased, which might be ascribed to the distinct configurations of PEG chains at different grafting densities (Kato et al., Citation2003). At low density, PEG chains were curled and adsorbed to the surface of the FA-LMC NPs, which formed mushroom configuration and thus resulted in the limited effects on the particle size. By contrast, PEG chains at moderate and high density were obliged to stretch away from the surface, which formed brush configuration and led to a significant increase in the particle size of FA-LMC NPs.
Figure 2. Particle size (a) surface charge (b) and contact angle (c) of LMC NPs, FA-LMC NPs and PEG-modified FA-LMC NPs. Indicated values were mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
As shown in , negative charge of FA-LMC NPs was slightly decreased compared to LMC NPs. The negative charge of LMC NPs arising from the ionization of carboxyl groups on the poly (β-malic acid) might be suppressed after FA modification, resulting in decreased surface charge of FA-LMC NPs. As for PEG-modified FA-LMC NPs, negative charge was gradually diminished with the increase of PEG grafting density, and positive charge was detected in the case of PEG2,FA-LMC NPs and PEG3,FA-LMC NPs. The brush configuration resulting from PEG chains at moderate and high grafting density might strongly shield the ionization of carboxyl groups of poly (β-malic acid), leading to the reversal of surface charge.
Notable improvement in the surface hydrophilicity was observed for FA-LMC NPs in comparison with LMC NPs, as evidenced by the significantly decreased contact angles () (Tziampazis et al., Citation2000). No significant difference of contact angle was noted between PEG1,FA-LMC NPs and FA-LMC NPs, whereas PEG modification with moderate and high grafting density could yield decreased contact angle, indicating that the hydrophilicity of FA-LMC NPs was enhanced with the increase of the PEG grafting density (Tziampazis et al., Citation2000). The enhanced hydrophilicity of the NPs after surface modification might be due to the good solubility of FA and PEG in the water.
Drug loading and EE
As listed in , the LC of LMC NPs was increased after FA modification, consistent with the previous result (Patil et al., Citation2009). The LC of FA-LMC NPs was significantly elevated after the conjugation of PEG at moderate and high density, suggesting that PEG modification with relatively high grafting density could improve the LC of the NPs. This might be due to the physical encapsulation effect of PEG chains on PTX (Yao et al., Citation2011). FA modification and high PEG grafting density resulted in the improved EE for PTX, whereas relatively low PEG grafting density contributed to the decreased EE.
Table 1. Particle size, LC and EE of PTX-loaded NPs.
In vitro drug release
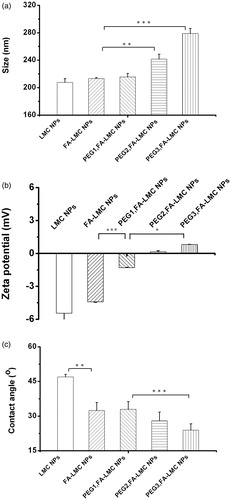
Drug release from the NPs was an important factor in determining the efficacy of targeted drug delivery (Ruenraroengsak et al., Citation2010). As shown in , all PTX-loaded NPs exhibited biphasic drug release with an initial burst release followed by a continuous and slow release. It was noted that the surface modification could decrease the release rate and initial burst release of PTX-loaded LMC NPs. The total amounts of PTX released from LMC NPs and FA-LMC NPs within 12 h were ∼79.8% and 69.7%, respectively. The release of PTX from PEG-modified FA-LMC NPs was further slowed down. These results were different from a previous report in which the drug release was speeded up by the surface modification (Sun et al., Citation2008). Such disparity might be related to the components of NPs and the properties of ligands adopted. The diffusion of drug, the erosion and swelling of polymer matrix, and the degradation of polymer were the main mechanisms for the drug release. Since the degradation of LMC is slow, the release of PTX from tested NPs would mainly depend on the drug diffusion and the matrix erosion (Zhao et al., Citation2009). In such case, the increased hydrophilicity of the outer surface as evidenced by the decreased contact angle () would decrease the release of PTX from the hydrophobic linoleic acid core of LMC NPs (Mu & Feng, Citation2002). Aside from the increased surface hydrophilicity, the brush configuration of PEG chains might also contribute to the decreased PTX release rate of PEG2,FA-LMC NPs and PEG3,FA-LMC NPs. Burst release associated with the NPs was considered as one of the barriers that limited the therapeutic efficacy of targeted drug delivery (Ruenraroengsak et al., Citation2010). The restrained initial release of PTX from LMC NPs with surface modification might partially contribute to their improved antitumor efficacy.
Figure 3. In vitro PTX release from LMC NPs, FA-LMC NPs and PEG-modified FA-LMC NPs in PBS (pH 7.4) containing 0.1% Tween 80 (w/v) at 37 °C. Indicated values were mean ± SD (n = 3).
Protein adsorption
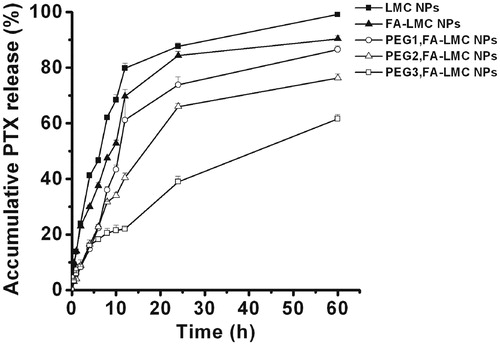
Protein adsorption occurred when the NPs came in contact with plasma, which induced the recognition of macrophages and reduction of the tumor accumulation of the NPs (Heldin et al., Citation2004; Owens & Peppas, Citation2006; Chen et al., Citation2008). Therefore, the protein adsorption of LMC NPs with various surface modifications was evaluated. As shown in , the protein adsorption of FA-LMC NPs was significantly lower than that of LMC NPs. This might be due to their improved surface hydrophilicity. For PEG-modified FA-LMC NPs, the protein adsorption decreased with the increase of PEG grafting density, which was in accordance with the previous result (Tziampazis et al., Citation2000). However, the protein adsorption of PEG1,FA-LMC NPs was higher than that of FA-LMC NPs, suggesting that PEG modification with low grafting density might not be qualified for providing enough coverage to avoid protein adsorption of ligand-modified NPs. Protein adsorption contained two phases: (i) movement of protein to the vicinity of the NPs which was mainly controlled by random Brownian motion and (ii) protein binding to the surface of the NPs via Van der Waal’s forces, electrostatic forces or hydrophobic/hydrophilic interactions (Chen et al., Citation2008). For PEG1,FA-LMC NPs, the PEG chains in mushroom configuration might be inability of preventing the protein movement in phase I and the negative surface charge was too low to form sufficient repulsive forces in phase II, thus resulting in higher BSA adsorption compared to FA-LMC NPs. The brush configuration of PEG chains with higher grafting density might effectively block protein movement in phase I, leading to the reduced BSA adsorption. Thus, PEG modification with appropriate grafting density was needed to provide enough protection for the NPs from protein adsorption in targeted drug delivery.
Figure 4. Protein adsorption of LMC NPs, FA-LMC NPs and PEG-modified FA-LMC NPs at 37 °C. Indicated values were mean ± SD (n = 3). ***p < 0.001.
In vitro cellular uptake
As shown in , the uptake of FA-LMC NPs by SMMC-7721 cells was 2.4-fold higher than that of LMC NPs, indicating that FA modification could be an efficient way of enhancing tumor cellular uptake of the NPs. The remarkable reduction in cellular uptake of FA-LMC NPs following the addition of free FA ligands further confirmed the specificity of FA-mediated endocytosis. However, it was worth noting that the uptake of FA-LMC NPs by L02 cells was also superior to LMC NPs (), which might be due to the extensive distribution of FA receptors in the body (Weitman et al., Citation1992). confirmed that the uptake of PEG-modified FA-LMC NPs by SMMC-7721 cells was correlated to the PEG grafting density, wherein an increase in the PEG grafting density strongly inhibited the internalization of the NPs, with ∼75.8% decrease for PEG3,FA-LMC NPs as compared to FA-LMC NPs. Similar tendency was also observed in L02 cells (). These results indicated that PEG chains might interfere with the interaction between FA and its receptors due to the non-specific steric shielding effect (Ruenraroengsak et al., Citation2010). The steric PEG on the surface of FA-LMC NPs might reduce their opportunities for adsorption to cell membrane, which resulted in decreased cellular uptake of PEG-modified FA-LMC NPs (Tziampazis et al., Citation2000). In addition, the larger particle size of PEG3,FA-LMC NPs might be adverse to the entry into cells (He et al., Citation2010). Thus, PEG modification with high grafting density might not be suitable for the active targeting of ligand-modified NPs.
Figure 5. Cellular uptakes in SMMC-7721 cells (a) L02 cells (b) and Raw 264.7 cells (c) following 2-h incubation with rhodamine B labeled NPs (200 µg/mL). Indicated values were mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001.
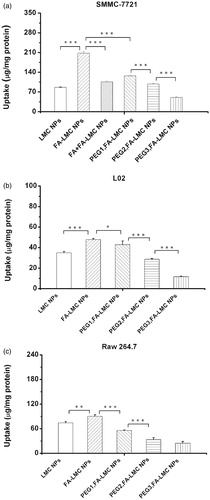
revealed that macrophage uptake was promoted after FA modification, suggesting that FA receptors might be present in Raw 264.7 cells (Turk et al., Citation2004). Compared to FA-LMC NPs, macrophage uptake of PEG-modified FA-LMC NPs was significantly suppressed and inversely correlated to the PEG grafting density. This result demonstrated that PEG modification could effectively avoid macrophage uptake of FA-LMC NPs, thereby prolonging blood circulation and enhancing tumor accumulation (Owens & Peppas, Citation2006). However, the penetration of FA-LMC NPs in tumor tissues could be limited after PEG modification, which was disadvantageous for tumor therapy (Heldin et al., Citation2004; Turk et al., Citation2004). Therefore, PEG grafting density of the NPs should be optimized for avoiding their endocytosis of macrophages and normal cells, while preserving their internalization into tumor cells and penetration in tumor tissues.
In vivo antitumor efficacy
The in vivo antitumor efficacy of PTX-loaded NPs was evaluated in mice bearing H-22 tumor. Natural body weight increase was observed among all groups throughout the experimental period (). With regard to TIR () and tumor volumes (), it was evident that PTX-loaded FA-LMC NPs had significantly enhanced antitumor efficacy compared to PTX-loaded LMC NPs (p < 0.05), which was correlated well with the results of in vitro cellular uptake. With the higher tumor cellular uptake, FA-LMC NPs could deliver more PTX into tumor cells than LMC NPs, thus resulting in their improved antitumor efficacy.
Figure 6. Antitumor effect achieved in H-22 tumor bearing mice following systemic administration of saline, PTX injection (3 mg/kg PTX eq.) and PTX-loaded NPs (3 mg/kg PTX eq.). The first administration day was recorded as 0 day and the administration was conducted at 0, 3 and 6 days. Indicated values were mean ± SD (n = 6). *Statistically significant difference from PTX-loaded FA-LMC NPs: *p < 0.05.
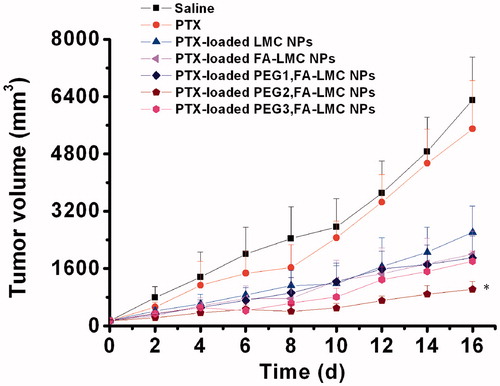
Table 2. TIR of saline, PTX injection and PTX-loaded NPs against H-22 tumor bearing mice.
The antitumor efficacy of PTX-loaded FA-LMC NPs was enhanced after PEG modification. Notably, the TIR of PTX-loaded PEG2,FA-LMC NPs was significantly higher than those of PTX-loaded PEG1,FA-LMC NPs and PTX-loaded PEG3,FA-LMC NPs (p < 0.01). Compared to FA-LMC NPs, PEG-modified FA-LMC NPs could enhance their passive targeting capacity owing to the less macrophage uptake (), resulting in their improved therapeutic efficacy. Nevertheless, PEG modification with high grafting density strongly suppressed the PTX release () and tumor cellular uptake of PTX-loaded FA-LMC NPs (), which might lead to the unsatisfactory antitumor efficacy of PTX-loaded PEG3,FA-LMC NPs. PEG modification with moderate grafting density might not influence the active targeting effect of FA ligand, thus resulting in the highest TIR of PTX-loaded PEG2,FA-LMC NPs.
PTX accumulation in tumor tissues
To elucidate the improved TIR of LMC NPs with surface modification, PTX accumulation in tumor tissues was determined by HPLC with limit of quantification (LOQ) of 4 ng (). At 2 and 24 h, drug accumulation of PTX-loaded FA-LMC NPs was both slightly higher than that of PTX-loaded LMC NPs. Such results were correlated well with the in vivo antitumor efficacy. Because of the reduced initial burst release, low protein adsorption and improved internalization into tumor cells, PTX-loaded FA-LMC NPs could enhance the accumulation of PTX in tumor tissues and the TIR compared to PTX-loaded LMC NPs (Roser et al., Citation1998; Ruenraroengsak et al., Citation2010).
Figure 7. PTX accumulation in tumor tissues of H-22 tumor bearing mice treated with saline, PTX injection (10 mg/kg, PTX eq.) and PTX-loaded NPs (10 mg/kg, PTX eq.) at 2- and 24-h post-injection. Indicated values were mean ± SD (n = 3). *p < 0.05, ***p < 0.001.
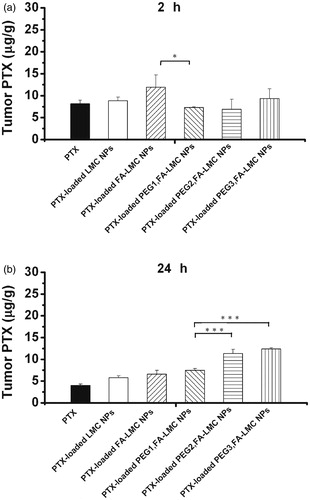
At 2 h, PEG-modified PTX-loaded FA-LMC NPs had lower drug accumulation in tumor tissues than PTX-loaded FA-LMC NPs, which might be caused by their decreased uptake by tumor cells. At 24 h, PEG-modified PTX-loaded FA-LMC NPs with low, moderate and high PEG grafting density led to 10%, 70% and 90% increase in PTX accumulation compared to PTX-loaded FA-LMC NPs, respectively, indicating that the PTX accumulation was remarkably increased with the increase of PEG grafting density. Due to their decreased macrophage uptake (), PEG-modified FA-LMC NPs could exhibit prolonged blood circulation and gradually accumulate in tumor tissues through the enhanced permeability and retention (EPR) effect, thus delivering more PTX to tumor tissues over 24 h. These might partly explain the promoted TIR of PTX-loaded PEG-modified FA-LMC NPs. It was noted that PTX-loaded PEG3,FA-LMC NPs possessed the higher PTX accumulation and lower TIR than those of PEG2,FA-LMC NPs. Though delivering more PTX to tumor tissues, the penetration of PTX in tumor tissues accompanied with PTX-loaded PEG3,FA-LMC NPs might still be poor due to the large particle size, slow PTX release and reduced tumor-associated macrophage uptake (Turk et al., Citation2004; Ruenraroengsak et al., Citation2010). In addition, the internalization of PTX-loaded PEG3,FA-LMC NPs into tumor cells was low as demonstrated by in vitro cellular uptake. These implied that PEG modification with high grafting density might interfere with the in vivo active targeting efficacy of FA-LMC NPs, resulting in reduced antitumor efficacy of PTX-loaded PEG3,FA-LMC NPs. By contrast, FA-LMC NPs with moderate PEG grafting density, combining the advantages of the hydrophilic polymer modification and ligand modification, might cooperate the drug accumulation in tumor tissues well with the uptake by tumor cells, leading to the high antitumor efficacy of PTX-loaded PEG2,FA-LMC NPs.
Conclusions
In the present work, FA-LMC NPs and FA-LMC NPs with various PEG grafting densities were developed. FA modification could enhance in vitro tumor cellular uptake and in vivo tumor targeting efficacy of LMC NPs. In vitro cellular uptake and in vivo drug accumulation in tumor tissues were PEG density dependent, wherein higher PEG grafting density was responsible for more drug accumulation in tumor tissues but lower cellular uptake. Notably, FA-LMC NPs with moderate PEG grafting density exhibited the highest antitumor efficacy. In conclusion, PEG modification with moderate grafting density was a promising strategy for improving the tumor targeting efficacy of ligand-modified NPs. These might provide insights into the development and application of targeted drug delivery systems for tumor therapy.
Declaration of interest
The authors report no declarations of interest. The authors are thankful for the financial support from the National Natural Science Foundation of China (No. 30873204), the Science and Technology Commission of Shanghai Municipality of China (No. 1052nm03500) and the National Science and Technology Major Project (No. 2009ZX09310-006).
Related Research Data
References
- Chen H, Hu XY, Zhang YX, et al. (2008). Effect of chain density and conformation on protein adsorption at PEG-grafted polyurethane surfaces. Colloid Surf B Biointerfaces 61:237–43
- Chen H, Yuan L, Song W, et al. (2008). Biocompatible polymer materials: role of protein–surface interactions. Prog Polym Sci 33:1059–87
- Danhier F, Vroman B, Lecouturier N, et al. (2009). Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with Paclitaxel. J Control Release 140:166–73
- Essa S, Rabanel JM, Hildgen P. (2011). Characterization of rhodamine loaded PEG-g-PLA nanoparticles (NPs): effect of poly(ethylene glycol) grafting density. Int J Pharm 411:178–87
- Fang C, Shi B, Pei YY, et al. (2006). In vivo tumor targeting of tumor necrosis factor-α-loaded stealth nanoparticles: effect of MePEG molecular weight and particle size. Eur J Pharm Sci 27:27–36
- Fang J, Nakamura H, Maeda H. (2011). The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 63:136–51
- Gu F, Zhang L, Teply BA, et al. (2008). Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci USA 105:2586–91
- He CB, Hu YP, Yin LC, et al. (2010). Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31:3657–66
- Heldin CH, Rubin K, Pietras K, Ostman A. (2004). High interstitial fluid pressure-an obstacle in cancer therapy. Nature 4:806–13
- Kato K, Uchida E, Kang ET, et al. (2003). Polymer surface with graft chains. Prog Polym Sci 28:209–59
- Koide H, Asai T, Hatanaka K, et al. (2008). Particle size-dependent triggering of accelerated blood clearance phenomenon. Int J Pharm 362:197–200
- Maruyama K. (2011). Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv Drug Deliv Rev 63:161–9
- Mu L, Feng SS. (2002). Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol®). J Control Release 80:129–44
- Owens DE, Peppas NA. (2006). Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm 307:93–102
- Patil YB, Toti US, Khdair A, et al. (2009). Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials 30:859–66
- Roser M, Fischer D, Kissel T. (1998). Surface-modified biodegradable albumin nano- and microspheres. II: effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur J Pharm Biopharm 46:255–63
- Ruenraroengsak P, Cook JM, Florence AT. (2010). Nanosystem drug targeting: facing up to complex realities. J Control Release 141:265–76
- Sawant RR, Sawant RM, Kale AA, Torchilin VP. (2008). The architecture of ligand attachment to nanocarriers controls their specific interaction with target cells. J Drug Target 16:596–600
- Shen Z, Yan L, Kohama K, et al. (2011). Improved drug targeting of cancer cells by utilizing actively targetable folic acid-conjugated albumin nanospheres. Pharmacol Res 63:51–8
- Sheng Y, Liu CS, Yuan Y, et al. (2009). Long-circulating polymeric nanoparticles bearing a combinatorial coating of PEG and water-soluble chitosan. Biomaterials 30:2340–8
- Sun B, Ranganathan B, Feng SS. (2008). Multifunctional poly(d,l-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by Trastuzumab for targeted chemotherapy of breast cancer. Biomaterials 29:475–86
- Turk MJ, Waters DJ, Low PS. (2004). Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett 213:165–72
- Tziampazis E, Kohn J, Moghe PV. (2000). PEG-variant biomaterials as selectively adhesive protein templates: model surfaces for controlled cell adhesion and migration. Biomaterials 21:511–20
- Wang M, Thanou M. (2010). Targeting nanoparticles to cancer. Pharmacol Res 62:90–9
- Weitman SD, Lark RH, Coney LR, et al. (1992). Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res 52:3396–401
- Yao HJ, Ju RJ, Wang XX, et al. (2011). The antitumor efficacy of functional paclitaxel nanomicelles in treating resistant breast cancers by oral delivery. Biomaterials 32:3285–302
- Zhao ZM, He M, Yin LC, et al. (2009). Biodegradable nanoparticles based on linoleic acid and poly(β-malic acid) double grafted chitosan derivatives as carriers of anticancer drugs. Biomacromolecules 10:565–72