
Abstract
Biphasic floating minitablets of cefuroxime axetil were prepared by melt granulation technique using two different grades of gelucire namely 50/13 and 43/01 to maintain constant plasma drug concentration. Loading dose of cefuroxime axetil was formulated as immediate release (IR) minitablets by using hydrophilic grade of gelucire 50/13. Maintenance dose was formulated as floating sustained release (SR) minitablets by using hydrophobic grade of gelucire 43/01. The prepared IR and SR granules were subjected to micromeritic studies and scanning electron microscopy. Fourier transform infrared spectroscopy (FT-IR) study revealed that drug and selected carriers were compatible. In vitro dissolution study of optimized IR minitablets showed more than 85% of loading dose dissolved within 30 min. Optimized SR minitablets showed zero lag time with floating duration more than 12 h. The drug release from SR minitablets was linear with square root of time with non-Fickian diffusion-controlled release. The optimized batch of minitablets was filled into 0 size hard gelatin capsule. In vitro dissolution study for capsule showed an immediate burst release followed by SR up to 12 h. There is no significant change in dissolution data after storage at 40 °C and 75% RH for three months. Microbiological assay of dissolution samples of optimized minitablets filled in capsules showed proportionate increase in inhibition of growth against Escherichia coli up to 12 h samples. In vivo bioavailability study in albino rabbits showed three times improvement in oral bioavailability.
Introduction
Modified-release formulations can be designed in single or multiple-unit dosage forms. Single-unit formulations contain the active pharmaceutical ingredient within the single tablet or capsule, whereas multiple-unit dosage forms comprise a number of discrete particles that are combined into one dosage unit. They may exist as pellets, granules, sugar seeds (non-pareil), minitablets, microspheres, ion-exchange resin particles, powders, and crystals with drugs being entrapped in or layered around cores. In the multiple-unit system, the total drug is divided into many units. Failure of few units may not be as consequential as failure of a single-unit system. This is apparent in sustained-release single-unit dosage form, where a failure may lead to dose-dumping of the drug (Abdul et al., Citation2010). Like other multiple unit systems, minitablets can be filled into hard gelatin capsules that release these subunits (minitablets) after disintegration of capsule. This concept can be used to produce a biphasic delivery system combining a fast release together with the slow release component of the drug, This approach can produce a rapid rise in plasma concentration for some drugs that are desired to produce the therapeutic effect promptly, followed by a prolonged release phase in order to avoid repeated administrations (Meka et al., Citation2008).
The above objectives of biphasic delivery can be achieved by using two different grades of gelucire, i.e. 50/13 and 43/01. Gelucire is a family of vehicles derived from mixtures of mono, di, and triglycerides with polyethylene glycol (PEG) esters of fatty acids. Gelucires are available with a range of properties depending on their hydrophilic–lipophilic balance (HLB) 1–18 and melting point (33–70 °C) range (Sheu et al., Citation2001). The gelucires containing only PEG ester 50/13 are generally used in the preparation of fast or rapidly dissolving formulations (Mahmoud et al., Citation2009). It is a class of hydrophilic lipid with HLB 13 and melting point 50 °C. That containing only glycerides or mixtures of glycerides and PEG esters such as 43/01 is used in the preparation of sustained release (Barker et al., Citation2003) and gastroretentive formulations (Jain & Jagdale, 2009). Gelucire 43/01 is a class of highly hydrophobic lipid with HLB 1 and melting point 43 °C. Gelucire 43/01 comprises of a mixture of saturated triglycerides of different fatty acids C8 (3%), C10 (2%), C12 (29%), C14 (2%), C16 (17%), and C18 (36%) melting at 43 °C. Extreme hydrophobicity of gelucire 43/01 is attributed to the absence of PEG esters, which in turn provide release-retarding ability.
Cefuroxime axetil is selected as a model drug as it satisfies all the requirements for the present research. Cefuroxime is a broad-spectrum, β-lactamase-stable cephalosporin antibiotic. It is used orally for the treatment of mild to moderate respiratory tract infections, acute bacterial otitis, pharyngitis, tonsillitis, mild to moderate uncomplicated skin infections, uncomplicated urinary tract infections, uncomplicated gonorrhea, and early Lyme disease (McEvoy, Citation1994). In humans, gastrointestinal absorption of cefuroxime is negligible, whereas the acetoxyethyl ester of cefuroxime (cefuroxime axetil), an oral prodrug, shows average bioavailability about 37% when given in the fasting state and 52% when given with or shortly after food (McEvoy, Citation1994). It is best absorbed from the proximal region of the GI tract (Sommers et al., Citation1984). It is rapidly hydrolyzed in intestine releasing cefuroxime which is poorly absorbed, thereby reducing its bioavailability. Cefuroxime axetil missing the absorption site in upper GI tract remains unabsorbed causing high concentration of antibiotic entering colon leading to antibiotic associated colitis (Dhumal et al., Citation2006). Cefuroxime axetil has a biological half-life of 1.2–1.6 h. The short biological half-life and absorption window in upper GI tract favor development of a sustained release (SR) and floating drug delivery system, respectively (Govikari et al., Citation2013). Hence the objective of the present research work is to prepare multiple unit IR minitablets for loading dose component using gelucire 50/13, SR floating minitablets for maintenance dose component using gelucire 43/01, and finally filling the required number of minitablets into hard gelatin capsule.
Materials and methods
Materials
Cefuroxime axetil was received as a gift sample from Aurobindo Pharma (Hyderabad, India). Gelucire 50/13 and 43/01 were obtained as gift samples from Gattefosse Pvt Ltd (Mumbai, India). Neusilin US2 was a gift sample from Gangwal Chemicals Pvt Ltd (Mumbai, India). All other chemicals were of reagent grade and used without further purification.
Methods
Calculation of loading dose and maintenance dose for cefuroxime axetil
The total dose of cefuroxime axetil for once daily sustained release formulation was calculated based on the available pharmacokinetic data (Hardman & Limbird, Citation2001; Rawlin, Citation2010). As per the zero-order release principle, the rate of delivery must be independent of the amount of drug remaining in the dosage form and constant over time. The release from the dosage form should follow zero-order kinetics, as shown by the equation: where
is the zero-order rate constant for drug release (amount per time), Ke is the first-order rate constant of overall drug elimination (per hour), Cd is the desired drug level in the body (amount per volume), and Vd is the volume in which the drug is distributed. The elimination half life of cefuroxime axetil is 1.6 h (
). The loading dose is given to attain therapeutic concentration range quickly after dosing
(1)
DL is the loading dose, Css is the average steady state plasma level, Vd is the apparent volume of distribution, and F is the fraction of dose absorbed.
(2)
H is the total desired time for sustained action in hours.
(3)
Ke is the overall elimination rate constant (per hour)
thus
(Rate of drug input) is 19.05 mg/h, which should also have been equal to the elimination constant so as to maintain the steady-state condition. For a system in which the maintenance dose releases drug by a zero-order process for a specified period of time, the total dose is as follows:
(4)
where
is the corrected loading dose.
If the maintenance dose begins the release of the drug at the time of dosing, it will add to that which is provided by the initial dose, thus increasing the initial drug level. In this case, a correction factor is needed to account for the added drug from the maintenance dose. This correction factor is the amount of drug provided during the period from t = 0 to the time of the peak drug level, Tp. If DL is 44 mg,
is 19.05 mg/h, and Tp is 1.05 h, then the corrected loading dose is
(5)
Thus, the total dose required
The total dose of cefuroxime axetil is rounded off to 252 mg, i.e. loading dose is 24 mg and maintenance dose is 228 mg.
Thus, the biphasic minitablets have loading dose for initial burst release (IR minitablet) and maintenance dose for sustained release floating minitablets (SR minitablet). Four numbers of IR minitablet and 19 numbers of SR minitablets each containing 6 mg and 12 mg of cefuroxime axetil, respectively, were filled in one “0” size capsule.
Preparation of biphasic release minitablets
Preparation of IR minitablets
The IR minitablets of cefuroxime axetil were prepared by melt granulation technique. The compositions of IR minitablets are shown in . Gelucire 50/13 was heated in a jacketed water bath at 60 °C while stirring. To the molten liquid of gelucire 50/13, cefuroxime axetil powder was added to the molten gelucire 50/13 and stirred for 15 min at 60 °C using a propeller blade at 100 rpm (RQ-129/D, Remi Laboratory Instruments, Mumbai, India). The molten mixture was then added drop-wise to preheated (90 °C) neusilin US2 with continued mixing for 15 min. The mixture was allowed to cool to room temperature by air-cooling followed by sieving through mesh no. # 40 to get uniform size granules. These granules are designated as formulation F1G. The above granules were compressed into minitablets (F1GT), i.e. IR minitablets without disintegrating agent crospovidone. In another IR minitablet formulation, granules (F1G) were mixed with crospovidone in a Cube mixer (Kalweka series, Karnavati Engineering Ltd, Ahmedabad, India). The final blend was compressed in to 3 mm flat, circular minitablets (F1) using multitip punches of Pacific Tools (Mumbai, India) fitted with Minipress-II (Karnavati, Ahmedabad, India).
Table 1. Composition of the cefuroxime axetil granules and minitablets.
Preparation of SR floating minitablets
The SR granules and minitablets were also prepared by a similar method as mentioned above. The composition of the above formulations is shown in . The proportion of neusilin US2 was kept as 50% of gelucire 43/01 in all the six SR formulations (F2G–F4G and F2–F4).
The biphasic minitablets comprising of a loading dose (IR minitablet) and maintenance dose (SR minitablets) of cefuroxime axetil were placed in “0” size hard gelatin capsules and evaluated.
Characterization of granules and minitablets
Micromeritic properties of granules
Pure drug cefuroxime axetil and granules of both IR and SR formulation were subjected to measurement of densities (bulk and tap densities), angle of repose, Carr’s Index, and Hausner’s ratio as per standard procedure (Wells & Aulton, Citation2007).
Kawakita analysis
Flowability was determined using the Kawakita analysis (Yamashiro et al., Citation1983). The pure drug cefuroxime axetil and granules (10 g) were poured into a 50 ml glass measuring cylinder, the heap of the particles in the cylinder was leveled off horizontally with a thin metallic spatula, and the bulk volume, Vo, was accurately measured. The cylinder was then tapped using Digital Tap density tester ETD-1020 (Electrolab, India) and the change in volume of the powder column, VN, was noted after tapping. The compactibility and cohesiveness of cefuroxime axetil and granules were evaluated using numerical constants obtained from the Kawakita plots based on the Kawakita equation, which is used for assessing the flow properties of powders
(6)
where a and b are constants; a describes the degree of volume reduction at the limit of tapping and is called compactibility; 1/b is considered to be a constant related to cohesion and is called cohesiveness, C. The degree of volume reduction was calculated from the initial volume (V0) and tapped volume (VN) as in the following equation:
(7)
Numerical values for constants a and 1/b were obtained from the slope of plots of N/C versus number of taps N (N = 10, 20, 30, up to 300).
Physical characteristics of minitablets
Compressed minitablets (IR and SR) were characterized for weight variation (n = 20). Crushing strength (n = 6) was measured with a Digital tablet hardness tester (Electrolab, Mumbai, India), friability (n = 6), with a Roche-type friabilator. The drug content in each formulation was determined by triturating 20 tablets in a mortar and powder equivalent to 100 mg of cefuroxime axetil was added in 100 mL of simulated gastric fluid (SGF) without enzyme, followed by shaking for 30 min. The sample was filtered, suitably diluted and analyzed by the UV–visible spectroscopy (Patel & Patel, Citation2006; Bomma & Veerabrahma, Citation2012) (UV-1800, Shimadzu, Tokyo, Japan).
Tablet density
The density of the SR minitablets (in g cm−3) was calculated from tablet height, diameter and mass using the equation (Bandari et al., Citation2010)
(8)
where W is the mass of a tablet, m is the tablet diameter, π is the circular constant, and h is the tablet height.
In vitro buoyancy and percent buoyancy
Buoyancy lag time and duration of buoyancy were determined in the USP dissolution apparatus II (USP, Citation2008) with simulated gastric fluid (without enzyme). The time between the SR minitablet introduction into the medium and its buoyancy in the medium was taken as the buoyancy lag time and the duration of the minitablet remaining buoyant was observed visually.
SR minitablets (19 numbers) were spread over the surface of a USP dissolution apparatus II) filled with 900 mL SGF without enzyme. The medium was agitated with a paddle rotating at 100 rpm for 12 h. The floating and the settled portions of SR minitablets were recovered separately. Buoyancy percentage was calculated as the ratio of the number of SR minitablets that remained floating and the total number of SR minitablets (Srivastava et al., Citation2005).
In vitro dissolution
The dissolution profile of cefuroxime axetil, IR (granules and minitablets), SR (granules and minitablet), and optimized minitablets filled into 0 capsules was determined using USP Apparatus II. The dissolution test was performed using 900 mL of SGF without enzyme at 37 ± 0.5 °C and 50 rpm. A sample (5 mL) of the solution was withdrawn from the dissolution apparatus at 10 min interval up to 1 h, 1 h interval up to 12 h, and 10 min interval up to 1 h followed by 1 h interval up to 12 h for IR formulations, SR formulations, and capsule filled with minitablets, respectively. The dissolution samples were replaced with fresh dissolution medium. The samples were filtered through a 0.45 μm membrane filter and analyzed at 280 nm using a UV/Vis double-beam spectrophotometer (UV-1800, Shimadzu, Japan).
Kinetic analysis of dissolution data
The percent dissolution efficiency (% DE) was computed for cefuroxime axetil and IR formulations to compare the relative performance of formulations. The % DE of a pharmaceutical dosage form is defined as the area under the dissolution curve up to a certain time, t, expressed as percentage of the area of the rectangle described by 100% dissolution at the same time (Khan & Rhodes, Citation1975). The % DE can be calculated from
(9)
where Y is the percent drug dissolved at time t.
The rate and mechanism of release of cefuroxime axetil from the prepared SR minitablets and capsules filled with both IR and SR minitablets were analyzed by fitting the dissolution data into the zero-order equation (Merchant et al., Citation2006)
(10)
where Q is the amount of drug released at time t, and K0 is the zero-order release rate constant, fitted to the first-order equation (Bourne, Citation2002)
(11)
where K1 is the first-order release rate constant.
The dissolution data were fitted to Higuchi’s equation (Higuchi, Citation1963). Higuchi tried to relate drug release to physical constants based on diffusion, which is shown in the following equation:
(12)
where C is the initial drug concentration, Cs is the drug solubility in matrix media, and D is the diffusivity of drug molecules in matrix substance. The simplified form of Higuchi equation is
(13)
where KH is the diffusion rate constant.
The dissolution data were also fitted to the well-known equation (Korsmeyer–Peppa’s equation), which is often used to describe the drug release behavior from polymeric systems (Korsmeyer et al., Citation1983)
(14)
where Mt is the amount of drug released at time t, M∞is the amount of drug release after infinite time, k is a release rate constant incorporating structural and geometric characteristics of the minitablet and n is the diffusional exponent indicative of the mechanism of drug release.
Fourier transform infrared spectroscopy (FT-IR)
FT-IR spectra of selected cefuroxime axetil, gelucire 50/13, gelucire 43/01 alone, and their formulations were recorded on IR Affinity-1 (Shimadzu, Japan) using KBr discs. The instrument was operated under dry air purge and the scans were collected at a scanning speed of 2 mm/s with resolution of 4 cm-1 over the region 4000–400 cm−1.
Scanning electron microscopy (SEM)
The SEM analysis of the samples was performed to investigate the surface morphology and homogeneity of surface of granules. The samples of optimized granules of IR and SR minitablets were sputter coated with gold at room temperature before examination to render the surface of particles electroconductive (Shrivastava et al., Citation2009). The SEM analysis of the granules was done by Jeol JSM-840 (Tokya, Japan) SEM.
Stability study
Optimized minitablets filled in capsules (F5C) were subjected to stability studies for a period of three months at 40 °C/75% RH in humidity controlled oven (TH90 S/G, Thermolab, India) and ambient condition. At the end of three months, samples were collected and evaluated for physicochemical parameters (Maurya et al., Citation2012). For the comparison of release profiles of initial and stability samples, “difference factor” f1 and “similarity factor” f2 were calculated (Moore & Flanner, Citation1996). Model-independent method developed by Moore was used to compare dissolution profiles using two factors, f1 and f2. The factor f2, known as the similarity factor, measures the closeness between the two dissolution profiles and f1, known as the difference factor, measures the dissimilarity between two dissolution profiles
(15)
(16)
where n is the number of time points, Rt is the dissolution value of the reference product at time t, and Tt is the dissolution value of the test product batch at time t. For curves to be considered similar, f1 values should be close to 0, and f2 values should be close to 100. Generally, f1 values up to 15 (0–15) and f2 values greater than 50 (50–100) ensure sameness or equivalence of the two curves and, thus the performance of the test (postchange) and reference (prechange) products.
Microbiological assay
For the purpose of this study, the antimicrobial activity of optimized minitablets F5C and marketed tablet was evaluated. Each experiment was carried out in triplicate. The in vitro release dissolution samples of IR and SR minitablets F5C and marketed tablet at different time intervals (0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 h) were tested against Escherichia coli ATCC25922, the most commonly found organism in patients with bacterial infections and urinary tract infections. Cefuroxime axetil (pure drug) at different concentrations in SGF without enzyme (0 µg/mL, 1 µg/mL, 2 µg/mL, 4 µg/mL, 8 µg/mL, 16 µg/mL, and 32 µg/mL) was also tested against the same strains. Molten agar media was transferred to sterilized petri dishes and allowed to solidify. The plates were swabbed with the culture of microorganism before 1 h of addition of each dissolution sample. Volume of inoculum was maintained constant for all the plates. Wells, equidistant from one another, were made in the solidified medium using a sterilized well borer. The samples 100 µL collected from the in vitro drug release aliquots from dissolution of optimized minitablets F5C and marketed tablet, filtered through a 0.45 µm nylon filter and carefully transferred into the wells. Samples were allowed to diffuse for 2 h at room temperature. The plates were then incubated for 48 h at 37 ± 0.5 °C (Govender et al., Citation2005). The diameter (mm) of zone of growth inhibition surrounding each agar well was measured using a caliper.
Bioavailability study
White albino rabbits having weight 2.5 kg were used for the study after acclimatization period of one week. All animal experiments were performed according to the guidelines of Institutional animal ethical committee [Registration no. 926/PO/ac/06/CPCSEA (approval no. 79)] of Roland Institute of Pharmaceutical Sciences, Berhampur, India. Bioavailability study was performed on co-administration of optimized formulation of IR and SR minitablets and pure drug cefuroxime axetil.
A simple rapid and precise reverse phase ultra fast liquid chromatographic method for analysis of cefuroxime axetil was optimized from the reported method (Nafiz et al., Citation2006; Raj et al., Citation2010). Chromatography was performed on a 25 cm × 4.6 mm i.d., 5 µm particle, C18 column with 80:20 (v/v) methanol:water as a mobile phase at a flow rate of 1 mL/min, PDA detection at 278 nm. Cefuroxime axetil was eluted with a retention time of 3.182 min. The method was found to be linear in the concentration range of 1–250 ng/mL of cefuroxime axetil. Coefficient of correlation was found to be 0.998.
Standard curve of cefuroxime axetil in rabbit serum was prepared by solvent extraction method. About 1 mL of blood was collected from the marginal ear vein of albino rabbit of average weight 2.5 kg in eppendorf tube. These blood samples were kept for 30 min to facilitate coagulation of blood. Then it was centrifuged for 10 min at 8000 rpm in a cooling centrifuge (4 °C). Approximately 600–700 µL of serum (supernatant yellow color layer) was separated for the further study. Then it was divided into six individual micro-centrifuge tube each containing 100 µL of serum. Previously drug was diluted with methanol to make different concentrations (1, 5, 10, 20, 50, 100, 150, 200, and 250 ng/mL). About 50 µL of drug solution of different concentrations were added and mixed by vortexing for 1 min. Then 150 µL of ethyl acetate was added to each sample and centrifuged (ELTech Lab, India) for 5 min at 2500 rpm. About 75 µL of supernatant layer was taken and dried at 70 ± 5 °C for the complete evaporation of ethyl acetate in a water bath. The dried samples were reconstituted with 1 mL of mobile phase. A blank serum sample was also prepared without drug solution. A volume of 20 µL from all the solution was injected into UFLC.
Twelve numbers of male albino rabbits of body weight 2.5 kg were selected from the stock of animal house. They were divided into two groups having six animals in each group for test (minitablet administration) and standard (cefuroxime axetil administration). They were treated with hygienic food and fresh water twice daily. All animals were maintained for a wash out period of 15 d prior to the study.
The dose for rabbit was calculated as follows:
Therefore, for rabbit, the total dose of cefuroxime axetil is round off to 30 mg, i.e. loading dose is 3 mg and maintenance dose is 27 mg.
Composition of IR and SR minitablet for in vivo administration are shown in . The ratio of drug to carrier is same as formulation F1 and F3 for IR and SR minitablets, respectively. Cefuroxime axetil and minitablets were administered to animals with the help of wood and feed tube (Ryle’s tube). About 1.5 mL blood was withdrawn from marginal ear vein of rabbit and transferred into eppendorf tubes at various time points of 0.5, 1, 2, 4, 6, 8, and 12 h after dose administration. For serum collection, the blood was left undisturbed in a slanting position for 30 min to allow coagulation. Then sample was centrifuged at room temperature for 10 min at 8000 rpm. The supernatant layer was separated using micropipette. After collection, the blood samples were treated in similar manner as described previously for the preparation of calibration curve in serum. Pharmacokinetic parameters were derived from the serum concentration vs time plot. The area under the curve (AUC), the peak plasma concentration (Cmax), and the time to attain the peak concentration (Tmax) were obtained. The elimination rate constants (k) for minitablet and pure drug cefuroxime axetil were determined from the semilogarithmic plot of serum concentration vs time.
Table 2. Composition of the cefuroxime axetil minitablets for in vivo bioavailability study.
Results and discussion
Micromeritic properties of granules
The IR and SR granules of cefuroxime axetil were prepared by the melt granulation technique using gelucire 50/13 and gelucire 43/01, respectively. Initial trial formulations were prepared by taking cefuroxime axetil and gelucire. The values of angle of repose, Carr’s index (CI) and Hausner’s ration (HR) for cefuroxime axetil and granules prepared only with gelucire revealed that the flowability and compressibility were not within the theoretical range for processing into tablet dosage form. But incorporation of molten mixture of cefuroxime axetil and gelucire to activated neusilin US2 showed significant improvement in micromeritics properties of granules (). Addition of neusilin US2 assisted in converting the gelucire based waxy dispersions into freely flowable granules. This was attributed to high oil adsorption capacity and high specific surface area of neusilin US2 (Fuji, Citation2008). Neusilin US2, 50% of the quantity of gelucire, was found to be in minimum quantity to achieve the desired flowability.
Table 3. Micromeritic properties of granules.
Kawakita analysis
The constants, a and 1/b, of the Kawakita equation for cefuroxime axetil and granules were resolved from the slope and intercept of the line plot of N/C versus N, are shown in . The lower value of a for the granules revealed better flowability than pure drug cefuroxime axetil. Whereas lower value of 1/b for granules showed that it is less cohesive than cefuroxime axetil (Patra et al., Citation2008). This can also be attributed to the properties of high oil adsorption capacity and high specific surface area of neusilin US2.
Physical characteristics of minitablets
Drug content values (96–97.8%) ensured uniform mixing of cefuroxime axetil, gelucires, and neusilin US2. Hardness of the tablets was in the range of 4.9–5.2 kg/cm2. Gelucire 50/13 and 43/01 were waxy materials and tend to stick to the punches during compression. This problem was resolved by incorporating Neusilin US2. Friability values were in the range of 0.38% and 0.43%, which ensures no loss of material from the surface or edge of tablets. This may be attributed to the waxy nature of gelucires. All the formulations passed weight variation test which is an indication of good flowability. Incorporation of 9% of cross povidone resulted in quick disintegration (5.5 min) of IR minitablets (F1). Crospovidone produces quick disintegration because of high capillary activity and pronounced hydration capacity, with little tendency to form gels (Rudnic et al., Citation1980). But it was observed that these IR minitablets did not disintegrate like conventional tablets rather they erode and flakes are produced from the minitablets. SR minitablets did not disintegrate during 2 h study of disintegration ().
Table 4. Physical characteristics of minitablets.
Bulk density
The results () show that SR minitablets remain floating up to 12 h and reflect excellent floating ability of minitablets. The floating properties of minitablets may be attributed to the low bulk density of minitablets, extreme hydrophobicity of gelucire 43/01, and density of gelucire 43/01 (true density 0.0856 g/cm3) (Jain & Gupta, 2009).
Table 5. Parameters related to buoyancy of minitablets.
In vitro buoyancy and percent buoyancy
No significant difference was observed in the floating ability of granules (F2G to F4G) containing different proportions of gelucire 43/01. The results showed that minitablets (F2–F4) remained floating up to 12 h and reflect excellent floating ability of minitablets (). In contrast to most conventional floating systems (including gas-generating ones), these minitablets floated immediately upon contact with the release medium showing zero lag time in floating behavior because the low density was prevailed from the beginning (t = 0). Thakkar et al. (Citation2009) reported that in case of a gelucire based floating tablet, incorporation of HPMC and sodium bicarbonate is essential to achieve desirable floating. But in the present research, we have achieved desirable floating without any gas generating agent by compressing these gelucire-based granules into minitablets of 3 mm diameter but when the same formulation was compressed into a tablet of 6 mm diameter they did not exhibit floating. Hence the size of the tablet also plays a significant role in design of gelucire 43/01 based floating drug delivery systems.
Figure 1. Photographs of floating of sustained release (SR) minitablets of formulation F3 at 0, 6, and 12 h.
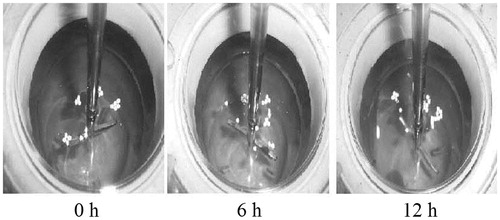
In vitro percent buoyancy studies reveal that, in spite of stirring the dissolution medium for more than 12 h, 80–90% of minitablets of formulation F2–F4 still continued to float (). The surface hydrophobicity imparted to the drug particle by the hydrophobic lipid coat was responsible for floating behavior (Shimpi et al., Citation2004). But all low HLB excipient did not ensure floating, as similar granules prepared using Compritol and glyceryl monosterate separately did not show floating property (Chauhan et al., Citation2005). Hence apart from hydrophobicity, density of gelucire 43/01 also played an important role in design of floating drug delivery systems using lipids. shows the buoyant minitablets of F3 at 0, 6, and 12 h.
In vitro dissolution
The dissolution profile of cefuroxime axetil, granules (F1G), compressed minitablet without crosspovidone (F1GT), and minitablet (F1) with 9% of crosspovidone are shown in . The dissolution of pure drug was very low due poor solubility of the drug. However, dissolution of the drug is enhanced significantly from the granules (F1G). Gelucire 50/13 has HLB value of 13 and is expected to solubilize the hydrophobic drug cefuroxime axetil in solid state. Simultaneous presence of neusilin US2 increases the effective surface area over which the drug is spread leading to rapid desorption of drug with exposure to dissolution medium (Planinisek et al., Citation2011). It appears that compression of the granules (F1GT) resulted in decreased dissolution of cefuroxime axetil. This may be attributed to decreased wettability due to pore-filling resulting in slow disintegration of the tablet. But when crospovidone was incorporated in formulation F1, minitablets disintegrated within 5 min with comparable dissolution profile as that of granules (F1G). Improvement in dissolution rate of IR minitablet (F1) can be attributed to quick disintegration. In vitro dissolution study of IR minitablets showed more than 85% of cefuroxime axetil dissolved within 30 min. The improved solubility of cefuroxime axetil from gelucire 50/13 based formulation can be explained by the improved wettability of cefuroxime axetil particles in aqueous solution from gelucire 50/13. Surfactive power improves the solubility and wettability of cefuroxime axetil (Potluri et al., Citation2011). Hence IR minitablet (F1) is selected as the optimized formulation
Figure 2. (A) In vitro dissolution profile of Immediate release (IR) formulations. (B) In vitro dissolution profile of sustained release (SR) formulations. (C) In vitro dissolution profile of optimized minitablets (IR and SR) filled in 0 size capsules.
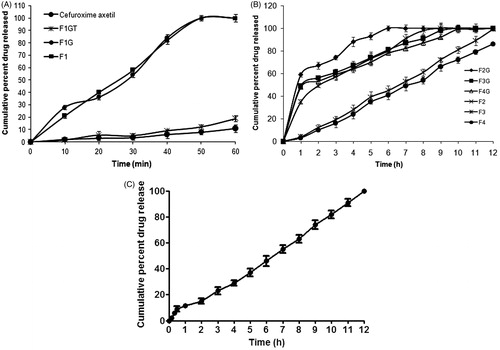
The dissolution profile of SR granules (F2G–F4G) and SR minitablets (F2–F4) are shown in . Drug release was retarded significantly with an increase in the amount of gelucire 43/01. All three granule formulations (F2G to F4G) showed uncontrolled burst release in the initial stage (more than 50% of drug released within 1 h), but compression of these granules into minitablets (F2–F4) caused significant sustained release of drug without any uncontrolled burst release making it suitable for sustained release. Patel et al. (Citation2007) have reported that in case of gelucire 43/01 based granules the drug release was sustained only up to 4 h for ranitidine hydrochloride and to sustain the release further release rate modifiers like ethylcellulose, methyl cellulose, and hydroxypropylmethyl cellulose were used. But in the present research, the drug release was sustained for 12 h by compressing the gelucire 43/01 based granules into minitablets. Hence compression of granules in to tablet plays a significant role in release modulation. SR minitablets also showed that as the proportion of gelucire 43/01 increased the rate of drug release decreased. In case of minitablet F2, F3, and F4 the drug release was sustained up to 7, 12, and 14 h, respectively. It has been suggested that, because of the high hydrophobicity of lipid materials, the release medium is not able to diffuse through the matrix and can progress in the dosage form by dissolving the grains of drug in contact with it. The dissolution of the drug particles on the surface of the matrix allows the formation of channels, from which the drug is slowly released (Sanker & Mishra, Citation2003). The pattern provides an idea about the effect of concentration of gelucire 43/01 on drug release from SR minitablets, i.e. the higher the gelucire 43/01 content, better the controlled drug release. This could be attributed to the increase of lipid matrix density and in the diffusion path length which the drug molecules have to transverse (Jain & Gupta, et al., 2009). Hence SR minitablet F3 is selected as the optimized sustained release minitablet formulation keeping in view the minimum amount of gelucire 43/01 required for sustaining and floating the minitablet up to 12 h from SR floating minitablets.
The dissolution profile of capsule filled with both IR (F1) and SR (F3) minitablets are shown in . The dissolution profile showed an immediate burst release (loading dose) followed by a sustained release (maintenance dose) up to 12 h (). From capsule, over 10% of the cefuroxime axetil was released within the first 30 min of the dissolution study. This initial high amount of cefuroxime axetil release can be attributed to drug release from IR minitablets. Further cefuroxime axetil was released in sustained manner up to 12 h.
Kinetic analysis of in vitro dissolution data
The DE10 and DE30 values for cefuroxime axetil were 1.1 and 1.8, respectively, which suggests very poor dissolution. Dissolution onset of F1GT was also very low as exhibited by their DE10 (1.3) and DE30 (2.4) values. Dissolution onset of granules (F1G) and IR minitablet (F1) at 10 min (DE10) were 14.2 and 10.1, respectively. This clearly indicates better dissolution onset with granules at 10 min. But at 30 min (DE30) values for F1G and F1 were 26.5 and 26.2, respectively, exhibiting similar dissolution profile. The difference in dissolution efficiency at 10 min for F1G and F1 was due to the additional step of disintegration of IR minitablet to granules but at 30 min no significant difference was observed.
In the optimized floating SR minitablet (F3), the calculated regression coefficients for zero-order, first-order, and Higuchi models were, 0.945, 0.835, and 0.992, respectively. The best fit with higher correlation was found with Higuchi equation (). However, this model fails to study the influence of swelling of the matrix and gradual erosion of the matrix. Therefore, the dissolution data were also fitted according to the well-known exponential Korsmeyer–Peppas equation, which is often used to describe the drug release behavior from polymeric systems. The value of release exponent (n) for the optimized formulation F3 was 0.875 indicating that the release was governed by non-Fickian diffusion (slope value was in the range of 0.5–0.89), i.e. coupled diffusion and polymer matrix relaxation (Abdelkader et al., Citation2008).
Table 6. In vitro drug release kinetics of formulations.
The dissolution efficiency for the capsule filled with both IR and SR minitablets (F5C) DE10 and DE30 were 14.5 and 32.4, respectively. This faster onset of dissolution can be attributed to four numbers of IR minitablets. The correlation coefficients after the initial burst release followed Higuchi kinetics (0.984) with non-Fickian diffusion controlled release mechanism (0.874).
Fourier transform infrared spectroscopy
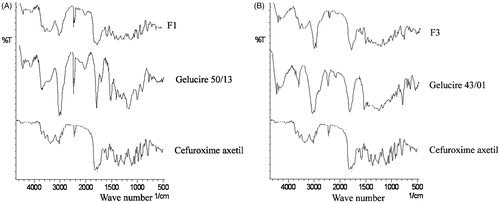
Fourier transform infrared spectroscopy (FT-IR) spectra of cefuroxime axetil, gelucire 50/13, and optimized IR minitablet (F1) are shown in . Similarly FT-IR spectra of cefuroxime axetil, gelucire 43/01 and optimized SR minitablet (F3) are shown in . Cefuroxime axetil shows two carbonyl absorption bands at 1681 cm−1, assigned to amide carbonyl stretching. There were two absorption peak at 3483 cm−1 and 1778 cm−1, assigned to secondary N–H stretching vibration and a C=O stretching of vinyl ester, which remained unchanged in case of formulations F1 and F3. Hence there is no interaction between cefuroxime axetil and carriers used in the present research. Major frequencies of functional groups of pure drug remain intact in IR and SR minitablets containing different carriers; hence, there is no major interaction between the drug and polymers used in the study.
Figure 3. (A) FT-IR spectra of cefuroxime axetil, gelucire 50/13, and optimized IR minitablet (F1). (B) FT-IR spectra of cefuroxime axetil, gelucire 43/01, and optimized SR minitablet (F3).
Scanning electron microscopy
SEM photograph of the melt granules of both IR and SR formulations, i.e. F1 and F3 has shown the presence of lipid on the surface (). The surface of the granules showed smooth patches of lipid on the surface. Hence this indicates that the drug is uniformly distributed in the meltable carriers like gelucire 50/13 and gelucire 43/01 for IR and SR granules, respectively.
Figure 4. (A) and (B) SEM photographs of granules of optimized IR minitablets (F1) and SR minitablets (F3).

Stability study
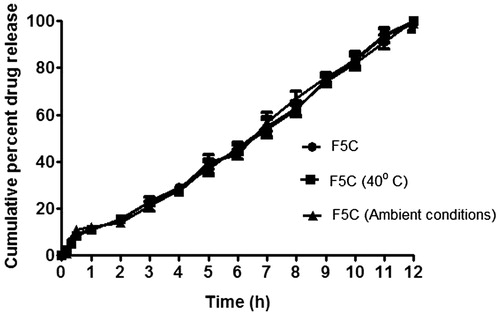
The analysis of dissolution data, after storage at 40 °C/75% RH and ambient temperature and humidity conditions for three months, is shown in (Maurya et al., Citation2012). The dissolution profile of all the three samples suggest that there is no statistically significant difference between the initial and stability samples (p > 0.05), indicating that the two dissolution profiles are considered to be similar (f 2 value is 72 and 81 and f 1 value is 6 and 4, respectively).
Figure 5. In vitro dissolution profile of optimized formulation F5C at 0 time and after three months of storage at 40 °C/75% RH and ambient conditions.
Microbiological assay
Inhibition of growth of pure drug cefuroxime axetil solutions at different concentrations against E. coli was studied. The zone of inhibition for various concentrations of pure drug cefuroxime axetil 0 µg/mL, 1 µg/mL, 2 µg/mL, 4 µg/mL, 8 µg/mL, 16 µg/mL, and 32 µg/mL was found to be 0 mm, 0.12 mm, 0.21 mm, 0.42 mm, 0.71 mm, 1.80 mm, and 3.13 mm, respectively. The in vitro antimicrobial activities of the optimized capsule filled with IR and SR minitablets (F5C) and marketed tablet were evaluated in SGF without enzyme against E. coli. In vitro dissolution samples of optimized minitablets F5C, showed proportionate inhibition of growth against E. coli up to 12 h samples. The results are in agreement with data of dissolution study. This indicates a good correlation between zone of inhibition and cumulative percent drug released (). The in vitro release samples for the marketed formulation showed proportionate increase in inhibition of growth up to 6 h and further samples showed similar growth of inhibition indicating complete drug release within 6 h.
Table 7. Inhibition of growth of in vitro dissolution samples against E. coli.
Bioavailability study
The linearity for standard curve of cefuroxime axetil in rabbit serum was seen in the concentration range of 1–250 ng/mL. The serum concentration-time profile obtained after oral administration of optimized minitablets and pure drug cefuroxime axetil to rabbits is shown in . Tmax for the cefuroxime axetil and minitablet was found to be 2 and 4 h, respectively, indicating controlled absorption of drug from minitablets. Moreover, serum drug concentration reduced to zero in 6 h for cefuroxime axetil but drug could be detected in serum up to 12 h in the case of optimized minitablets corroborating the in vitro sustained release behavior of optimized minitablets. Cmax was found to be significantly (p < 0.05) higher for optimized minitablets (3.678 ± 0.32) than cefuroxime axetil (3.125 ± 0.21 µg/mL) indicating better absorption of drug from IR and SR floating minitablets. The AUC in case of optimized minitablets (25.854 µg/mL × h) was found to be significantly higher (p < 0.05) than pure drug (8.491 µg/mL × h) which indicates increased (three times) bioavailability from optimized minitablets. Lower value of elimination rate constant (K) and higher value of mean residence time (MRT) suggest sustained release of drug from minitablets. The pharmacokinetic parameters were summarized in . Thus comparative pharmacokinetic studies of modified release minitablets and cefuroxime axetil in albino rabbits revealed that optimized minitablets seem to be a potential candidate as a multiple unit biphasic drug delivery system.
Figure 6. In vivo bioavailability studies for pure drug cefuroxime axetil and optimized minitablet formulation.
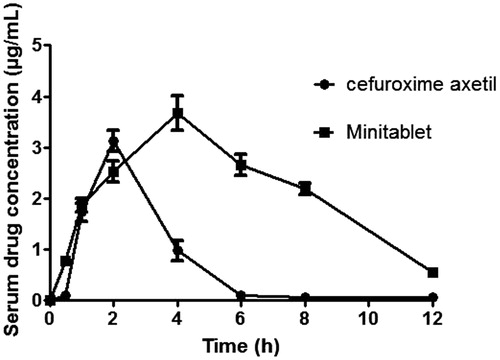
Table 8. Pharmacokinetic parameters for pure drug cefuroxime axetil and optimized minitablet formulation.
Conclusions
From the above research, it was observed that gelucire 50/13 based minitablets can be used as a hydrophilic carrier to achieve desired release of cefuroxime axetil. On the basis of buoyancy and in vitro release kinetics, it was concluded that gelucire 43/01-based minitablets (3 mm) were buoyant and sustained release up to 12 h. Biphasic release in capsules filled with optimized IR and SR minitablets is characterized by the initial burst release followed by sustained release that shows high flexibility in modulating the delivery of the initial loading dose and the sustained dose. In vivo studies revealed that oral bioavailability of the drug increased three times by formulating the drug into multiple unit biphasic floating minitablets. Thus, the formulated multiple unit biphasic minitablets using gelucire 50/13 and 43/01 seem to be a potential approach.
Acknowledgements
The corresponding author J. Sruti gratefully acknowledges Department of Science & Technology, Government of India, New Delhi, India, for the award of the Inspire Fellowship as Senior Research Fellow during her Ph.D. programme.
Declaration of interest
All the authors report no declaration of interest.
References
- Abdelkader H, Abdalla OY, Salem H. (2008). Formulation of controlled-release Baclofen matrix tablets II: influence of some hydrophobic excipients on the release rate and in vitro evaluation. AAPS Pharm Sci Tech 9:675–83
- Abdul S, Chandeswar AV, Jaiswal SB. (2010). A flexible technology for modified-release drugs: multiple-unit pellet system (MUPS). J Controlled Release 147:2–16
- Bandari S, Eaga CM, Thadishetty A, Yamsani MR. (2010). Formulation and evaluation of multiple tablets as a biphasic gastroretentive floating drug delivery system for fenoverine. Acta Pharm 60:89–97
- Barker SA, Yap SP, Yuen KH, et al. (2003). An investigation into the structure and bioavailability of a-tocopherol dispersion in gelucire 44/14. J Controlled Release 91:477–88
- Bomma R, Veerabrahma K. (2012). Development of gastroretentive drug delivery system for cefuroxime axetil: in vitro and in vivo evaluation in human volunteers. Pharm Dev Technol 18:1230--37
- Bourne DW. (2002). Pharmacokinetics. In: Banker GS, Rhodes CT, eds. Modern pharmaceutics, 4th ed. New York: Marcel Dekker Inc, 67–92
- Chauhan B, Shimpi S, Mahadik KR, Paradkar A. (2005). Preparation and evaluation of floating risedronate sodium–gelucire 1 43/01 formulations. Drug Dev Ind Pharm 31:851–60
- Dhumal RS, Rajmane ST, Dhumal ST, Pawar AP. (2006). Design and evaluation of bilayer floating tablets of cefuroxime axetil for bimodal release. J Sci Ind Res 65:812–6
- Fuji Chemical Industry co. Ltd. (2008). Technical News letter, May, Available from: www.fujichemical.co.jp/english/neusilin.html [last accessed 11 Dec 2011]
- Govender S, Pillay V, Chetty DJ, et al. (2005). Optimisation and characterisation of bioadhesive controlled release tetracycline microspheres. Int J Pharm 306:24–40
- Govikari KR, Mandapalli PK, Manthri R, Reddy VP. (2013). Development and in vivo evaluation of gastroretentive delivery systems for cefuroxime axetil. Saudi Pharm J 21:53–9
- Hardman JG, Limbird LE. (2001). In: Goodman & Gilman’s, 10th ed. Pharmacological basis of therapeutics. McGraw Hill, 1938
- Higuchi T. (1963). Mechanism of sustained action medication. J Pharm Sci 52:1145–9
- Jain SK, Gupta A. (2009). Development of gelucire 43/01 beads of metformin hydrochloride for floating delivery. AAPS Pharm Sci Tech 10:1128–36
- Jain SK, Jangde M. (2009). Lectin conjugated gastro-retentive multiparticulate delivery system of clarithromycin for the effective treatment of helicobacter pylori. Mol Pharm 6:295–304
- Khan CA, Rhodes CT. (1975). The concept of dissolution efficiency. J Pharm Pharmacol 27:48–9
- Korsmeyer RW, Gurny R, Docler E, et al. (1983). Mechanism of solute release from porous hydrophilic polymers. Int J Pharm 15:25–35
- Mahmoud EB, Gihan F, Mohamed F. (2009). Improvement of solubility and dissolution rate of indomethacin by solid dispersions in gelucire 50/13 and PEG4000. Saudi Pharm J 17:217–25
- Maurya SK, Bali V, Pathak K. (2012). Bilayered transmucosal drug delivery system of pravastatin sodium: statistical optimization, in vitro, ex vivo, in vivo and stability assessment. Drug Deliv 19:45–57
- McEvoy GK. (1994). Cephalosporins: cefuroxime sodium and cefuroxime axetil. In: AHFS Drug Information. Bethesda, USA: American Society of Hospital Pharmacists, 99–108
- Meka L, Thadisetty A, Vobalaboina V, Yamsani MR. (2008). Design and evaluation of a novel matrix type multiple units as biphasic gastroretentive drug delivery systems. AAPS Pharm Sci Tech 9:1253–61
- Merchant HA, Shoaib HM, Tazeen J, Yousuf RI. (2006). Once-daily tablet formulation and in vitro release evaluation of cefpodoxime using hydroxypropyl methylcellulose: a technical note. AAPS Pharm Sci Tech 7:E178–83
- Moore JW, Flanner HH. (1996). Mathematical comparison of dissolution profiles. Pharma Tech 20:64–74
- Nafiz OC, Göksel A, Hassan YAE. (2006). Determination of cefuroxime axetil in tablets and biological fluids using liquid chromatography and flow injection analysis. Anal Chim Acta 576:246–52
- Patel VF, Patel NM. (2006). Intragastric floating drug delivery system of cefuroxime axetil: in vitro evaluation. AAPS Pharm Sci Tech 7:E118--E124
- Patel DM, Patel NM, Patel VF, Bhatt DA. (2007). Floating granules of ranitidine hydrochloride-gelucire 43/01: formulation optimization using factorial design. AAPS Pharm Sci Tech 8:E1–7
- Patra CN, Pandit HK, Singh SP, Devi MV. (2008). Applicability and comparative evaluation of wet granulation and direct compression technology to Rauwolfia serpentina root powder: a technical note. AAPS Pharm Sci Tech 9:100–4
- Planinisek O, Kovacic B, Vrecer F. (2011). Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int J Pharm 406:41–8
- Potluri RHK, Bandari S, Jukanti R, et al. (2011). Solubility enhancement and physicochemical characterization of carvedilol solid dispersion with gelucire 50/13. Arch Pharm Res 34:51–7
- Raj KA, Yada D, Yada D, et al. (2010). Determination of cefixime trihydrate and cefuroxime axetil in bulk drug and pharmaceutical dosage forms by HPLC. Int J Chem Tech Res 2:334–6
- Rawlin EA. (2010). Bentley’s text book of pharmaceutics, 8th ed. Elsevier: Philadelphia
- Rudnic EM, Lausier JM, Chilamkurti RN, Rhodes CT. (1980). Studies of the utility of cross linked polyvinylpolypyrrolidine as a tablet disintegrant. Drug Dev Ind Pharm 6:291–309
- Sanker C, Mishra C. (2003). Development and in vitro evaluations of gelatin microspheres of ketorolac tromethamine for intranasal administration. Acta Pharm 53:101–10
- Sheu MT, Hsia A, Ho HO. (2001). Polyglycolized saturated glycerides as carrier and enhancer for drug penetration. Chin Pharm J 53:107–18
- Shimpi S, Chauhan B, Mahadik KR, Paradkar A. (2004). Preparation and evaluation of diltiazem hydrochloride-gelucire 43/01 floating granules prepared by melt granulation. AAPS Pharm SciTech 5:51–6
- Shrivastava AR, Ursekar B, Kapadia CJ. (2009). Design, optimization, preparation and evaluation of dispersion granules of valsartan and formulation into tablets. Curr Drug Delivery 10:28–37
- Sommers DK, Van WM, Moncrieff J, Schoeman HS. (1984). Influence of food and reduced gastric acidity on the bioavailability of bacampicillin and cefuroxime axetil. Br J Clin Pharmacol 18:535–9
- Srivastava AK, Ridhurkar DN, Wadhwa S. (2005). Floating microspheres of cimetidine: formulation, characterization and in vitro evaluation. Acta Pharm 55:277–85
- Thakkar VT, Shah PA, Soni TG, et al. (2009). Goodness-of-Fit Model-Dependent approach for release kinetics of levofloxacin hemihydrates floating tablet. Dissol Technol 16:35–9
- The United States Pharmacopoeia 31. (2008). United State Pharmacopeial Convention INC: Asian edition, 1698–700
- Wells JI, Aulton ME. (2007). Pharmaceutical preformulation. In: Aulton ME, ed. Aulton’s pharmaceutics: the design and manufacture of medicines, 3rd ed. USA, Churchill Livingstone: Elsevier, 355–7
- Yamashiro M, Yuasa Y, Kawakita K. (1983). An experimental study on the relationships between compressibility, fluidity and cohesion of powder solids at small tapping numbers. Powder Technol 34:225–31