
Abstract
The aims of this study were to prepare fine famotidine-containing floating-bioadhesive cooperative minitablets and to investigate the possibility of using those minitablets as a delivery system for promoting the oral bioavailability of famotidine. Nine minitablet formulations were designed using hydroxypropylmethylcellulose (HPMC K4M) as release-retarding polymers, Carbopol 971P as bioadhesive materials and sodium bicarbonate (NaHCO3) as gas formers. The prepared 3 ± 0.02 mm minitablets were evaluated in terms of their swelling ability, floating behavior, bioadhesion test and in vitro release. The optimized minitablets (F6) containing HPMC K4M (50.00%, w/w), Carbopol 971P (10.00%, w/w) and NaHCO3 (10.00%, w/w) were found to float in 1 min and remain lastingly buoyant over a period of 8 h in vitro, with excellent bioadhesive properties (20.81 g) and sustained drug release characteristics (T50% = 46.54%) followed one-order model. In addition, plasma concentration–time profiles from pharmacokinetic studies in rats dosed with minitablets showed 1.62-fold (p < 0.05) increased absorption of famotidine, compared to the market tablets XinFaDing®. These studies demonstrated that the multiple-unit floating-bioadhesive cooperative minitablets may be a promising gastro-retentive delivery system for drugs that play a therapeutic role in the stomach.
Introduction
Rapid gastrointestinal (GI) transit could lead to incomplete drug release from the drug delivery device in the absorption zone and diminished efficacy of the administered dose (Jivani et al., Citation2009). For many drugs that are absorbed mainly from the proximal small intestine, controlled release in the stomach would improve their bioavailability (Sato et al., Citation2004). However, the real challenge in the development of an oral controlled-release drug delivery system is not just to sustain the drug release but also to prolong the residence of the dosage form within the gastrointestinal tract (GIT) until all the drug is completely released at the desired period of time. Over the years, various approaches have been proposed to control the presence of drug delivery systems in the upper part of the gastrointestinal tract, like mucoadhesive systems (Wang et al., Citation2001; Bernkop-Schnürch, Citation2005), swellable systems (Fix et al., Citation1993), floating systems (Atyabi et al., Citation1996; Iannuccelli et al., Citation1998) and other delayed gastric emptying devices (Gröning & Heun, Citation1984, Citation1989).
However, conventional stomach floating formulations will be emptied without sufficient water intake after oral administration. Similarly, conventional bioadhesive formulations will be emptied as a result of gastric motility, sticky membrane shedding constantly or intragastric pH changes; they cannot even come in contact with the mucosa when the stomach is filled with liquid contents. Floating-bioadhesive cooperative drug delivery system overcomes the shortcomings of a single floating or bioadhesive type, increases the residence time of drugs in the stomach via floating and bioadhesion mechanism, thereby increasing the efficacy of the administered dose. It provides a more reliable efficacy compared with a single floating or bioadhesive type. Currently, floating-bioadhesive cooperative drug delivery system has become a new way to regulate drug release and absorption effectively (Liu et al., Citation2011).
Minitablets are tablets with a diameter equal to or smaller than 2–3 mm (Lennartz & Mielck, Citation1998). As multiple-unit dosage forms, the minitablets present several advantages over conventional single-unit dosage forms, such as dose flexibility, high degree of dispersion in the gastrointestinal tract, and thus minimizing irritation due to high local drug concentrations, reproducible bioavailability (Graffner et al., Citation1986) and uniform plasma levels. Due to the manufacturing process, smooth surface, defined size, regular shape and strengths can easily be produced, with small variability within and between batches. Furthermore, it is an attractive alternative to the production of pellets, particularly by avoiding the presence of a solvent and by maintaining high production yields like the ones observed in extrusion or spheronization (Munday & Fassihi, Citation1989; Munday, Citation1994). Indeed, minitablets is also a potentially suitable dosage form for pediatric drug delivery systems and a recent study has assessed the use and safety of minitablets for children 2–6 years old (Thomson et al., Citation2009). Like other multiple-unit dosage forms, several minitablets can be either filled into hard capsules (Bredenberg et al., Citation2003) or compacted into bigger tablets (Roberts et al., Citation2012). DILACOR XR of Phone Poulene-RORer, a device for the 24 h extent release of Diltiazem Hydrochloride was launched in 1992 in the USA. The capsule is formed by a plurality of minitablets of different doses, which is flexible for different people to take under different situations.
Famotidine is approved for use in the treatment of peptic ulcer and duodenum ulcer as a histamine H2-receptor antagonist (Kumar et al., Citation2009). The low oral bioavailability (40–45%) and short biological half-life (2.5–4.0 h) of famotidine following oral administration favors development of a sustained-release formulation (Jaimini et al., Citation2007). Since the absorption site of famotidine is proximal small bowel, it is necessary to optimize both the residence time of the system in the gastrointestinal (GI) tract and the rate of release of the drug in order to develop a desired sustained-release dosage form for famotidine (Baki et al., Citation2011). Currently, the main dosage forms of famotidine are conventional tablet (Gaster®) and injection (XinFaDing®).
In this article, a gastro-retentive delivery system with multiple-unit minitablets was developed in order to increase the overall bioavailability of the drug in rats. Floating-bioadhesive cooperative minitablets with hydrogel matrix and sodium bicarbonate as the gas-generating agents, were prepared by direct compression. The matrix was a mixture using the combination of HPMC K4M (Siepmann et al., Citation1999) and Carbopol 971 P. Upon contact with acidic environment of the stomach, the matrix hydrated rapidly at the surface, retaining the bubbles developed from sodium bicarbonate which led to a bulk density lower than 1 g/mL. Drug release from hydrophilic matrix tablets is controlled by the formation of a hydrated viscous layer which acts as a barrier to drug release. The penetration of water into tablet and diffusion of dissolved solutes out of the matrix tablet are hindered by this viscous layer (Korsmeyer et al., Citation1983). The prepared minitablets were evaluated in terms of their swelling ability, floating behavior, bioadhesion characteristic and in vitro release profiles. Finally, the optimized formula was evaluated by in vivo release in rats, compared with market tablets XinFaDing®.
Materials and methods
Materials
Famotidine (FMTD) was obtained from Jianyuan Chemical Co., Ltd. (Hubei, China). Hydroxypropylmethylcellulose (HPMC) in three different viscosity grades (K4M, K15M and K100M) were used as matrix-former (Orpington, UK). Carbopol (CP) was obtained from BF Goodrich (Cleveland, America). Sodium bicarbonate was obtained from Lingfeng Chemical Reagent Co., Ltd. (Shanghai, China). Magnesium stearate was obtained from Sunhere Pharmaceutical Excipients Co., Ltd. (Anhui, China). Aerosil was obtained from Sunhere Pharmaceutical Excipients Co., Ltd. (Anhui, China). Microcrystalline cellulose was obtained from Avicel® PH 102 (Tokyo, Japan). Immediate release tablet, 20 mg, as a reference product was obtained from Xinyi Pharmaceutical Co., Ltd. (Shanghai, China). All other materials were of reagent grade and used as received.
Preparation of famotidine minitablets
Minitablets were prepared by the direct compression method according to the formulations shown in . All were passed through 80-mesh sieve and thoroughly mixed. Magnesium stearate was mixed with the ingredients for 5 min. Tableting was performed using a single press tableting machine (Shanghai Pharmaceutical Machinery Factory, China) with a flat-face punch and 3 mm diameter weighing 15.0 ± 1.0 mg. Every 15 minitablets would be filled into hard capsules. The minitablets were white, round and convex. The hardness of the minitablets was kept constant using a Monsanto hardness tester (Shanghai Huanghai Drug Test Instrument Co., Shanghai, China).
Table 1. The composition of the investigated different formulations of famotidine minitablets.
Determination of famotidine minitablets swelling ability
The swelling property of the formulations has been determined by various techniques (Mohammed & Khedr, Citation2003). The swelling study of the minitablets was done with USP dissolution apparatus II. The medium used was 0.1 M HCl, rotated at 50 rpm. The minitablets were withdrawn, blotted to remove excess water and weighed after selected time intervals. Swelling characteristics of the minitablets were expressed in terms of water uptake (WU) (Gerogiannis et al., Citation1993) as
Famotidine minitablets floating behavior studies
The buoyancy of the minitablets was studied in 150 mL of gastric fluid at pH 1.2. The floating lag times (the duration of the period between the placing of the miniablets in the medium and the minitablets floating) and durations of miniablets floating were determined by visual observation.
Famotidine minitablets bioadhesion tests
In vitro bioadhesion of the minitablets was examined by the modified procedure which using the gastric mucosa of healthy male rats that fasted overnight for at least 12 h; the mucosa was washed with and kept in physiological saline solution. A square piece (surface area 1 cm2) of mucosa was glued to the surface of plexiglass with cyanoacrylate adhesive. Similarly, the minitablet was glued to another plexiglass. Then, the mucosa and minitablet were put in contact with each other with constant pressure for 5 min. After a preload time of 5 min, water was added to the polypropylene bottle through an intravenous infusion set at a rate of 5 mL/min until the minitablet detached from the mucosa. The water collected in the bottle was measured and expressed as weight (g) required for the detachment (Agarwal & Mishra, Citation1999).
In vitro drug release studies
Dissolution study was carried out using USP Dissolution Tester Apparatus, type-II (Paddle method) with 900 mL of 0.1 M HCl at 37 ± 0.5 °C and the paddle speed was 50 rpm. Samples through a 0.45 μm filter were taken in the preparatory time point and assayed by HPLC (Shimadzu, Kyoto, Japan) at a wavelength of 266 nm. At each time of withdrawal, 5 mL of fresh medium was replaced into the dissolution flask (Sriamornsak et al., Citation2007).
Kinetic modeling of drug release profiles
The dissolution profiles of F6 in 0.1 M HCl were fitted to different kinetic models, including zero-order, first-order and Higuchi kinetic models (Costa & Sousa Lobo, Citation2001), The largest value of the correlation coefficient (R) indicated a superiority of the release profile fitting to the mathematical equation.
Oral administration of famotidine minitablets to rats
The usage of animals was agreed by China Pharmaceutical University Animal Management and Ethics Committee. Male Wistar rats were supplied by the Qinglong Mountain Animal Centre (Nanjing, China) and acclimatized for 7 days. The weights of the rats were 200–250 g. The rats were fasted overnight for 17 h before dosing. At time zero, 20 mg/kg of sample were given to rats by gavage (n = 6). XinFaDing® as a conventional tablet was suspended with purified water to a concentration of 0.4 mg/mL and given to rats by intragastric administration.
Half, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 h after intragastric administration, the rats were anesthetized with ether. Blood samples (0.5 mL) were collected from the eye ground vein in heparinized plastic centrifuge tubes using 0.8–1.1 mm capillary glass tube. Individual plasma samples were prepared by centrifugation at 2600 × g for 15 min in an Nr.12154 rotor (Sigma 3K30). All plasma samples were immediately frozen at −20 °C until analysis.
Plasma samples were prepared by liquid–liquid extraction. 180 μL of plasma sample were pipetted into an Eppendorf tube and fortified with 200 μL of the internal standard (acetaminophen) working solution (400 ng/mL). 7.5% hydroxylamine hydrochloride 50 μL and 0.3 mL saturated solution of potassium carbonate were added and subjected to 0.5 min vortex oscillation. The content of the tube was gently mixed and then extracted with 5 mL ethyl acetate. This involved thorough vortex-mixing for 10 min and subsequent centrifugation at 1300 × g for 10 min. The upper organic phase was transferred into another Eppendorf tube and evaporated to dryness in a preheated concentrator-evaporator. The residue was reconstructed with 200 μL of 0.01 mol/L KH2PO4: acetonitrile = 93:7 (v/v). 20 μL was injected into the HPLC system described below. This method is simple and fast, which can be used for the determination of famotidine in plasma.
Then, the resulting aqueous solution was injected into an HPLC apparatus (Shimadzu, Kyoto, Japan) with 0.01 mol/L KH2PO4:acetonitrile = 93:7 (v/v) as a mobile phase at a flow rate of 5.0 mL/min. The column was a reversed-phase column (Inertsil ODS-3, 4.6 × 250 mm, i.d. 5 μm, GL Sciences, Japan), and UV detection was performed at 266 nm. Methodological studies, such as linearity, specificity, recovery, precision of with-in and between days, could satisfy the requirements of the methodology.
Statistical analysis
Individual plasma concentrations and nominal sampling times were used to assess the pharmacokinetic parameters by non-compartmental analysis using HPLC (Shimadzu, Kyoto, Japan). Cmax refers to the peak plasma level and Tmax refers to the time at which Cmax occurred. The area under the curve AUC0-t (i.e. area under the concentration-time curve calculated from zero up to time corresponding to the last measurable concentration) was calculated using the linear trapezoidal rule using individual concentrations.
All results were expressed as mean ± SD. Differences between two related parameters were considered statistically significant for p values of or less than 0.05. Pharmacokinetics parameters were determined from the plasma concentration–time data by means of a model-independent method using a computer program, DAS.
Results and discussion
Swelling indices of famotidine minitablets
The swelling of the polymers used (HPMC K4M and Carbopol 971 P) could be determined by water uptake of the minitablets. The percent swelling of the minitablets was determined by the method described in USP dissolution apparatus II at different time intervals.
As reported by Bertram & Bodmeier (Citation2006), the ability of hydrogels to absorb water is due to the presence of hydrophilic groups. The hydration of these functional groups results in water entry into the polymer network leading to expansion and consequently an ordering of the polymer chains. The hydration ability of the formula is important because it influences: (i) tablet buoyancy, (ii) adhesion ability of swellable polymers as HPMC K4M and Carbopol 971 P in contact with the test fluid, (iii) drug release kinetics (Tadros, Citation2010).
The percent swelling of F6 gradually increased up to 6 h and then gradually decreased till 8 h. However, the minitablets without Carbopol 971P (F10) had the maximum swelling at 2 h after which polymer started eroding slowly in the medium (). This could be the reason that the high initial uptake of water may be due to the faster hydration rate of HPMC K4M and the property of carbopol to retain water and form a thick swollen mass. This showed that HPMC hydrated more rapidly than Carbopol 971P in the presence of 0.1 M HCl solution. The water content of the tablets determine the diffusion of drug significantly. The swelling rates of these formulas are corresponding with the studies in vitro release.
Figure 1. Effect of different kinds of hydrogel matrix material (HPMC K4M and Carbopol 971 P) on swelling behavior showing swelling indices (%) of famotidine minitablets under the conditions of 0.1 M HCl and 37 °C (apparatus II, 50 rpm) (mean ± SD, n = 6).
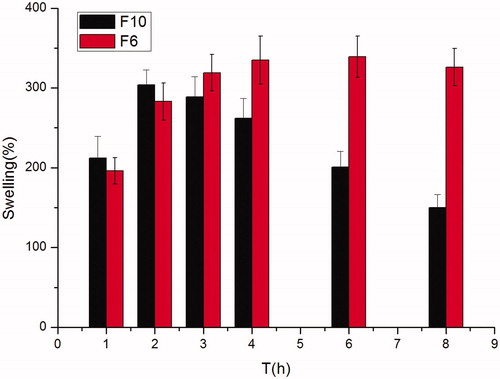
An anisotropic swelling phenomena (i.e. more swelling in the axial direction than in the radial direction on exposure to water) was seen during the swelling of the minitablets, similar phenomena was observed by Papadimitriou et al. (Citation1993) who related the predominantly axial relaxation of the HPMC compacts to the relief of stress induced during unidirectional swelling to the orientation of molecules during compression. The reason for such preferential swelling in axial direction might be due to the need for directional stresses.
Influence of formulation on the floating lag time and duration of famotidine minitablets
There are two main influencing factors about the floating lag time and duration: the percentage of HPMC and NaHCO3.
As a gas forming agent, sodium bicarbonate develops carbon dioxide after reacting with hydrochloride acid. In this investigation, the in vitro testing revealed the ability to maintain buoyant more than 8 h of most formulations ( and ).
Table 2. The influence of different concentrations of HPMC K4M on the floating lag time and duration of minitablets.
Table 3. The influence of different concentrations of NaHCO3 on the floating lag time and duration of minitablets.
Floating lag time and duration were determined visually. showed photographs taken during in vitro buoyancy study of F6 in 150 mL 0.1 M HCl at different time intervals. The formulations containing higher NaHCO3 ratios – 10 % w/w (F6) and 20 % w/w (F7) had significantly shorter floating lag time (1 min) than other formulations. Moreover, a similar conclusion was noted by Gambhire et al. (Citation2007) who concluded that as the percentage of NaHCO3 increases, the floating lag time decreases. This phenomenon might be due to the generation of larger amounts of effervescence with higher NaHCO3 percentage which led to an increase in the rate of pore formation and consequently rapid hydration of the minitablets’ matrices. Furthermore, sodium bicarbonate less than 22.5 mg was not sufficient to keep minitablets floating for 8 h.
Figure 2. Photographs taken during in vitro buoyancy study of F6 in 150 mL 0.1 M HCl at different time intervals (10 s, 30 s, 60 s, 1 h, 4 h, 8 h).

Similarly, reduction in HPMC level in the formulations prolonged the floating lag time and shortened the total floating time. The floating lag time and total floating time of F1 with 10% HPMC K4M were 10 min and 3 h. This phenomenon might be due to low HPMC level could not form sufficient gel matrix that enabled efficient entrapment of the generated gas bubbles.
In vitro bioadhesion study of famotidine minitablets
The adhesive property of minitablets containing varying proportions of Carbopol 971P was investigated and the results were that as the percentage of Carbopol 971P increased, the bioadhesive strength increased. The bioadhesive strength of HPMC K4M is lower than Carbopol 971P.
Influence of formulation on the drug release
The use of hydrophilic polymers is the most common method for controlling the release of drugs in the formulation of oral pharmaceutical dosage forms. Hydroxypropylmethylcellulose (HPMC) is a polymer frequently used in the formulation of sustained-release dosage forms (Cedillo-Ramirez et al., Citation2006). The influence of the formulation was evaluated for the 3 mm minitablets at pH 1.2 with a rotational speed of 50 rpm.
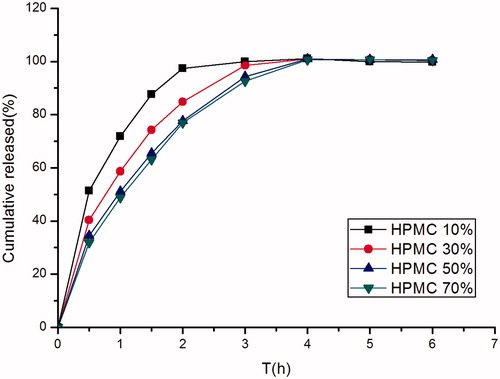
The influence of viscosity grade and concentration of the HPMC K4M on drug release rate was investigated. Profiles with different viscosity grades did not have a significant difference, this phenomenon might be due to the fact that minitablets had a higher relative surface area, which was a more significant factor than viscosity grades. As shown in , as expected, the rate of drug release decreased with the increase in the concentration of HPMC K4M, which could be attributed to the formation of a thick gel structure that delayed famotidine release from the tablet matrix. When the proportion added up to 50%, the higher proportion could not decrease the release rate. F6 had better swellable, floating properties and release rate, so F6 was selected to evaluate the influence of the tablet diameter on the drug release.
Figure 3. Effect of different concentrations of HPMC K4M (10.00%, w/w; 30.00%, w/w; 50.00%, w/w; 70.00%, w/w) on in vitro release of famotidine minitablets under the conditions of 0.1 M HCl and 37 °C (apparatus II, 50 rpm) (mean ± SD, n = 6).
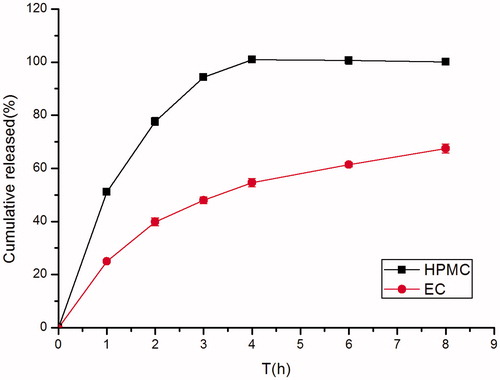
As was shown in , in identical quantities, EC produced a greater sustaining release effect than HPMC. As described by Lopes et al. (Citation2006), even at a lower level, 15% presence of EC in the formulation has shown a larger impact over the release of ibuprofen compared to the HPMC. However, water-soluble polymers that retain the gas and float in an acidic environment of the stomach could not be developed from EC, it might be due to its insoluble matrix materis nature.
Figure 4. Effect of different kinds of matrix material (HPMC K4M and EC) on in vitro release of famotidine minitablets under the conditions of 0.1 M HCl and 37 °C (apparatus II, 50 rpm) (mean ± SD, n = 6).
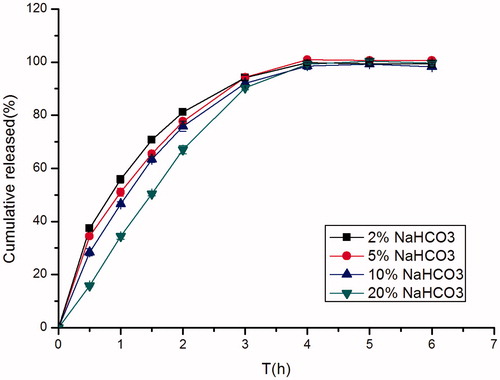
The effect of sodium bicarbonate concentration on in vitro drug release of famotidine is shown in . In such systems, sodium bicarbonate acted as a gas generating agent. It generated gas when it came into contact with the acidic environment of the stomach. This gas entrapped into the matrix of water-soluble polymers and the systems floated in an acidic environment of the stomach. As the concentration of sodium bicarbonate increased from 4.5 mg (F5) to 45 mg (F7) per capsule, the drug release decreased. This might be due to the solubility characteristic of famotidine. Famotidine has a good solubility in acid solution while it is sparingly to slightly soluble in alkaline solution. Sodium bicarbonate being alkaline in nature created an alkaline microenvironment around the minitablets and famotidine was less soluble in alkaline pH which decreased the drug release from the tablet matrix (Chavanpatil et al., Citation2006).
Figure 5. Effect of different concentrations of NaHCO3 (2%, w/w; 5%, w/w; 10%, w/w; 20%, w/w) on in vitro release of famotidine minitablets under the conditions of 0.1 M HCl and 37 °C (apparatus II, 50 rpm) (mean ± SD, n = 6).
Influence of tablet diameter on the drug release
A simple but very effective way to modify the drug release kinetic from tablets was to change their diameters. As was shown in , the release rate of the drug increased proportionally with the decrease in diameter. This could be attributed to the higher relative surface area of the dosage form when the diameter decreased, this result to a proportionately greater area of contact between the tablets and the solution (Goole et al., Citation2007). Photographs taken of different diameters of famotidine tablets were shown in .
Figure 6. Effect of different diameters (3 mm, 5 mm, 7 mm, 9 mm) on in vitro release of famotidine tablets under the conditions of 0.1 M HCl and 37 °C (apparatus II, 50 rpm) (mean ± SD, n = 6).
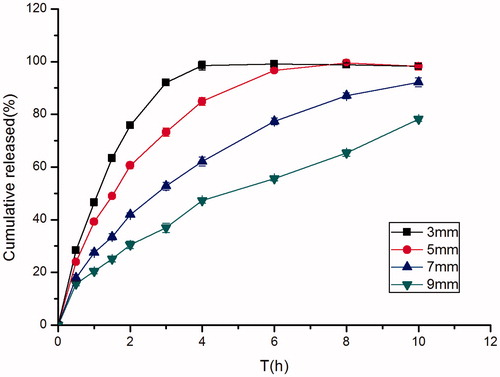
Figure 7. Photographs taken of different diameters (3 mm, 5 mm, 7 mm, 9 mm) of famotidine tablets.

Effect of different pH dissolution mediums on the drug release
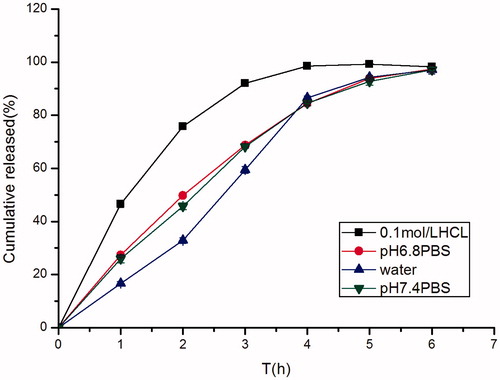
The release profiles of famotidine from minitablets were influenced by pH of release media. In order to investigate the impact of different pHs on drug release behaviors of the minitablets, a dissolution study at different pHs was carried out. showed the release profiles in different release media (0.1 M HCl solution, pH 6.8 PBS, water, pH 7.4 PBS). Drug release from some formulations was significantly influenced by the release media. Drug release in 0.1 M HCl was more rapid than in water, this suggested that famotidine was a drug with a pH-dependent solubility.
Figure 8. Effect of different dissolution mediums (0.1 M HCl, distilled water, pH 6.8 PBS, pH 7.4 PBS) on in vitro release of famotidine minitablets (apparatus II, 50 rpm) (mean ± SD, n = 6).
Effect of different agitation rates on the drug release
The effect of agitation rate on the famotidine dissolution rate was examined at pH 1.2 by using the paddle method. The release rate of the drug increased as the stirring rate increased, however, there was no significant difference between them. It could be concluded that our famotidine minitablets showed a good level of robustness.
F6 containing HPMC K4M (50.00%, w/w), Carbopol 971P (10.00%, w/w) and NaHCO3 (10.00%, w/w) was a promising system exhibiting excellent floating properties, bioadhesive strength and sustained drug release characteristics. The dissolution profile of the optimized formula was fitted to zero-order, first-order and Higuchi equation. The R-square showed in the indicated that one-order equation was a superior to fit to the release data.
Table 4. R-square of the optimized formulation fitted to zero-order, first-order and Higuchi equations.
Bioavailability of multiple-unit floating-bioadhesive cooperative minitablets
The mean plasma level profiles of famotidine obtained following oral administration were compared in , and the main pharmacokinetic parameters were shown in . The mean value of Cmax was 717.30 ± 68.44 ng/mL and the Tmax was 3 h after oral administration of minitablets. However, after oral administration of market tablets the average value of Cmax was 921.92 ± 111.95 ng/mL and the Tmax was 1.5 h. The AUC0–∞ of the minitablets was approximately 1.6 times higher than the AUC0–∞ of market tablets. Famotidine minitablets can improve drug absorbent and bioavailability by increasing the residence time after oral administration. So the bioavailability of famotidine floating-bioadhesive cooperative minitablets is significantly improved compared with the market tablets.
Figure 9. Plasma concentration-time profiles of famotidine in rats given famotidine minitablets and reference tablets (XinFaDing®) (mean ± SD, n = 6).
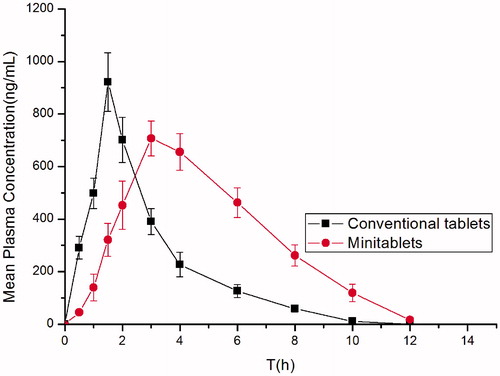
Table 5. The main pharmacokinetic parameters obtained following oral administration of XinFaDing® and the minitablets containing famotidine to rats.
Conclusion
The innovation of this article is the combination of minitablets and Gastro-retentive drug delivery system (GRDDS). There is almost no literature reported this dosage form. Floating-bioadhesive cooperative minitablets not only have the advantages of the multiple-unit delivery system, but also can prolong the drugs in gastric retention time and improve the bioavailability of drugs.
In conclusion, a new multiple-unit Gastro retentive drug delivery system had been developed. F6 containing HPMC K4M (50.00%, w/w), Carbopol 971P (10.00%, w/w) and NaHCO3 (10.00%, w/w) was a promising system exhibiting excellent floating properties, bioadhesive strength and sustained drug release characteristics. These minitablets floated after contacted with gastric fluid in 1 min and remained buoyant over an extended period of time (more than 8 h). Their robustness under varying pH or agitation rates was also demonstrated.
The parameter AUC0–∞ of the minitablets and the market tablets was 4096.49 ± 520.06 h ng/mL and 2514.06 ± 279.59 h ng/mL, while the relative bioavailability was 162.94 ± 31.64. The optimized gastric floating-bioadhesive cooperative minitablets containing famotidine did improve the oral bioavailability of famotidine in rats.
Declaration of interest
This work was financially supported by the Major State Basic Research Development Program of the National Science and Technology of China for new drugs development (Program No. 2012CB724002) and Natural Science Foundation of Jiangsu Province (No. BK20130663). The authors report no conflict of interest.
References
- Agarwal V, Mishra B. (1999). Design, development, and biopharmaceutical properties of buccoadhesive compacts of pentazocine. Drug Dev Ind Pharm 25:701–9
- Atyabi F, Sharma H, Mohammad H, et al. (1996). In vivo evaluation of a novel gastric retentive formulation based on ion exchange resins. J Controlled Release 42:105–13
- Baki G, Bajdik J, Pintye-Hódi K. (2011). Evaluation of powder mixtures and hydrophilic gastroretentive drug delivery systems containing zinc acetate and sodium bicarbonate. J Pharm Biomed Anal 54:711–16
- Bernkop-Schnürch A. (2005). Mucoadhesive systems in oral drug delivery. Drug Discov Today Tech 2:83–7
- Bertram U, Bodmeier R. (2006). In situ gelling, bioadhesive nasal inserts for extended drug delivery: in vitro characterization of a new nasal dosage form. Eur J Pharm Sci 27:62–71
- Bredenberg S, Nyholm D, Aquilonius SM, et al. (2003). An automatic dose dispenser for microtablets—a new concept for individual dosage of drugs in tablet form. Int J Pharm 261:137–46
- Cedillo-Ramirez E, Villafuerte-Robles L, Hernandez-Leon A. (2006). Effect of added Pharmatose DCL11 on the sustained-release of metronidazole from Methocel K4M and Carbopol 971P NF floating matrices. Drug Dev Ind Pharm 32:955–65
- Chavanpatil MD, Jain P, Chaudhari S, et al. (2006). Novel sustained release, swellable and bioadhesive gastroretentive drug delivery system for ofloxacin. Int J Pharm 316:86–92
- Costa P, Sousa Lobo JM. (2001). Modeling and comparison of dissolution profiles. Eur J Pharm Sci 13:123–33
- Fix JA, Cargill R, Engle K. (1993). Controlled gastric emptying. III. Gastric residence time of a nondisintegrating geometric shape in human volunteers. Pharm Res 10:1087–9
- Gambhire MN, Ambade KW, Kurmi SD, et al. (2007). Development and in vitro evaluation of an oral floating matrix tablet formulation of diltiazem hydrochloride. AAPS PharmSciTech 8:E166–74
- Gerogiannis V, Rekkas D, Dallas P, et al. (1993). Floating and swelling characteristics of various excipients used in controlled release technology. Drug Dev Ind Pharm 19:1061–81
- Goole J, Vanderbist F, Amighi K. (2007). Development and evaluation of new multiple-unit levodopa sustained-release floating dosage forms. Int J Pharm 334:35–41
- Graffner C, Josefsson K, Stockman O. (1986). Intra- and intersubject variation of erythromycin absorption from single-unit and multiple-unit enteric-coated products. Biopharm Drug Dispos 7:163–71
- Gröning R, Heun G. (1984). Oral dosage forms with controlled gastrointestinal transit. Drug Dev Ind Pharm 10:527–39
- Gröning R, Heun G. (1989). Dosage forms with controlled gastrointestinal passage – studies on the absorption of nitrofurantoin. Int J Pharm 56:111–16
- Iannuccelli V, Coppi G, Sansone R, et al. (1998). Air compartment multiple-unit system for prolonged gastric residence. Part II. In vivo evaluation. Int J Pharm 174:55–62
- Jaimini M, Rana A, Tanwar Y. (2007). Formulation and evaluation of famotidine floating tablets. Curr Drug Deliv 4:51–5
- Jivani RR, Patel CN, Jivani NP. (2009). Design and development of a self correcting monolithic gastroretentive tablet of baclofen. Sci Pharm 77:651–67
- Korsmeyer RW, Gurny R, Doelker E, et al. (1983). Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm 15:25–35
- Kumar R, Patil M, Patil SR, et al. (2009). Formulation and evaluation of effervescent floating tablet of famotidine. Int J Pharmnt Res 1:754–63
- Lennartz P, Mielck JB. (1998). Minitabletting: improving the compactibility of paracetamol powder mixtures. Int J Pharm 173:75–85
- Liu Y, Zhang J, Gao Y, et al. (2011). Preparation and evaluation of glyceryl monooleate-coated hollow-bioadhesive microspheres for gastroretentive drug delivery. Int J Pharm 413:103–9
- Lopes CM, Sousa Lobo JM, Costa P, et al. (2006). Directly compressed mini matrix tablets containing ibuprofen: preparation and evaluation of sustained release. Drug Dev Ind Pharm 32:95–106
- Mohammed FA, Khedr H. (2003). Preparation and in vitro/in vivo evaluation of the buccal bioadhesive properties of slow-release tablets containing miconazole nitrate. Drug Dev Ind Pharm 29:321–37
- Munday D. (1994). A comparison of the dissolution characteristics of theophylline from film coated granules and mini-tablets. Drug Dev Ind Pharm 20:2369–79
- Munday D, Fassihi A. (1989). Controlled release delivery: effect of coating composition on release characteristics of mini-tablets. Int J Pharm 52:109–14
- Papadimitriou E, Buckton G, Efentakis M. (1993). Probing the mechanisms of swelling of hydroxypropylmethylcellulose matrices. Int J Pharm 98:57–62
- Roberts M, Vellucci D, Mostafa S, et al. (2012). Development and evaluation of sustained-release Compritol® 888 ATO matrix mini-tablets. Drug Dev Ind Pharm 38:1068–76
- Sato Y, Kawashima Y, Takeuchi H, et al. (2004). In vitro and in vivo evaluation of riboflavin-containing microballoons for a floating controlled drug delivery system in healthy humans. Int J Pharm 275:97–107
- Siepmann J, Kranz H, Bodmeier R, et al. (1999). HPMC-matrices for controlled drug delivery: a new model combining diffusion, swelling, and dissolution mechanisms and predicting the release kinetics. Pharm Res 16:1748–56
- Sriamornsak P, Thirawong N, Korkerd K. (2007). Swelling, erosion and release behavior of alginate-based matrix tablets. Eur J Pharm Biopharm 66:435–50
- Tadros MI. (2010). Controlled-release effervescent floating matrix tablets of ciprofloxacin hydrochloride: Development, optimization and in vitro–in vivo evaluation in healthy human volunteers. Eur J Pharm Biopharm 74:332–9
- Thomson SA, Tuleu C, Wong IC, et al. (2009). Minitablets: new modality to deliver medicines to preschool-aged children. Pediatrics 123:235–8
- Wang J, Tabata Y, Bi D, et al. (2001). Evaluation of gastric mucoadhesive properties of aminated gelatin microspheres. J Controlled Release 73:223–31