
Abstract
This study aims to develop oseltamivir phosphate (OP) liposomes as inhalation powders by spray-drying based on the single factor investigation, which was mainly composed of lactose, l-leucine and mannitol. It was found that the ratio of OP and liposomes (1:10), inlet temperature (110 °C) and airflow rate (2.3 mL/min) showed optimized physical properties of OP liposomes. Deposition was evaluated after the aerosolization of powders at 600 L/h via the Aerolizer® into a twin-stage impinger. The concentrations of OP and oseltamivir carboxylate (OSCA) in rats plasma using LC-MS have been determined and performed via pharmacokinetic software DAS 2.0 package. The liposomal OP dry powders displayed an average particle size around 3.5 µm with fine particle fraction (FPF = 35.40%). In vitro evaluation demonstrated a sustained release pattern accounting for 20% drug release compared to that of OP solution up to 90% drug release in 2 h. And the cumulative release percentage was up to 50% in 20 h. Atrioventricular fitting results indicated that all preparations were best fitted with a two-compartment model. There was a significant difference in MRT, Cmax and Tmax (p < 0.01) between the two groups of liposomal OP dry powders and OP solution with t-test, which indicated that the drug released slowly from liposomal OP dry powders in the lung. To sum up, dry powders formulation of liposome-encapsulated OP for inhalation was suitable for pulmonary administration, which offering the opportunity to reduce dosing frequency.
Introduction
Pandemic influenza is considered to be one of the most serious potential global public health threats caused by influenza virus. Oseltamivir phosphate (OP) was recommended by the World Health Organization (WHO) for both treatment and prophylaxis of influenza. As an important first-line defense in the event of a flu pandemic, it has been licensed to Hoffmann-La Roche Ltd. under the trade name Tamiflu® (Davies, Citation2010).
As a pro-drug, OP can be readily absorbed from the gastrointestinal tract whose absolute bioavailability is about 75–80% and extensively converted by hepatic carboxylesterases to the active metabolite oseltamivir carboxylate (OSCA), a potent and selective inhibitor of influenza virus neuraminidase (Heinig & Bucheli, Citation2008; Dutkowski et al., Citation2010) that is essential for the release of progeny influenza viruses from infected host cells, which in turn prevents infection of new host cells and viral spread throughout the respiratory tract (Ghosh et al., Citation2010). A wide-ranging program of clinical studies demonstrated that OP has a good overall tolerability and safety profile (Dutkowski et al., Citation2003). However, besides nausea, vomiting and diarrhea, some adverse reactions of central nervous system, such as dizziness, headache, fatigue and agrypnia affected some of patients after oral delivery of OP, which remains a major concern in the clinical use of OP (Maxwell, Citation2007; Dalvi et al., Citation2011).
Pulmonary delivery is a promising route for drug administration, both for effective local therapy and for systemic administration (Al-Hallak et al., Citation2011). Inhalation powders are widely accepted and currently the most convenient dosage form among all the pulmonary dosage forms (Mansour et al., Citation2009; Harush-Frenkel et al., Citation2010). Zanamivir (Relenza®) has been developed as dry powder inhalation and marketed by GlaxoSmithKline as another neuroaminidase inhibitor.
Furthermore, it was also documented that liposomes offer several attractive features for pulmonary drug delivery (Drulis-Kawa & Dorotkiewicz-Jach, Citation2010; Swaminathan & Enrhardt, Citation2012). They can serve as a solubilization matrix for poorly soluble agents and act as sustained release reservoir, showing good compatibility with lung surfactant components and low local irritation to lung tissue (Drulis-Kawa & Dorotkiewicz-Jach, Citation2010; Chen et al., Citation2012). And, the development of liposomal dry powder inhalers also has been reported in the literature (Huang et al., Citation2010).
OP is commercially available as oral capsules (35, 40 and 75 mg) and granules (25 mg). A dry powder formulation of liposome-encapsulated OP for inhalation will exhibit many advantages over the oral formulations. First, inhalational OP has a targeted effect without diffusing to other unnecessary sites in the body, consequently attaining higher concentrations in the lung especially the epithelial lining fluid, resulting in a reduced drug dose via pulmonary delivery. Second, oral OP therapy is commonly associated with gastrointestinal symptoms and long-term use has been linked with central nervous system impairment, which can be avoided by the delivery of inhalational OP directly to the lung.
This article describes the preparation of a dry powder formulation of liposome-encapsulated OP for inhalation using active loading technique and spray-drying process (Harrigan et al., Citation1993). The physical properties of liposomal OP dry powders, including entrapment efficacy (EE), zeta potential, particle size and distribution, morphology, yield of the powder, angle of repose (θ), powder density and the aerosol performance were all investigated in this study. In vitro release evaluation, metabolism in rat lung, in vivo pharmacokinetic study and tissue distribution were also carried out, which showed that a dry powder formulation of liposome-encapsulated OP for inhalation has the ability of lung targeting.
Materials and methods
Materials
Oseltamivir phosphate was provided by Yuanchenggongchuang Technologies Ltd., Company (Wuhan, China, Batch no. YC20110302-A). OSCA (purity 99%) was synthesized by Pharmaceutical Institute, China Pharmaceutical University (Nanjing, China). Berberine hydrochloride (I.S.) was purchased from National Institute for Food and Drug Control (Beijing, China). Methanol (HPLC grade) was purchased from Nanjing Chemical Reagent Co., Ltd (Nanjing, China). Ovelecithin (PL-100 M injection) was obtained from Evister Pharmaceutical Technologies Ltd. Company (Shanghai, China). Cholesterol (ELISA), l-leucine and mannitol were purchased from Huixing Reagent Co. Ltd. (Shanghai, China). Sephadex G50 was purchased from Luqiaosijia Biochemical Plastic Factory (Taizhou, China). Dichloromethane, acetonitrile, ammonium sulfate, ethanol and triethylamine were HPLC grade and purchased from Hanbang Science and Technology Co. Ltd. (Jiangsu, China). All other chemicals used were analytical grade.
Methods
Preparation of liposomes
Ovelecithin 0.2 g and cholesterol 0.030 g were added to a round flask and dissolved with 10 mL organic solvent composed of dichloromethane/methanol (8/2, v/v). Then uniform film was prepared using film dispersion method (Gregoriadis & Perrie, Citation2010) and hydrated for 1 h. Then it was added to a warm ammonium sulfate solution of 0.3 mol/L (no bulky grains were observed by naked eyes) at certain temperature. The solution prepared above was transferred into a bottle, processed with ultrasonic probe and filtrated with filter membrane of 0.45 and 0.22 µm successively for three times to prepare the blank liposomes. The dialysis sac was loaded with the blank liposomes and immersed into physiological saline with 100-folds volume. Following dialysis for 12 h, it was transferred into a bottle again, mixed with OP solution (10 µg/mL), and then incubated. Then the liposomes loaded with OP were obtained. And the factors most significant affecting the entrapment efficiency of OP liposomes, such as the ratio of OP and blank liposomes, the concentration of ammonium sulfate, the temperature and the time of incubation (labeled as A, B, C and D) were all investigated by orthogonal experimental design as shown in and .
Table 1. Levels and factors in the orthogonal design.
Table 2. The test of orthogonal design for optimization of OP liposome formulation.
Preparation of liposomal dry powder formulations
The prepared formulations were subsequently spray-dried using a Büchi B-191 mini spray dryer equipped with a high performance cyclone (Büchi Laboratory Techniques, Switzerland) under spray flow rate 600 L/h and 100% aspirator setting. The following operating conditions were optimized based on the single factor design: inlet temperature (100, 110 and 120 °C), pump rate (1.15, 2.3 and 3.45 mL/min), the mole ratio of l-leucine and liposome (1:1, 2:1 and 3:1) were investigated respectively.
Characteristics of OP liposomes
Determination of the entrapment efficiency
The entrapment efficiency (EE) of liposomes was determined using micro-column centrifugation method (Gu et al., Citation2011). About 0.2 mL solution of OP liposomes was kept on the Sephadex G-50 gel micro-column for 2 min. Then centrifuged at 500 rpm for 5 min and eluted with 0.5 mL PBS (pH = 7.4) by being centrifuged at 1000 rpm for 5 min. The elution collected was diluted to 5 mL with methanol and determined at 215 nm using ultraviolet spectrophotometry.
Morphology
The morphology of OP liposomes was observed with transmission electron microscope (TEM) and atomic force microscope (AFM).
Determination of particle size and zeta potential
The particle size and zeta potential of OP liposomes were determined by Zeta PALS-Zeta potential measurement analyzer (Brookhaven, USA).
Characteristics of liposomal OP dry powders
Particle size and distribution
The volume mean diameter of inhalation powders was determined using a Malvern Mastersizer 2000 (Malvern Instruments Ltd., UK). The powders sample was dispersed in 10 mL isopropanol and vortex mixed for 3 min. Then the sample was added to the sample cell dropwise, which filled with 500 mL isopropanol. Each sample was analyzed in triplicate.
Microscopic morphology
Microscopic morphology of inhalation powders was evaluated by scanning electron microscope (SEM). The acceleration voltage during observation was set to 15 kV. The sample was placed on copper and gilded, then placed in the sample cell. The microscopic morphology was observed.
Spray-drying yield and drug content
Yield is one important index for evaluating powders, and the high drying temperature may affect the stabilities of excipients and the final yield (Li et al., Citation2013). The yield was calculated by the following formula, where W1 is the weight of product collected and W is the weight of drug and the excipients.
Three batches of liposomal OP dry powders were prepared following the optimized methods described above. About 10 mg from each batch was dissolved in 2 mL deionized water and diluted to 10 mL with methanol. The concentration of drug was determined using HPLC which was described in the experiment of “In vitro release” section. Content uniformity of OP in liposomal dry powders was evaluated by comparison between actual quality and theoretical quality.
Bulk density and angle of repose
The bulk density and the angle of repose as the most frequently used index for evaluating powder characteristics were measured. The bulk density could be obtained from the following formula. As the bulk density is within the range of 0.1–0.4 mg/cm3, the powders can be used for pulmonary drug delivery.
Generally, the flowability of powders is considered to be good when the angle of repose is less than 40°. The angle of repose was measured as described elsewhere (Huang et al., Citation2010).
X-ray diffraction measurement
X-ray diffraction (XRD) of l-leucine, OP and liposomal OP dry powders were carried out with a Siemens D 5000 (Siemens AG, Berlin, Germany). The diffraction intensity was recorded with an angle of 2θ from 5° to 80°. The voltage and current were set to 40 kV and 40 mA, respectively. The total time of the diffraction scan was 19 min and each sample was examined in triplicate. The data was analyzed by Evaluation (EVA) software.
Fine particle fraction measurement
Fine particle fraction (FPF) was determined using the twin-stage impinger (TI, British Pharmacopoeia) to evaluate the deposition of OP in vitro.
Characteristics of rehydration of liposomal OP dry powders
Morphology, particle size, zeta potential and entrapment efficiency
The liposomal OP dry powders were rehydrated with deionized water. Then morphology, particle size, zeta potential and entrapment efficiency were determined as the methods described above.
In vitro release
High performance liquid chromatography
The chromatographic separation was performed on a SinoChrom ODS-BP column (200 × 4.6 mm, 5 μm, Elite Analytical Instrument Co., Ltd., Dalian, China). The mobile phase consisted of PBS and methanol (v/v, 35:65) and pumped at a flow rate of 1 mL/min with a constant column temperature of 40 °C.
Sample preparation
In vitro release of OP was conducted by dialysis in a dialysis sac (10 000–14 000 Mw cut off; Sigma-Aldrich) with PBS (pH 7.4) in 100 mL Erlenmeyer flask at 37 ± 0.5 °C according to the method published previously (Du et al., Citation2010; Panwar et al., Citation2010). Three sacs were prepared containing OP solution, OP liposomes and the solution of liposomal OP dry powders. A 2 mL sample containing OP (1 mg/mL) was poured into the dialysis sac and two ends of the dialysis sac were tightly bound with threads. Then the sacs were hung inside a conical flask. The flask was placed in a shaker with the rotate rate of 100 rpm at 37 °C. At each predetermined time (0.5, 1, 2, 3, 4, 5, 7, 9 and 20 h), 1 mL sample was collected. Subsequent filtrate was transferred to 1 mL centrifugal tube and 20 μL sample was assayed for drug cumulative release at various time points using HPLC as above. Then the curve of drug cumulative release percentage was plotted over time for different preparations.
Metabolism of OP in rat lung
Preparation of tissue homogenates
To investigate whether OP could be hydrolyzed by esterases in the lung, a metabolism study in vitro was proposed using rat liver and lung homogenates. The tissue homogenates were prepared according to the following method. Briefly, a male rat weighing 220 ± 20 g was anesthetized and exsanguinated. The lung and liver were quickly excised, rinsed with physiological saline solution and weighted. They were then chopped with scissors and added to 100 mL of 100 mM Tris–KCl buffer (1.15%). The mixture was then homogenized by a FJ-200 homogenizer (Shanghai Specimens & Models Factory, Shanghai, China) for 3 s in ice bath. The lung and liver homogenates were stored at 1–4 °C until analysis.
High performance liquid chromatography
The chromatographic separation was performed on a SinoChrom ODS-BP column (200 × 4.6 mm, 5 μm, Elite Analytical Instrument Co., Ltd., Dalian, China). The mobile phase consisted of PBS and methanol (v/v, 35:65) and pumped at a flow rate of 1 mL/min with a constant column temperature of 40 °C. The detection wavelength was 295 nm.
Determination of homogenates protein content
Concentrations of protein in the liver and lung homogenates were determined by the Coomassie Blue Staining Method using bovine serum albumin as the standard. The 1000 μL solution prepared by tissue homogenates and distilled water (v/v, 1/9) was added to 5 mL Coomassie Brilliant Blue and placed for 5 min. Then the absorbance of solution was detected by ultraviolet–visible spectroscopy with the detection wavelength of 590 nm. The concentrations of protein in tissue homogenates were calculated using the absorbance and the standard curve established. Analysis was carried out on a 752 UV–visible spectrophotometer (Jinghua Instruments, Shanghai, China).
Determination of Vmax and Km in liver and lung homogenates
A 500 μL lung homogenates, liver homogenates and Tris–KCl buffer solution (control group) were added to 100 μL OP solution with different concentration of 44, 66, 132, 264 and 396 μg/mL and incubated at 37 °C, respectively. Then 100 μL homogenates were added to 10 μL berberine hydrochloride (I.S.) and 190 μL acetonitrile, then vortex mixed for 3 min. After refrigerated centrifugation at 10 000 rpm for 5 min (4 °C), 50 μL supernatant was analyzed by HPLC.
For there is only one metabolite of OP (Nicholson et al., Citation2000; Dutkowski et al., Citation2010; Lindemann et al., Citation2010), the velocity of the reaction (v) was measured as the appearance of OSCA. The metabolism velocity of OP was calculated according to the following Equation (Equation1(1) ), where C and V are the concentration of OSCA after incubation and the volume of incubated solution, respectively. MOSCA is the molecular weight of OSCA. S is the concentration of protein in tissue homogenates. T is the time of incubation.
(1)
Then the curve was plotted for different tissues. Km and Vmax were calculated according to the Equation (Equation2(2) ).
(2)
Pharmacokinetics in rats
Liquid chromatography–mass spectrometry
The chromatographic separation was performed on a Hypersil 300A C18 column (250 × 4.6 mm, 5 μm, Elite Analytical Instrument Co., Ltd., Dalian, China). The mobile phase consisted of methanol/water/formic acid (v/v/v, 50/50/1) and pumped at a flow rate of 0.5 mL/min with a constant column temperature of 30 °C. The precursor ions and product ions were ascertained for use in MRM. Meanwhile the electrospray interface (ESI) was employed for good sensitivity and fragment ion were obtained. The ion spray voltage was 30 kV and the source temperature was 350 °C. The multiple reaction monitoring (MRM) analysis was applied in a positive ionization mode to detect ion transitions of OP (m/z 313 → 166), vanillin (m/z 153 → 93) and OSCA (m/z 285 → 138), respectively.
Plasma sample preparation
Ten adult male SD rats weighing 200–220 g were maintained under normal conditions with free access to food and water and fasted for 12 h before the experiments. Then they were randomly divided into two groups (five per group) which were labeled as group a and group b. Group a was administered with OP solution orally at a dose of 12 mg/kg. Group b was administered with liposomal OP dry powders by endotracheal route via a tubing-needle-syringe arrangement with some modification at a dose of 12 mg/kg. After administration, 0.5 mL blood samples of 10 rats were collected by retro-orbital sinus subsequently at each predetermined time (0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6 and 8 h) into heparin-treated tubes. After being processed, the samples were determined using LC–MS (C18 column; mobile phase, methanol/water/formic acid (v/v/v, 50/50/1); flow rate, 0.5 mL/min; column temperature, 30 °C). The pharmacokinetic modeling to obtain the “best” parameters was performed as judged by non-linear least-squares criterion with Akaike’s information criterion (AIC) values using software program DAS 2.0 package (BioGuider Co., Shanghai, China).
Rats tissue distribution
Sixteen adult male SD rats weighing 200–220 g were randomly divided into two groups (eight per group) labeled as group c and group d. Group c was administered with OP solution of 12 mg/kg orally and group d was administered with liposomal OP dry powders by endotracheal route via a tubing-needle-syringe arrangement with some modification at a dose of 12 mg/kg. At each predetermined time (1, 1.5, 3, 5, 8 and 12 h) after administration, the rats were immediately sacrificed by exsanguinations. Then the lungs of rats were isolated. Tissue samples were rinsed with physiological saline solution, weighed accurately and stored at −20 °C until analysis. The tissue samples were analyzed using LC–MS in a manner described above. The content of OSCA (Clung) was calculated according to the following Equation (Equation3(3) ), where mlung is the weight of lung and Chomogenate, Clung are the OP concentration of lung homogenate and lung, respectively. Then Clung and AUC (area under the tissue concentration–time curve) were compared between the two groups.
(3)
Results and discussion
Preparation of liposomes
Factors affecting the EE of OP liposomes have been investigated based on orthogonal experimental design as shown in and . The ranking of the four factors in this experiment was A > B > D > C. For all the experiments in the orthogonal design, the encapsulation efficiencies were within a range of 23–91%. Accordingly, the optimized formulation was selected. And, the optimal preparation and formulation was A3B3C1D3: the ratio of OP and liposomes was 1:10, the concentration of (NH4)2SO4 was 0.3 mol/L, the incubation time and temperature were 25 min and 40 °C, respectively.
Table 3. Results of the orthogonal design of OP liposomes.
Table 4. The entrapment efficiency of liposomes (n = 3).
Microscopic morphology of OP liposomes
The surface topography of OP liposomes was observed with TEM and AFM as displayed in , respectively. The size distribution of the liposomes before and after drug-loaded was around 100 nm. Both the blank liposomes and OP liposomes were uniformly distributed and good dispersibility as indicated in the images of TEM. The particle size, zeta potential and polydispersity index were demonstrated in . The zeta potential was −13.65 ± 1.22 mV. Liposomes presented a negative surface charge that would enhance the colloidal stability of the nanoparticles (Du et al., 2010). l-Leucine could be efficiently adsorbed at the nanoparticle surface, which contributes to conceal recombination of nanoparticles.
Figure 1. TEM image of blank liposomes.

Figure 2. TEM image of OP liposomes.
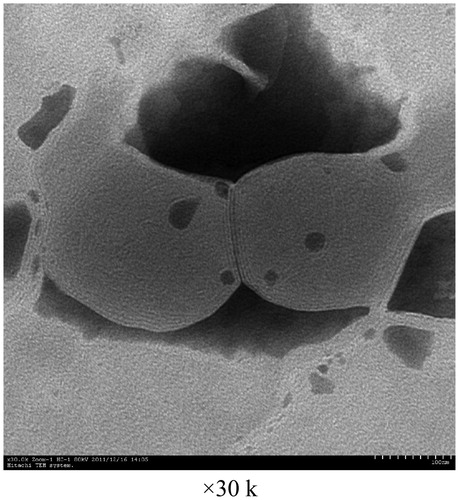
Figure 3. AFM image of blank liposomes.
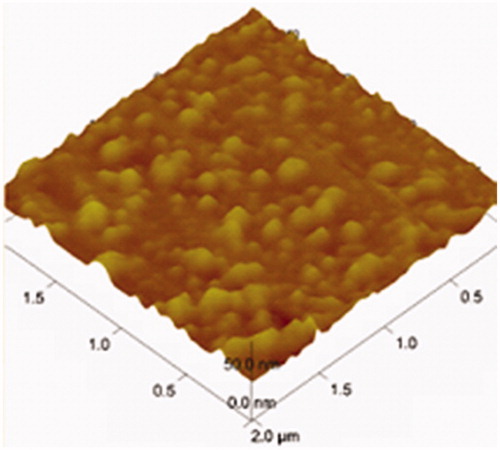
Figure 4. AFM image of OP liposomes.
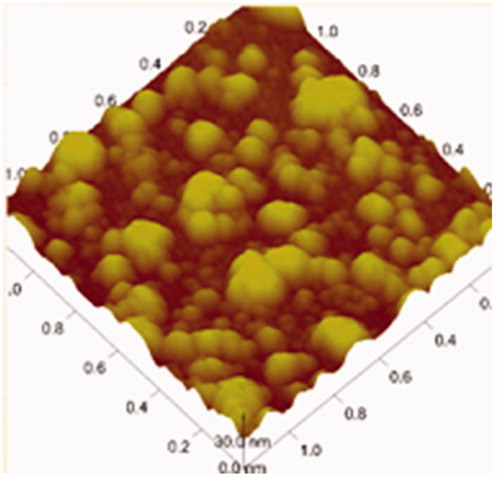
Table 5. Particle size, polydispersity index and zeta potential of OP liposomes.
Optimization of preparation and formulation
Based on the single-factor experimental design, the optimized formulation was selected. According to the results of pre-experiments, when inlet temperature was 110 °C and the pump rate was 2.3 mL/min, the EE, yield and the angle of repose were optimized. As shown in , the formulation was optimal when the ratio of l-leucine and solid content was 1:1.
Table 6. The physical and chemical properties of liposomal OP dry powders as different ratio of l-leucine and solid content liposome.
Properties of liposomal OP dry powders
The microscopic morphology of liposomal OP dry powders was imaged by SEM as shown in . The yield, bulk density and angle of repose were displayed in . The liposomal OP dry powders displayed an average particle size around 3.5 µm and the angle of repose was 29.3°. As shown in , some difference was found between the solution of liposomes and liposomal OP dry powders at 4 °C for 30 days. The results demonstrated that the storage stability of OP liposomes can be improved when prepared as dry powders by spray-drying. As shown in , there was no significant difference between theoretical content and measured content (RSD = 0.012), which demonstrated OP was dispersed in powder uniformly.
Figure 5. SEM images liposomal OP dry powders.

Table 7. Properties of optimized liposomal OP dry powders formulation.
Table 8. The solution of liposomes before and after spray-dried at 4 °C.
Table 9. Drug content uniformity of liposomal OP dry powders.
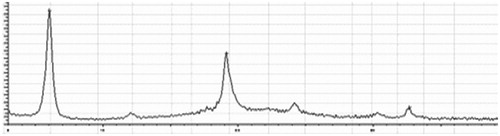
X-ray diffraction spectrum
The spectrums of OP, l-leucine and liposomal OP dry powders were shown . No crystal diffraction of drug and only diffraction of l-leucine were shown in the spectrum of liposomal OP dry powders as compared with the other figures, which illustrated that OP was almost completely encapsulated in liposomes.
Figure 6. X-ray diffraction spectrum of OP.
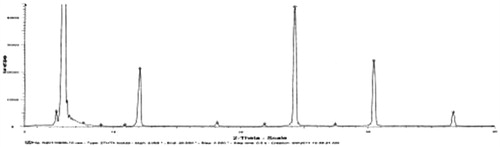
Figure 7. X-ray diffraction spectrum of L-Lecuine.
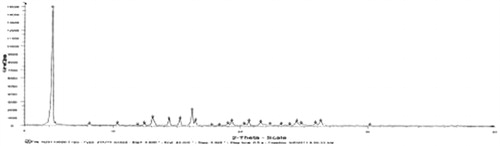
Figure 8. X-ray diffraction spectrum of liposomal OP dry powders.
Microscopic morphology of liposomes resolution
Liposomes using TEM and AFM were observed as shown in , which shown the liposomes had spherical shapes. showed that there were some irregular, globular shapes with rough surfaces on the blank liposomes. When the OP liposomes were spray-dried, the resulting particles () were surprisingly distinct, forming non-aggregating spherical particles. And, the surface of particles appeared dimpled or corrugated, which have been reported as appropriate morphologies for reducing particle–particle cohesion.
Figure 9. TEM image of blank liposomal dry powders.

Figure 10. TEM image of liposomal OP dry powders.
Figure 11. AFM image of blank liposomal dry powders.

Figure 12. AFM image of OP liposomal dry powders.
Fine particle fraction
Fine particle fraction (FPF) characterized the efficiency of drug deposited in the lower respiratory tract. The flowability of the dry powders is an important factor affecting its in vitro deposition performance (Huang et al., Citation2010). The value of FPF was 35.4% for this formulation, which demonstrated liposomal OP dry powders could be distributed in the lung well.
Evaluation of drug release in vitro
The result of the accumulative release profile of OP was shown in . OP was released from solution was investigated as control. OP was continuously released from the liposomal dry powders and liposomes in PBS (pH 7.4) for 20 h at 37 ± 0.5 °C. The initial release of around 20% of the drug from liposomes was observed in the first 2 h, which probably due to the portion of the drug that leaked out of liposomes and the unloaded drug (Glavas-Dodov et al., Citation2005). Subsequently, the release of drug from liposomes was slower and about 50% of the drug remained encapsulated in liposomes after 20 h of dialysis. The delayed release might be attributed to the liposomal membrane made of cholesterol and phospholipids and the drug encapsulated in lipid membrane that released mainly through dissolution and diffusion from the lipid bilayer (Cocera et al., Citation2000). Furthermore, some lipophilic drugs in liposomes show a sustained release character. In vitro release of a drug is an indirect method to predict its release action in vivo from a formulation (Du et al., 2010). The correlation analysis using SPSS program exhibited a relative good correlation in vivo/in vitro and the r was 0.95 (p < 0.05). Obviously, the solution of OP liposomes and liposomal OP dry powders can be sustained-release and prolong the action of drug in vivo.
Figure 13. The curve of drug cumulative release percentage over time for different preparations.
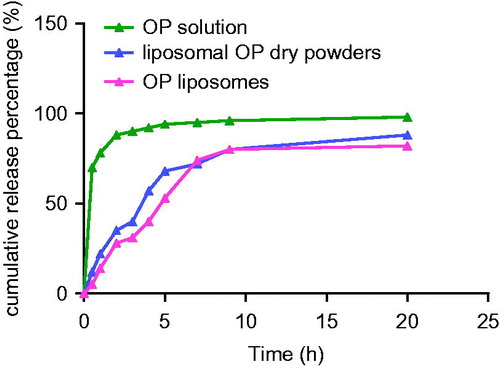
OSCA was released from OP slowly, which was good for treatment of influenza. The reasons are listed as follows: first, dosage of OP which was recommended for treatment of influenza is 75 mg twice daily for 5 days (Moscona, Citation2005). Second, short-term prevention of OP (10–14 days) can be considered for outbreak control (Gubareva et al., Citation2000). In a word, OP is administered for 5 days for treatment of influenza and 10–14 days for prevention of influenza. As the administration frequency can be reduced, the slow release of OP from liposomal powder is good for OP administration.
Metabolism of OP in rat lung
Determination of homogenates protein content
The concentration of protein in rat lung homogenates was 2.5 mg/mL. And, the concentration of protein in rat liver homogenates was 16.7 mg/mL.
HPLC
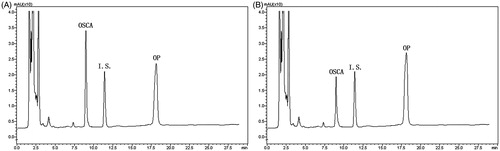
The chromatogram of OP in rat liver homogenates (A) and lung homogenates (B) was displayed in , which showed that OP could be transformed to OSCA not only in the liver homogenates but also in the lung homogenates.
Figure 14. Chromatography of OP in liver homogenates (A) and lung homogenates (B).
Vmax and Km
Vmax and Km were determined in the lung homogenates and liver homogenates. Since liver is the main metabolism organ, liver homogenates are chose as control. Vmax is the maximum velocity of OP metabolism in tissues. Km is the concentration of OP when the velocity is a half of Vmax, which illustrates the enzyme species and the affinity of different enzymes in the study.
showed the relationship of v and v/[s], the slope of the line was −Km, and the intercept was Vmax. Maximum enzyme activity (Vmax) for metabolite formation was 37.2 and 19.4 nmol/h g protein, respectively, for rat liver and lung homogenates. Km for rat liver and lung homogenates were 67.0 and 36.4 nmol/mL, respectively. The results indicated that OP could be hydrolyzed in rat lung, despite the absence of liver first-pass effect.
Figure 15. Regression line of v and v/[s] of liver homogenates and lung homogenates.
![Figure 15. Regression line of v and v/[s] of liver homogenates and lung homogenates.](/cms/asset/7f74516d-8d6d-4822-8f7b-d20665dc0eb6/idrd_a_863526_f0015_b.jpg)
Pharmacokinetics of liposomal OP dry powders in rats
Atrioventricular fitting
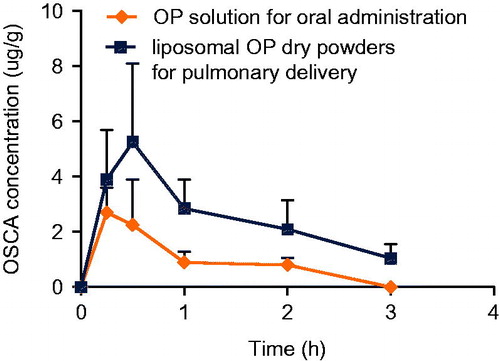
Plasma concentrations of each group were performed via pharmacokinetic software DAS 2.0 package with non-compartmental pharmacokinetic analysis. Atrioventricular fitting results indicated that the data of plasma OSCA from all formulations in rats were best fitted with a two-compartment model. The concentration–time (C–t) profiles of active metabolites OSCA were presented in . By analyzing the pharmacokinetic parameters which were obtained from group a and group b, there was a significant difference in MRT, Cmax and Tmax (p < 0.01) between the two groups using t-test, which was shown in . MRT of group a and group b were 4.652 and 5.608 h, respectively. AUC0–∞ in different tissues of group b was 1.14-fold higher than that of group a.
Figure 16. OSCA plasma concentration-time profile of group a (OP solution for oral administration) and group b (liposomal OP dry powders for pulmonary delivery).
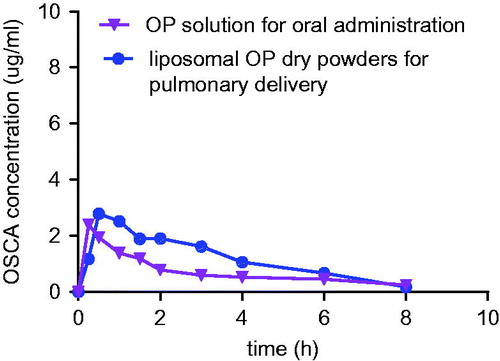
Table 10. Pharmacokinetic analysis of OSCA following different administration (n = 5).
The tissue distribution of rats
As shown in , the tissue distribution of active metabolites in the lungs, and the feasibility of pulmonary delivery of OP were investigated. OP can be transformed to OSCA in lung homogenates. When OP was administered orally, OSCA was transformed from OP in the liver and distributed throughout the body including the lung. When OP was administered by pulmonary delivery, OSCA was transformed from OP in the lung directly, thus Cmax of OSCA was higher than that of oral administration in rat lung.
Figure 17. OSCA concentrations in the lung of group c (OP solution for oral administration) and group d (liposomal OP dry powders for pulmonary delivery).
Conclusion
This study brings a new idea to the development of liposomal dry powder for inhalation containing OP by delivering the effective drug directly to the lung. The results obtained from metabolism of OP in rat lung showed that OP could be transformed to OSCA in the lung. And, in vitro release of liposomal OP dry powders presented a sustained-release pattern. According to the results of pharmacokinetics and tissue distribution, liposomal OP dry powders administered by pulmonary delivery can be targeted to the lung. Consequently, liposomal OP dry powders for inhalation may be a substitution for oral administration. However, the clinical test and productive technology should be further studied.
Acknowledgements
This study was supported by the Fundamental Research Funds for the Central Universities (Program No. JKP2009009).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Al-Hallak MH, Sarfraz MK, Azarmi S, et al. (2011). Pulmonary delivery of inhalable nanoparticles: dry powder inhalers. Ther Deliv 2:1313–24
- Chen X, Huang W, Wong BC, et al. (2012). Liposomes prolong the therapeutic effect of anti-asthmatic medication via pulmonary delivery. Int J Nanomedicine 7:1139–48
- Cocera M, Lopez O, Coderch L, et al. (2000). Alterations in stratum corneum lipid liposomes due to the action of Triton X-100: influence of the level of ceramides on this process. J Control Release 68:387–96
- Dalvi PS, Singh A, Trivedi HR, et al. (2011). Adverse drug reaction profile of oseltamivir in children. J Pharmacol Pharmacother 2:100–3
- Davies BE. (2010). Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J Antimicrob Chemother 65:ii5–10
- Drulis-Kawa Z, Dorotkiewicz-Jach A. (2010). Liposomes as delivery systems for antibiotics. Int J Pharm 387:187–98
- Du B, Li Y, Li X, et al. (2010). Preparation, characterization and in vivo evaluation of 2-methoxystradiol-loaded liposomes. Int J Pharm 384:140–7
- Dutkowski R, Smith JR, Davies BE. (2010). Safety and pharmacokinetics of oseltamivir at standard and high dosages. Int J Antimicrob Agents 35:461–7
- Dutkowski R, Thakrar B, Froehlich E, et al. (2003). Safety and pharmacology of oseltamivir in clinical use. Drug Saf 26:787–91
- Ghosh GC, Nakada N, Yamashita N, Tanaka H. (2010). Oseltamivir carboxylate, the active metabolite of oseltamivir phosphate (Tamiflu), detected in sewage discharge and river water in Japan. Environ Health Perspect 118:103–7
- Glavas-Dodov M, Fredro-Kumbaradzi E, Goracinova K, et al. (2005). The effects of lyophilization on the stability of liposomes containing 5-FU. Int J Pharm 291:79–86
- Gregoriadis G, Perrie Y. (2010). Liposomes. Enc Life Sci. 1–8. DOI: 10.1002/9780470015902.a0002656.pub2
- Gu X, Zhang W, Liu J, et al. (2011). Preparation and characterization of a lovastatin-loaded protein-free nanostructured lipid carrier resembling high-density lipoprotein and evaluation of its targeting to foam cells. AAPS PharmSciTech 12:1200–8
- Gubareva LV, Kaiser L, Hayden FG. (2000). Influenza virus neuraminidase inhibitors. Lancet 355:827–35
- Harrigan PR, Wong KF, Redelmeier TE, et al. (1993). Accumulation of doxorubicin and other lipophilic amines into large unilamellar vesicles in response to transmembrane pH gradients. Biochim Biophys Acta 1149:329–38
- Harush-Frenkel O, Bivas-Benita M, Nassar T, et al. (2010). A safety and tolerability study of differently-charged nanoparticles for local pulmonary drug delivery. Toxicol Appl Pharmacol 246:83–90
- Heinig K, Bucheli F. (2008). Sensitive determination of oseltamivir and oseltamivir carboxylate in plasma, urine, cerebrospinal fluid and brain by liquid chromatography–tandem mass spectrometry. J Chromatogr B 876:129–36
- Huang WH, Yang ZJ, Wu H, et al. (2010). Development of liposomal salbutamol sulfate dry powder inhaler formulation. Biol Pharm Bull 33:512–7
- Li L, Yi T, Lam CW. (2013). Effects of spray-drying and choice of solid carriers on concentrations of Labrasol® and Transcutol® in solid self-microemulsifying drug delivery systems (SMEDDS). Molecules 18:545–60
- Lindemann L, Jacobsen H, Schuhbauer D, et al. (2010). In vitro pharmacological selectivity profile of oseltamivir prodrug (Tamiflu) and active metabolite. Eur J Pharmacol 628:6–10
- Mansour HM, Rhee Y-S, Wu X. (2009). Nanomedicine in pulmonary delivery. Int J Nanomed 4:299–319
- Maxwell SR. (2007). Tamiflu and neuropsychiatric disturbance in adolescents. BMJ 334:1232–3
- Moscona A. (2005). Neuraminidase inhibitors for influenza. N Engl J Med 353:1363–73
- Nicholson KG, Aoki FY, Osterhaus AD, et al. (2000). Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Lancet 355:1845–50
- Panwar P, Pandey B, Lakhera PC, Singh KP. (2010). Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int J Nanomed 5:101–8
- Swaminathan J, Enrhardt C. (2012). Liposomal delivery of proteins and peptides. Expert Opin Drug Deliv 9:1489–503