
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Intranasal thermosensitive gel for rasagiline mesylate (RM) was developed for effective treatment of Parkinson’s disease. Intranasal gels were prepared by combination of poloxamer 407 and poloxamer 188 (1:1) with mucoadhesive polymers (carbopol 934 P and chitosan). The formulations were evaluated for sol–gel transition temperature, in-vitro drug release and in-vivo mucociliary transit time. Further, optimal intranasal gel formulations were tested for in-vivo pharmacokinetic behavior, nasal toxicity studies and brain uptake studies. It was found that optimal formulations had acceptable gelation temperature (28–33 °C) and adequate in-vitro drug release profile. Pharmacokinetic study in rabbits showed significant (p < 0.05) improvement in bioavailability (four- to six-folds) of the drug from intranasal gels than oral solution. Chronic exposure studies in Wistar rats showed that these intranasal gels were non-irritant and non-toxic to rat nasal mucosa. Estimation of RM in rat brain tissue showed significant (p < 0.01) improvement in uptake of RM form intranasal gel formulations than nasal solution.
Introduction
In the past decade, nasal route for drug delivery has established itself as an important alternative to parenteral route. This route provides higher degree of patient compliance and makes the patient more tractable. Moreover, drugs can be painlessly self-administered by the patient, which adds to the popularity of this route (Illum, Citation2003).
Drugs administered through nasal route absorb rapidly and reach therapeutically effective plasma levels quickly due to rich vasculature and highly permeable membranes in nasal cavity (Majithiya et al., Citation2006). Moreover, nasal route offers additional advantage over oral route, particularly for those drugs that have poor oral bioavailability due to pH instability and enzyme degradation in the gastrointestinal tract (GIT). Further, drugs that undergo gut wall metabolism, P-glycoprotein efflux and high hepatic first pass metabolism are also benefited from nasal route of administration (Ugwoke et al., Citation2001).
For a few drugs, plasma drug profiles from intranasal delivery are akin to that of intravenous drug profile of the same drug (Romeo et al., Citation1998; Ugwoke et al., Citation2001; Arora et al., Citation2002).
In recent times, interest in intranasal route to target drugs to brain and cerebrospinal fluid circumventing blood–brain barrier has gained impetus. Intranasal formulation of drugs for treatment of Parkinson’s disease (PD) (Khan et al., Citation2010), Alzheimer’s disease (AD) (Zhang et al., Citation2004) and psychosis (Kumar et al., Citation2008) have been developed and their therapeutic effectiveness over conventional oral formulations has been demonstrated. Comprehensive studies have shown the role of olfactory pathway in transport of nasally administered drugs to central nervous system (CNS) (Illum, Citation2000; Pardeshi & Belgamwar, Citation2013).
Rasagiline mesylate (RM) is a potent inhibitor of enzyme monoamine oxidase (MAO) type B that has major role in the inactivation of biogenic amines in the CNS. MAO-B is primarily responsible for inactivation of dopamine in CNS. As dopamine deficiency is primarily responsible for clinical manifestations of PD, inhibition of MAO-B tends to restore levels of dopamine towards normal, thus, improving symptoms of PD (Chen et al., Citation2007). RM is available for oral use in the form of immediate release tablets (Azilect 1 mg) and was approved for use in USA in May 2006 (Azilect, Citation2010).
Upon oral administration, RM is rapidly absorbed from GIT, reaching peak plasma concentrations within an hour. Because RM undergoes high hepatic first-pass metabolism, it demonstrates an absolute oral bioavailability (BA) of about 36% in humans (Guay, Citation2006). RM is completely bio-transformed in liver by cytochrome P450 system. CYP1A2 isoenzyme is a major contributor in the metabolism of RM (Guay, Citation2006).
Due to low bioavailability and its inability to reach high concentration in brain tissue after oral administration, it was excogitated to develop a novel dosage form for RM that could address these problems.
A look into existing literature revealed few novel formulation approaches to improve BA of RM. Bukka et al. (Citation2010) have reported buccal patches for RM using carbopol 940P and sodium alginate as polymers. However, lack of in - vivo evidence in improvement of BA of RM limits the scope of this work.
Several patents for delivery of RM through transdermal route are available (Deng et al., 2007; Lin et al., 2009), however, adequate in vivo and clinical evidence for usefulness of transdermal route in delivery of RM needs to be established.
Delivery of RM through intranasal route could improve its BA (by avoidance of first-pass metabolism). Further, higher concentrations of RM in brain tissue could be possibly achieved after intranasal administration than after oral administration.
However, owing to the anatomical and physiological conditions of nasal cavity, intranasally administered drugs have low residence time at the site of administration. Rapid mucocilliary clearance (MC) also limits the time available for drug absorption (Ugwoke et al., Citation2000, Citation2001).
Therefore, in design of intranasal formulations, one must address the problem of low residence time in nasal cavity. Possible strategy to improve nasal residence time is to use formulations with higher viscosity, and preferably, with mucoadhesive character. Designing an intranasal dosage form with mucoadhesive character is particularly helpful because it provides intimate contact between dosage form and nasal epithelium thereby maximizing the chances of absorption for a drug molecule.
Mucoadhesive gel formulations can be a solution, but they suffer from problems of accuracy in dosing. Mucoadhesive powder particles (Ugwoke et al., Citation1999; Jain et al., Citation2004; Belgamwar et al., Citation2011) are not favored due to their irritation potential. Thermosensitive gels provide a suitable alternative; they are easy to administer and have suitable viscosity at body temperature to counter MC.
Poloxamers are triblock copolymers of poly (ethylene oxide) and poly (propylene oxide). Using hydrophilic excipients, gelation temperature of poloxamer blends can be modulated so that they form in - situ gels at body temperature (Zhou & Donovan, Citation1996). Further, several researchers have reported the use of thermosensitive poloxamer gels in intranasal administration of drugs like anti-emetics (Zaki et al., Citation2007), anti-migraine agents (Majithiya et al., Citation2006) and vitamin B12 (Pisal et al., Citation2004). Poloxamers have poor inherent mucoadhesive properties (Dumortier et al., Citation2006). Therefore, some mucoadhesive polymer is added along with poloxamers to make effective intranasal gel formulation.
The objective of this study was to develop and evaluate thermosensitive mucoadhesive gels of poloxamer for delivery of RM. For the study, carbopol 934 P (CP) and chitosan were selected as anionic and cationic mucoadhesive polymers, respectively.
Materials and methods
Materials
RM (Gifted by Apotex Research Pvt. Ltd., Bangalore, India), acetonitrile high-performance liquid chromatography (HPLC) grade (ACN), ammonium acetate (AA), benzalkonium chloride (BKC), glacial acetic acid (GAA), methanol HPLC grade (Sigma-Aldrich Co., Mumbai, India); Poloxamer 407 (P407), Poloxamer 188 (P188) (Sigma chemical Co., St. Louis, MO); Carbopol 934 P (CP, molecular weight 3 × 106 Da) (Tama, Japan); chitosan (molecular weight 1–3 × 105 Da and 75–85% degree of acetylation); sodium chloride (NaCl), potassium chloride (KCl) and calcium chloride dihydrate (CaCl2 ċ 2H2O) (Hi-media Laboratories Pvt. Ltd., Mumbai, India) were used in the study. Water purification system (Millipore®, MA, USA) was used to obtain high quality water.
Methods
Preparation of thermosensitive mucoadhesive gels (TM gel)
The cold method described by Choi et al. (Citation1998) was followed with slight modification. Briefly, calculated quantities of the drug and mucoadhesive polymer were stirred in de-ionized water (for CP) or acetic acid solution prepared in de-ionized water (2% v/v) (for chitosan) maintained at 25 °C using a thermostatically controlled magnetic stirrer (IKA® India Private Limited, Bengaluru, India). This dispersion was then cooled to 4 °C in a refrigerator. Calculated quantities of P407 and P188 () were added to the cold dispersion under continuous stirring (on magnetic stirrer) at 1200 rpm. The dispersions were again stored under refrigeration overnight to obtain a clear and transparent gel. In all the cases, pH of the TM gels was adjusted to 5.5 ± 0.2 using suitable volume of 0.1N sodium hydroxide solution.
Table 1. Sol–gel temperatures of intranasal TM gels of rasagiline mesylate.
Measurement of sol–gel transition temperature
The magnetic-bar method, previously described by Ryu et al. (Citation1999) was adopted for determination of sol–gel transition temperature (T sol→gel). Briefly, 10 g of the prepared TM gel was placed in 25 mL glass beaker which in turn was placed in a water bath. The water bath was placed on a thermostatically controlled magnetic stirrer (IKA, Baden-Wurttemberg, Germany). A small magnetic bar (1.5 cm in length), weighing 1.5 g was placed into the TM gel sample and was left to equilibrate at 25 °C ± 0.5 °C. After equilibration, the actual initial temperature was noted using digital temperature recorder (Labindia, Mumbai, India). Then, the magnetic bar was moved at 50 rpm. Temperature of external water bath was gradually increased at an increment of 2 °C per min (or 1 °C/min in region of sol–gel transition temperature). The TM gels were left to equilibrate for few minutes at each new setting. When movement of the magnetic bar ceased (due to increased viscosity of the system), the temperature was recorded. This temperature was considered as sol–gel transition temperature (T sol→gel). All measurements were done in triplicates and values were reported as mean ± standard deviation.
In - vitro drug release studies
Drug release studies were performed by dialysis bag method (Spectra/Por 3 dialysis membrane, MWCO 3500 Da). The sealed dialysis bag contained 1.5 g of TM gel that was immersed into a dialysis reservoir containing 150 mL phosphate-buffered saline (PBS, pH 6.4). PBS was pre-conditioned and maintained at 34 ± 0.5 °C on a water bath. The samples (1 mL) were withdrawn at pre-determined time intervals (5, 15, 30, 45, 60, 90, 120, 150, 180 and 240 min) and same amount of fresh PBS was added to maintain sink conditions. All the samples were analyzed using validated HPLC method that was previously developed in our lab. The data obtained from the in
-
vitro release study was analyzed using following Peppas equation (Peppas, Citation1985):
Where M t /M∞ is fraction of drug released at time‘t’; ‘k’ is the release rate constant and ‘n’ is the release exponent determining drug release mechanism. For acquisition of ‘n’ value, only the portion of M t /M∞ < 0.6 in the drug release curve was used.
Measurement of mucociliary transit time (MTT)
All the animal experiments were carried out after prior approval of the protocol by institutional animal ethics committee (Approval number: IAEC-04/12-13). The method reported by Lale et al. (Citation1998) for measurement of in - vivo mucociliary transport time (MTT) was adopted with slight modifications. Male Wistar rats (n = 6), weighing 250–320 g were anesthetized using intra-peritoneal injection of urethane solution (1200 mg/kg). Using a micropipette, 5 µL of TM gel containing a dye, xylene cyanol (3 mg/mL), was instilled 0.5 cm deep into one of the nostrils of the rats. The pharyngeal cavities of the dosed rats were swabbed with cotton-tipped applicators. The swab samples were collected at a minute’s interval for first 30 min and then at an interval of 5 min for next 90 min (Lale et al., Citation1998). As controls, 5 µL of dye solution (prepared in normal saline) and 5 µL dye-loaded plain poloxamer gel (without mucoadhesive polymer) were administered to rats. After suitable dilution in water, dye content from each swab was estimated using a validated UV-visible spectrophotometric method.
Morphological study of rat nasal epithelium
The method followed was similar to that previously described by Tengamnuay et al. (Citation2000) and Miyamoto et al. (Citation2001) Briefly, right nostril of male Wistar rats (n = 3) was dosed with 20 µL of TM gel formulation daily, for period of two weeks. Before dosing, intra-peritoneal injection of thiopental (45 mg/kg) was given to all the rats to facilitate easy administration. After two weeks, the rats were sacrificed and nasal septum with epithelial membrane was carefully separated from the bone. The septum was fixed in 10% formalin solution, sliced with rotary microtome (Leica, Wetzlar, Germany) and stained with hematoxylin–eosin dye. In all the cases, left nostril epithelium from the same rat was used as a control. The prepared slides were observed under light microscope (Olympus, Tokyo, Japan) fitted with a camera and associated software.
Pharmacokinetic studies in rabbits
Animal handling and administration of nasal gel
For this study, prior clearance from institutional animal ethics committee (Approval number: IAEC-05/12-13) was obtained. Briefly, female New Zealand white rabbits (n = 3), weighing 2.4 ± 0.2 kg were housed individually in stainless steel cages, fed with commercial rabbit diet and provided drinking water ad l ibitum. The rabbits were fasted for 18 h before and also during the pharmacokinetic study. All the animals were conscious throughout the duration of experiment. They were held in commercially available rabbit restrainers during collection of blood samples.
A cross-over study protocol (with 1 week washout period) was followed. A micropipette (10–100 µL capacity), with its tip placed 1 cm deep inside the right nostril of rabbits, was used to administer 200 µL of TM gel (equivalent to dose of 4 mg/kg of RM). For oral pharmacokinetic study, 5 mL oral solution of RM in water (equivalent to dose of 10 mg/kg of RM) was administered to rabbits by oral gavage.
Collection of blood samples and analysis
Post dosing, blood samples (1.5 mL) were collected by marginal ear vein puncture at following time intervals: 5, 15, 30, 45, 60, 90, 120, 150 and 180 min. Blood samples were collected into 2 mL sample collection tubes containing 4% w/v sodium citrate solution as an anti-coagulant. Plasma samples were obtained by centrifuging blood samples in a cooling centrifuge at 905 × g for 10 min at 4 °C. Supernatant clear plasma was carefully collected and frozen at −20 °C till further analysis. For analysis of RM plasma samples, a validated HPLC method (Ravi et al., Citation2013) previously developed in our lab was used. Briefly, frozen plasma samples were thawed to room temperature; to 100 µL of each sample, 150 µL of ACN was added as a protein precipitating agent. Further, vortex-mixing (5 min) followed by centrifugation at 8500 × g for 20 min at 4 °C gave clear supernatant; 100 µL of clear supernatant was collected and 20 µL of this was injected into HPLC system (Shimadzu LC-20AD, Prominence Liquid Chromatograph, Shimadzu Corporation, Tokyo, Japan). Separation of RM was achieved using endcapped C18 column (Kromasil C18, 250 mm long and 4.6 mm internal diameter, particle size 5 µm, AkzoNobel, Brewster, NY) equipped with a guard column of same packing material. Mobile phase consisted of 10 mM ammonium acetate buffer and acetonitrile in the ratio of 40:60. Prior to analysis, the HPLC system was stabilized for 1 h at 1 mL/min flow rate, through baseline monitoring. RM was monitored at wavelength of 265 nm. Retention time for RM was 5.64 ± 0.19 min with an acceptable tailing factor of 1.11 ± 0.01. Calibration curves (n = 6) were plotted by spiking RM solution in blank rabbit plasma. The mean slope of the curve was calculated as 2006.06 ± 12.51, mean intercept as 151.81 ± 33.51 and regression co-efficient (r 2) as 0.997. Limit of quantification was 0.472 µg/mL. Recovery studies indicated near full recovery (99.47 ± 1.61) of RM form plasma samples, thus precluding the use of an internal standard. All plasma samples were analyzed at ambient temperature.
Estimation of RM in brain tissue of rats
Male Wistar rats (n = 9), aged 4–5 weeks, weighing 200–250 g were selected for this study. They were divided into three groups (two test groups and one control group) with equal number of rats in each group (n = 3). Under shallow ether anesthesia, rats from test group were administered either nasal solution of RM in saline (equivalent to dose of 4 mg/kg) or RM-loaded TM gel (equivalent to dose of 4 mg/kg), intra-nasally with a micropipette (10–100 µL) inserted 0.5 cm into the nostril. Control group received similar treatment with drug-free physiological saline. Rats were held from the back in slanted position during and up to 5 min after nasal administration. They were then humanely sacrificed at different time intervals (30, 60 and 120 min; n = 3 per time point) and blood collected by cardiac puncture. Brain samples were isolated from rats, washed twice with physiological saline and weighed. Equal weight of drug-free, high purity water was added and samples were homogenized using a tissue homogenizer (Remi, Mumbai, India). Two hundred microliters of brain homogenate were then collected and treated with 400 µL of ACN, vortex-mixed for 5 min and centrifuged at 8500 × g for 20 min at 4 °C. The clear supernatant (20 µL) was collected and injected into HPLC system without further processing; concentration of RM was estimated using a previously developed, validated method. Chromatogram obtained from blank processed brain sample was used to ensure absence of interfering peaks. The limit of quantification for RM in brain tissue matrix was 1 µg/mL and retention time of RM was 27.3 min.
Statistical analysis and treatment of data
Following pharmacokinetic parameters were determined from the in - vivo studies in rabbits: maximum plasma drug concentration (C max), time taken to achieve maximum drug concentration (T max), total area under the curve – AUC0 –∞ (by linear trapezoid rule) and elimination rate constant (K e). The values of AUC0 –∞ for nasal gels and oral solution were used to determine the difference in bioavailability by these routes. Other parameters like elimination half-life (T 1/2) were suitably calculated.
All the results are expressed as mean ± standard deviation (SD) of three determinations. Statistical tests of significance were performed using one-way analysis of variance (ANOVA) with multiple comparisons by Dunnett's multiple comparison test method. Significance value was set at p < 0.05. Unpaired Student’s t test was also used for comparison between two data sets.
Results
Sol–gel transition temperature (T sol→gel)
Sol–gel transitions play a critical role in establishing effectiveness of the formulations. Initial investigations showed that addition of RM did not significantly alter the sol–gel temperature of TM gels. RM gels with different weight ratios of P407:P188 (1:1, 2:1 and 1:2) were prepared and evaluated for T sol→gel (data not shown). Apparently, ratio of P407 to P188 affected gelation temperature of the formulations. Gels formulated with P407:P188 (1:1; each polymer used at 15% w/v in formulation) showed T sol→gel in the range of 27.2–29.8 °C. However, formulations with 1:2 ratios were still fluid in this temperature range. Conversely, formulations with 2:1 ratio underwent T sol→gel transitions below room temperature. Therefore, only formulations with P407:P188 (1:1) were selected for further study. From the results, proportion of P407 in the formulation inversely affected gelation temperature. Moreover, increase in P407 also reduced the concentration of P188 required to form stable gels. In summary, combination of P407 and P188 in equal proportions (15% w/v) yielded T sol→gel in the required range. Similar observations have been previously reported (Choi et al., Citation1998).
Effect of mucoadhesive polymer on the T sol→gel was established by evaluating two different types of polymers (CP and chitosan) at two levels (0.1% and 0.3% w/v). The polymers evaluated were chitosan (cationic polymer) and CP (anionic polymer). While CP had no significant (p < 0.05) impact on gelation temperature, chitosan significantly (p < 0.05) increased T sol→gel temperature of poloxamer gel at both the evaluated concentrations (0.1% and 0.3% w/v). This data is presented in . Similar results have also been previously reported by Zaki et al. (Citation2007).
In - vitro release studies from TM gels
This study was performed to understand the in - vitro drug release behavior of different formulations. The release profiles for RM from different TM gels are shown in . Time period of 4 h was selected because maximum residence time for formulations in nasal cavity is reported to be 4 h (Ugwoke et al., Citation2001). However, within 4 h, complete drug release was not achieved for some formulations. Therefore, mean dissolution time (MDT) upto 4 h was considered for comparison of drug release profiles. The goodness of fit and drug release mechanism were calculated using Microsoft Excel Ad-in program, DDSolver (Zhang et al., Citation2010). The drug release profiles of pure RM solution and gels containing 0.3% w/v mucoadhesive polymer (CP/chitosan) are shown in . It took less than 30 min for solution formulation to completely release RM into the media. In general, it was found that, compared to control (gel without mucoadhesive polymer), addition of mucoadhesive polymer retarded drug release from gels (data not shown). The MDT values for TM gels containing CP (0.3% w/v) and chitosan (0.3% w/v) were 147.21 ± 2.41 min and 146.24 ± 1.68 min, respectively. Drug release data and other related parameters are summarized in . The n values between 0.57 and 0.54 indicate non-Fickian release mechanism from TM gels. This type of release behavior may be attributed to hydrogel network (with aqueous pores) formed by poloxamer. The drug is thought to be released by diffusion mechanism by extra-micellar water channels in the gel matrix (Nair & Panchagnula, Citation2003). It is also reported that along with diffusion, dissolution of gel can additionally contribute to drug release (Moore et al., Citation2000).
Figure 1. In-vitro drug release profile of RM from intranasal TM gels with 0.3% w/v of different mucoadhesive polymers in pH 6.4 phosphate-buffered saline solution. Values are expressed as the mean of three measurements.
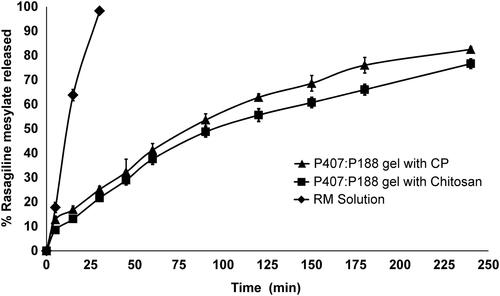
Table 2. Drug release data and mechanism of release from intranasal TM gels of rasagiline mesylate.
In - vivo MTT of TM gels
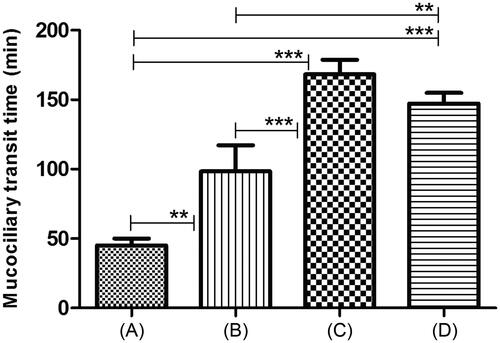
This study was performed to assess and compare in - vivo mucoadhesive properties of different formulations. From the results (), plain poloxamer gel showed significant increase (p < 0.05) in MTT value than control (aqueous dye solution). Type of mucoadhesive polymer also affected the MTT values. Both formulations containing mucoadhesive polymers displayed significantly (p < 0.05) higher MTT than plain poloxamer gels (). However, no significant difference was observed in mean MTT values of formulations containing different mucoadhesive polymers.
Figure 2. In-vivo MTT of intranasal TM gels. (A) Control: aqueous dye solution (3 mg/mL). (B) Plain poloxamer (P407:P188) gel without mucoadhesive polymer. (C) Poloxamer (P407:P188) gel with carbopol 934 P (0.3% w/v). (D) Poloxamer (P407:P188) gel with chitosan (0.3% w/v). **Indicates a very significant difference in the compared values at p < 0.05. ***Indicates extremely significant difference in the compared values at p < 0.01.
Nasal mucosal integrity by morphological observation
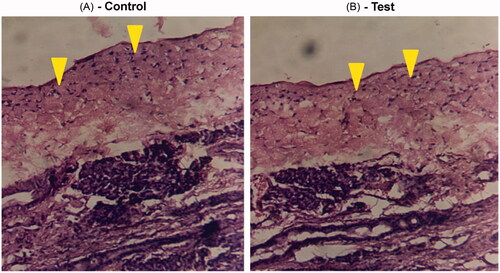
This study was performed to assess the extent of nasal irritation/damage that could be caused by chronic use of TM gel formulation. shows stained photomicrographs taken from anterior cross-section of rat nasal cavity following two-week exposure to TM gel formulations. Examination of control side and dosed side shows intact ciliary epithelium and normal goblet cells. No signs of irritation or edema were observed. Signs of epithelial necrosis, sloughing or hemorrhage were undetected indicating that TM gels did not cause toxicity or tissue necrosis upon chronic administration.
Figure 3. Light photomicrograph of anterior cross-section of rat nasal cavity following fourteen day exposure to intranasal TM gel. (A) Photomicrograph from left nostril (control) of the rat that was not dosed. (B) Photomicrograph from right nostril of the same rat which was dosed with intranasal gel. Arrows show intact epithelium in both the cases.
Pharmacokinetic studies in rabbits
To assess the in - vivo performance of TM gels, pharmacokinetic studies were performed in New Zealand white rabbits. From the plasma concentration profile (), apparently, both intranasal TM gel formulations demonstrated better bioavailability for RM than oral solution. In both these formulations, maximum plasma concentration (C max) was significantly (p < 0.05) higher than oral solution. However, no change was observed in T max values of the formulations. Dose normalized AUC0 –∞ values () for RM were significantly higher (p < 0.05) in TM gel formulations than oral solution. The bioavailability (F rel) of RM from TM gel formulation containing chitosan and CP was 6.05-folds and 4.35-folds higher than oral solution, respectively.
Figure 4. Mean plasma concentration–time profiles following administration of oral solutions and intranasal TM gels of rasagiline mesylate in rabbits. Values are expressed as the mean of three measurements.
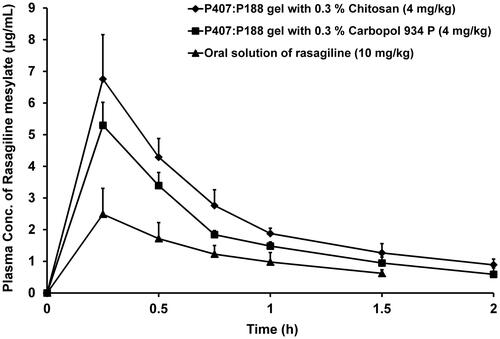
Table 3. Comparative pharmacokinetic parameters of rasagiline mesylate following the administration of oral solution and intranasal TM gels in rabbits.
Estimation of RM in brain tissue of rats
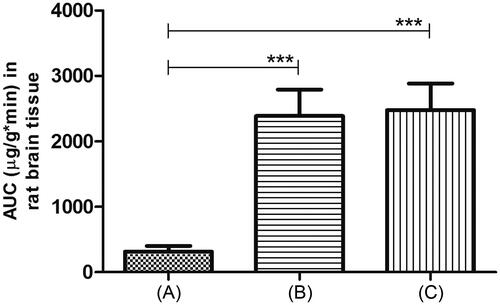
This study was done to examine uptake of RM into brain tissue after intranasal dosing of TM gels. We compared AUCs of RM in rat brain tissue after intranasal administration of these gels. Aqueous intranasal RM solution was used as a control. The results are illustrated in . From the figure, apparently, both TM gels showed significantly (p < 0.01) higher AUCs than control solution.
Figure 5. Localization of rasagiline mesylate in brain tissue after intranasal administration in rats. (A) Control: drug solution in saline. (B) RM-loaded poloxamer (P407:P188) gel with carbopol 934 P (0.3% w/v) and (C) RM-loaded poloxamer (P407:P188) gel with chitosan (0.3% w/v). In all cases, dose was 4 mg/kg. Values are expressed as the mean of three measurements. ***Indicates extremely significant difference in the compared values at p < 0.01.
Discussion
The nasal mucosal temperature has been reported to be between 32 and 35 °C (Dahl & Mygind, Citation1998; Merkus et al., Citation1998) which is significantly different from overall body temperature. Therefore, the aim of our experiment was to develop in - situ gelling systems that can remain as liquid at room temperature (for ease of administration) and rapidly form a gel after touching the nasal mucosa. The acceptable gelling temperature range for in-situ TM gels was set as 28–32 °C. Drug loading into gels was adjusted such that each 100 µL of gel contained 2 mg of RM. As RM is highly water soluble, it was expected to affect the gelling mechanism by interfering with micellar association of in - situ TM gels. This in turn could have impact on gelation temperature (Yun et al., Citation1999; Abd ElHady et al., Citation2003). However, from our experiment, apparently, RM did not alter gelation temperature of poloxamer gels significantly. It is also reported that mucoadhesive polymers can have significant impact on gelation temperature (Choi et al., Citation1999). In our study, only chitosan caused a significant increase in the gelation temperature. This was attributed to the use of acetic acid as solvent for dissolving chitosan. Acetic acid is known to weaken the hydrogen bonds between close packed arrangements of poloxamer gels causing an increase in gelation temperature (Choi et al., Citation1999).
In - vitro drug release profiles indicated decrease in release of RM with increasing mucoadhesive polymer concentration. As mucoadhesive polymer concentration increases, viscosity of the gel increases. This decreases diffusion of the drug through gel matrix, thereby reducing release rate. Our conclusions agree with some previously reported works (Desai & Blanchard, Citation1998). Squeezing effect of mucoadhesive polymers (Ryu et al., Citation1999) on the aqueous channels of poloxamer micelles (through which the drug has to diffuse) also contributes to retardation in drug release. The non-Fickinan release kinetics as evident from the n values (between 0.57 and 0.54) indicates that release of drug is both due to diffusion of drug through gel matrix and swelling of the gel matrix (Ritger & Peppas, Citation1987; Chang et al., Citation2002).
The in - vivo MTT studies were used to decipher in - vivo adhesion behavior of TM gels. Addition of mucoadhesive polymer to plain poloxamer gel helped in retaining the formulation in nasal cavity for longer time period. It is reported that, positive spreading co-efficient and rheology of the liquid play important role in spread and retention of gels on the mucosae. To spread on the mucosae, formulation should be sufficiently mobile. However, too mobile formulations get easily dislodged from the site of application resulting in failure of delivery system. Hence, formulations demonstrating in - situ gelling can be retained at the site of administration for extended period of time (Andreas, Citation2005). It is also known that, in dilute solutions, mucoadhesive polymers (CP and chitosan) can remain bound to mucous membrane for at least 2 h (Kockisch et al., Citation2001). Mucoadhesive polymers can collect at gel–mucus interface and form many weak bonds. In case of cationic polymer like chitosan, the positive charge could also favor binding to negatively charged mucosal surface (Fiebrig et al., Citation1995).
Nasal mucosal integrity studies showed no damage to nasal mucosa of rats even after chronic administration of TM gels. However, these results must be viewed with sagacity. A complete evaluation of nasal toxicity must consider irritation of nasal mucosa, damage to nasal epithelial cells, and also the effects on nasal ciliary function (motility). For this, simultaneous evaluation with animal models like in - situ toad palate model and rat nasal mucociliary model has been suggested (Cai et al., Citation2011).
From the pharmacokinetic study in rabbits, apparently, both intranasal TM gels showed significant (p < 0.05) improvement in bioavailability of RM. It is reported that, upon oral administration, RM is rapidly absorbed and undergoes extensive first pass metabolism by CYP 450 family of enzymes. This limits its bioavailability when administered by oral route. However, intranasal administration of the same drug bypasses the first-pass effect, thus improving its bioavailability. Moreover, longer residence time and intimate contact with nasal epithelium (due to mucoadhesive behavior of TM gels) provided better chance for drug absorption from nasal cavity. While higher C max values could be attributed to rapid absorption of the drug, higher AUC0 –∞ values indicate near complete absorption of drug from TM gel formulations.
The brain distribution study was performed in rats. Distribution of RM from intranasal gels was found to be significantly (p < 0.01) higher than control nasal solution of the drug. Apparently, this increase in AUC0 – t can be correlated with MTT values of the formulations. It was observed that, for formulations with higher MTT (greater residence time), AUC0 – t increased. In case of control (RM nasal solution), much of the dissolved drug was lost from the site of administration by mucociliary clearance (Ugwoke et al., Citation2005). It is thought that the drug reaches brain tissue via olfactory pathway (Wu et al., Citation2008). So, if the retention time of drug in nasal cavity is adequate, chances for the drug to access olfactory pathway increases. However, we need more comprehensive studies to discern the drug’s path to the brain. Moreover, distribution pattern of the drug in brain tissue can significantly affect the overall effectiveness of these intranasal TM gel formulations.
Conclusion
We have demonstrated that, for the anti-Parkinson drug RM, intranasal thermosensitive mucoadhesive gels could prove to be useful alternatives to oral formulations. The intranasal gels showed four- to six-folds increase in bioavailability of the drug than oral solution. It was also shown that these gels were non-irritant to nasal mucosa even after chronic use. From the brain distribution studies in rats, we have shown that significant amount of drug could be delivered to the brain tissue by administering the drug through intranasal gels.
Declaration of interest
The authors declare that they have no conflicts of interest to disclose.
References
- Abd ElHady SS , Mortada N , Awad GAS , et al . (2003). Development of in-situ gelling and mucoadhesive mebeverine hydrochloride solution for rectal administration. Saudi Pharm J 11:159–71
- Andreas B . (2005). Mucoadhesive polymers: strategies, achievements and future challenges. Adv Drug Deliv Rev 57:1553–5
- Arora P , Sharma S , Garg S . (2002). Permeability issues in nasal drug delivery. Drug Discov Today 7:967–75
- Azilect T . (2010). AZILECT; Product Monograph, Control no: 13427; Israel: Teva Pharmaceutical Industries Ltd
- Belgamwar VS , Patel HS , Joshi AS , et al . (2011). Design and development of nasal mucoadhesive microspheres containing tramadol HCl for CNS targeting. Drug Deliv 18:353–60
- Bukka R , Prakasam K , Patel CD . (2010). Preparation and evaluation of intraoral drug delivery system for rasagiline mesylate. Int J Pharm Sci Drug Res 2:294–301
- Cai Z , Song X , Sun F , et al . (2011). Formulation and evaluation of in situ gelling systems for intranasal administration of gastrodin. AAPS PharmsciTech 12:1102–9
- Chang JY , Oh Y , Choi HG , et al . (2002). Prolonged antifungal effects of clotrimazole-containing mucoadhesive thermosensitive gels on vaginitis. J Control Release 82:39–50
- Chen JJ , Swope DM , Dashtipour K . (2007). Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson's disease. Clin Ther 29:1825–49
- Choi H , Jung J , Ryu J , et al . (1998). Development of in situ-gelling and mucoadhesive acetaminophen liquid suppository. Int J Pharm 165:33–44
- Choi HK , Lee M , Kim M , Kim C . (1999). Effect of additives on the physicochemical properties of liquid suppository bases. Int J Pharm 190:13–9
- Dahl R , Mygind N . (1998). Anatomy, physiology and function of the nasal cavities in health and disease. Adv Drug Deliv Rev 29:3–12
- Deng J, Lin J, Xiao J. (2007). Transdermal patch containing rasagiline for treatment or prophylaxis of nervous system disease and its preparation process. WIPO Patent WO2007101400 A1
- Desai SD , Blanchard J . (1998). In vitro evaluation of pluronic F127-based controlled-release ocular delivery systems for pilocarpine. J Pharm Sci 87:226–30
- Dumortier G , Grossiord JL , Agnely F , Chaumeil JC . (2006). A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res 23:2709–28
- Fiebrig I , Harding SE , Rowe AJ , et al . (1995). Transmission electron microscopy studies on pig gastric mucin and its interactions with chitosan. Carbohydr Polym 28:239–44
- Guay DR . (2006). Rasagiline (TVP-1012): A new selective monoamine oxidase inhibitor for Parkinson's disease. Am J Geriatr Pharmacother 4:330–46
- Illum L . (2000). Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci 11:1–18
- Illum L . (2003). Nasal drug delivery: Possibilities, problems and solutions. J Control Release 87:187–98
- Jain SK , Chourasia MK , Jain AK , et al . (2004). Development and characterization of mucoadhesive microspheres bearing salbutamol for nasal delivery. Drug Deliv 11:113–22
- Khan S , Patil K , Bobade N , et al . (2010). Formulation of intranasal mucoadhesive temperature-mediated in situ gel containing ropinirole and evaluation of brain targeting efficiency in rats. J Drug Target 18:223–34
- Kockisch S , Rees G , Young S , et al . (2001). A direct-staining method to evaluate the mucoadhesion of polymers from aqueous dispersion. J Control Release 77:1–6
- Kumar M , Misra A , Babbar AK , et al . (2008). Intranasal nanoemulsion based brain targeting drug delivery system of risperidone. Int J Pharm 358:285–91
- Lale AM , Mason JD , Jones NS . (1998). Mucociliary transport and its assessment: a review. Clin Otolaryngol 23:388–96
- Lin J, Li Q, Li H, et al. (2009). A stable and release-controlled rasagiline transdermal patch and method of preparation thereof. WIPO Patent WO2009152777 A1
- Majithiya R , Ghosh P , Umrethia M , Murthy R . (2006). Thermoreversible-mucoadhesive gel for nasal delivery of sumatriptan. AAPS PharmsciTech 7:E80–6
- Merkus FW , Verhoef JC , Schipper NG , Marttin E . (1998). Nasal mucociliary clearance as a factor in nasal drug delivery. Adv Drug Deliv Rev 29:13–38
- Miyamoto M , Natsume H , Iwata S , et al . (2001). Improved nasal absorption of drugs using poly-L-arginine: effects of concentration and molecular weight of poly-L-arginine on the nasal absorption of fluorescein isothiocyanate-dextran in rats. Eur J Pharm Biopharm 52:21–30
- Moore T , Croy S , Mallapragada SK , Pandit NK . (2000). Experimental investigation and mathematical modeling of Pluronic F127 gel dissolution: drug release in stirred systems. J Control Release 67:191–202
- Nair V , Panchagnula R . (2003). Poloxamer gel as vehicle for transdermal iontophoretic delivery of arginine vasopressin: evaluation of in vivo performance in rats. Pharmacol Res 47:555–62
- Pardeshi CV , Belgamwar VS . (2013). Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: an excellent platform for brain targeting. Expert Opin Drug Deliv 10:957–72
- Peppas NA . (1985). Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv 60:110–11
- Pisal SS , Paradkar AR , Mahadik KR , Kadam SS . (2004). Pluronic gels for nasal delivery of vitamin B12. Part I: preformulation study. Int J Pharm 270:37–45
- Ravi PR , Aditya N , Cherian L , Patil S . (2013). LC method for determination of rasagiline mesylate in different plasma matrices and its application to oral pharmacokinetic study in rabbits. J Chrom Sci 51:1–7
- Ritger PL , Peppas NA . (1987). A simple equation for description of solute release I. Fickian and non-fickian release non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release 5:23–36
- Romeo VD , deMeireles J , Sileno AP , et al . (1998). Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev 29:89–116
- Ryu J , Chung S , Lee M , et al . (1999). Increased bioavailability of propranolol in rats by retaining thermally gelling liquid suppositories in the rectum. J Control Release 59:163–72
- Tengamnuay P , Sahamethapat A , Sailasuta A , Mitra AK . (2000). Chitosans as nasal absorption enhancers of peptides: comparison between free amine chitosans and soluble salts. Int J Pharm 197:53–67
- Ugwoke MI , Verbeke N , Kinget R . (2001). The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J Pharm Pharmacol 53:3–21
- Ugwoke MI , Agu RU , Verbeke N , Kinget R . (2005). Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev 57:1640–65
- Ugwoke MI , Exaud S , Van Den Mooter G , et al . (1999). Bioavailability of apomorphine following intranasal administration of mucoadhesive drug delivery systems in rabbits. Eur J Pharm Sci 9:213–19
- Ugwoke MI , Agu RU , Vanbilloen H , et al . (2000). Scintigraphic evaluation in rabbits of nasal drug delivery systems based on carbopol 971P® and carboxymethylcellulose. J Control Release 68:207–14
- Wu H , Hu K , Jiang X . (2008). From nose to brain: Understanding transport capacity and transport rate of drugs. Expert Opin Drug Deliv 5:1159–68
- Yun M , Choi H , Jung J , Kim C . (1999). Development of a thermo-reversible insulin liquid suppository with bioavailability enhancement. Int J Pharm 189:137–45
- Zaki NM , Awad GA , Mortada ND , Abd Elhady SS . (2007). Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharm Sci 32:296–307
- Zhang Q , Jiang X , Jiang W , et al . (2004). Preparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brain. Int J Pharm 275:85–96
- Zhang Y , Huo M , Zhou J , et al . (2010). DDSolver: an add-in program for modeling and comparison of drug dissolution. AAPS J 12:263–71
- Zhou M , Donovan MD . (1996). Intranasal mucociliary clearance of putative bioadhesive polymer gels. Int J Pharm 135:115–25