
Abstract
Trans-resveratrol (t-RVT) is a potent antioxidant. By virtue of extensive pre-systemic metabolism and existence of enterohepatic recirculation, t-RVT bioavailability is almost zero. The current study aimed to develop self-nanoemulsifying drug delivery systems (SNEDDS) using long-chain triglycerides (LCTs) of t-RVT in an attempt to circumvent such obstacles. Equilibrium solubility studies indicated the choice of Lauroglycol FCC as lipid, and of Labrasol and Transcutol P as surfactants, for formulating the SNEDDS. Ternary phase diagrams were constructed to select the areas of nanoemulsions, and the amounts of lipid (X1) and surfactant (X2) as the critical factor variables. The SNEDDS were optimized using 32 central composite design (CCD) and the optimized formulation (OPT) located using overlay plot. The nanometer size range and high negative values of zeta potential depicted non-coalescent nature of the SNEDDS. Optimized formulation indicated marked improvement in drug release profile vis-à-vis pure drug. Cloud point determination and accelerated stability studies ascertained the stability of OPT. Augmentation in the values of Ka (3.29-fold) and AUC (4.31-fold) indicated significant enhancement in the rate and extent of bioavailability by the OPT compared with pure drug. In situ perfusion (SPIP) studies in Wistar rats construed remarkable enhancement in the absorptivity and permeability parameters of SNEDDS vis-à-vis the pure drug. Successful establishment of level A of in vitro/in vivo correlation substantiated the judicious choice of the in vitro dissolution milieu for simulating the in vivo conditions. The present study, therefore, reports the successful development of SNEDDS with distinctly enhanced bioavailability of t-RVT.
Introduction
Self-nanoemulsifying drug delivery systems (SNEDDS) are lipid-based nanocarriers that have recently exhibited an intriguing role in oral delivery of drugs (Elsheikh et al., Citation2012). SNEDDS are newer and novel technological innovations with immense potential in oral bioavailability enhancement of lipophilic drugs (Elnaggar et al., Citation2009; Basalious et al., Citation2010). Of late, SNEDDS have emerged as an effective delivery system owing to their inherent meritorious visages.
Several potential advantages of SNEDDS include capability of bypassing hepatic portal route and promoting the lymphatic transport of lipophilic drugs, reducing metabolism by cytochrome-P450 family of enzymes present in the gut enterocytes (Porter et al., Citation2001; Balakrishnan et al., Citation2009; Date et al., Citation2010) and overcome enterohepatic recirculation (Elsheikh et al., Citation2012). This hypothesis was believed to be attained chiefly, by lymphatic transport via Peyer’s patches along the GI tract (Balakumara et al., Citation2013). Self-emulsifying drug delivery system is one among the lipid-based drug delivery systems that has been currently investigated for its advantages, providing a large interfacial area for partitioning the drug between oil and GI fluid (Jeevana et al., Citation2011).
Of the excipients employed for the formulation of SNEDDS, lipids have an enormous role for the biological fate of drug. Studies have shown that the type of absorption pathway and subsequent transportation of drug is drastically influenced by the long-chain triglycerides (LCTs) (Porter et al., Citation2007; Bandyopadhyay et al., Citation2012). The LCTs are transported via the intestinal lymphatics. Furthermore, bypassing hepatic first-pass metabolism was also reported as a result of oral lymphatic targeting of drugs (Nielsen et al., Citation2008; Date et al., Citation2010; Elsheikh et al., Citation2012). The LCTs are likely to augment the lymphatic transport of a lipophilic drug substance leading to enhanced oral bioavailability (Nielsen et al., Citation2008).
The drug, i.e. trans-resveratrol (t-RVT), chosen in the present study is a BCS class II drug with poor water-solubility and high permeability (log P of 3.1) (Amri et al., Citation2012). Besides these, it undergoes rapid first-pass metabolism by CYP3A4 in the liver and suffers enterohepatic recirculation (Marier et al., Citation2002; Singh et al., Citation2013), leading eventually to marked reduction in the drug oral bioavailable fraction (almost zero) in humans and animals like rats (Amri et al., Citation2012). Under the aforementioned circumstances, various formulation approaches of t-RVT have been reported, such as liposome (Hung et al., Citation2006), solid dispersions (Wegiel et al., Citation2013), β-cyclodextrins inclusion complex (Troche-Pesqueira et al., Citation2013), microspheres (Nam et al., Citation2005), suspensions, but all with limited fruition.
The literature lacks any data about the use of SNEDDS for the improvement of the dissolution and oral absorption of t-RVT. The current work endeavors to design an OPT SNEDDS system of t-RVT, resulting in enhanced oral bioavailability potential. It is anticipated that this study not only offers a good example of augmenting the oral bioavailability of t-RVT by the use of SNEDDS but also provides a promising oral formulation of t-RVT for clinical application.
Materials and methods
t-RVT was provided ex-gratis by M/s Sami Labs, Bangalore, Karnataka, India. Labrafil, olive oil, and Lauroglycol FCC were received as the gift samples from M/s Gattefosse, Mumbai, India. Tween 80 was procured from M/s HIMEDIA Laboratories Pvt. Ltd., Mumbai, India. Labrasol and Transcutol P were received as the gift samples from M/s Gattefosse, Mumbai, India. All other materials and chemicals procured for the studies were of analytical grade, and were used as such as obtained.
Preliminary investigation
Solubility studies
The solubility of t-RVT was determined in various LCTs, namely Labrafil, olive oil, and Lauroglycol FCC and surfactants, namely Transcutol P, Labrasol, and Tween 80 by placing an excess amount of t-RVT (approximately 100 mg) into 10 ml of each vehicle. Then, the mixture was vortexed and kept for three days at 37 ± 1 °C in a shaking water bath to facilitate the solubilization. The samples were centrifuged at 5000×g for 10 min to remove the undissolved t-RVT. The supernatant was taken and diluted with methanol for the quantification of t-RVT by the HPLC method as previously reported by us earlier (Singh et al., Citation2013).
Construction of ternary phase diagram
The existence of self-emulsifying lipid formulation fields that could self-emulsify under dilution and gentle agitation was identified from ternary phase diagrams of systems containing lipids and surfactants. Compositions with different oil-to-surfactant ratios, within the range of 1:9 and 9:1, were selected and titrated with water at ambient temperature. The mixtures were observed visually following equilibration. Subsequently, a series of ternary phase diagrams were constructed using PCP Disso software ver 3.0 (M/s Pune College of Pharmacy, Pune, India). The phase diagrams were so constructed to delineate the boundaries of various phases, i.e. nano/microemulsion, emulsion, emulgel, and microgel (Bandyopadhyay et al., Citation2012). The generated samples, clear or slightly bluish in appearance, were taken as the nanoemulsions.
Preparation of SNEDDS as per CCD design
Once the self-emulsifying region was identified, the desired component ratios of SNEDDS were selected for drug incorporation. Lipidic and emulsifying excipients were chosen on the basis of the formation of maximal nanoemulsion region in the ternary-phase diagram. The SNEDDS formulation was prepared by dissolving the formulation amount of t-RVT (100 mg) in the lipid(s) at 37 °C and the surfactant, in the predetermined ratio, was added to the lipidic drug solution, while stirring at high speed using a magnetic stirrer (M/s Remi, Bangalore, India) maintained at 37 °C.
For the preparation of SNEDDS, Lauroglycol FCC as lipid (X1) and Transcutol P as surfactant (X2) were chosen and finally selected as the two critical influential factors for further formulation optimization work (Singh et al., Citation2012a). A CCD with α = 1 was employed, where the amounts of oil and surfactant were studied at three levels each (Singh et al., Citation2012b). Overall, a set of 13 experimental runs each were studied as per the experimental design matrix as depicted in . The formulation at the intermediate coded factor levels (i.e. 0,0) was studied in quintuplicate. The response variables considered for the current DoE optimization studies encompassed, globule size (Dnm), amount permeated in 45 min (Perm45 min), and %dissolution efficiency in 30 min (%DE30 min).
Table 1. Preparation of t-RVT SNEDDS as per the experimental design.
Optimization data analysis
The response variables, which were considered for systematic DoE optimization studies, included Dnm, Perm45 min, and %DE30 min. Polynomial regression results were demonstrated for the studied responses. Finally, the prognosis of OPT formulation was conducted using overlay plots, drawn using the Design Expert software (M/s Stat-Ease, MN) (Singh & Pai (2013). Overlay plot was obtained by superimposing the values of various response variables in order to locate the OPT formulation. The observed and predicted responses were critically compared and the percent bias (i.e. prediction error) was also calculated with respect to the observed responses.
Characterization of OPT formulation
Determination of the emulsification time
In order to determine the emulsification time (the time needed to reach the emulsified and homogeneous mixture, upon dilution), 1 g of OPT formulation was added to 200 ml of 0.1 N HCl at 37 °C with gentle agitation using a magnetic stirrer. The formulation was assessed visually according to the rate of emulsification and the final appearance of the emulsion.
Determination of globule size and zeta potential
Aliquots (1 ml) of the OPT formulation, serially diluted 100-fold with purified water, were employed to assess the globule size and zeta potential using particle size analyzer (ZS 90, M/s Malvern, Worcesershine, UK).
Thermodynamic stability studies
The OPT formulation was subjected to various thermodynamic studies in order to assess the phase separation and/or stability of the nanoemulsion (Lawrence et al., Citation2000).
Centrifugation study
The OPT formulation was centrifuged at 2000 × g for 15 min. The resultant formulation was then checked for any instability problem, such as phase separation, creaming, or cracking.
Heating and cooling cycle
The contents of the OPT formulation were subjected to six heating/cooling cycles between 4 °C and 40 °C with storage at each temperature for not less than 48 h. The resultant formulation was assessed for their physical instability like phase separation and precipitation.
Cloud point measurement
The OPT formulation was diluted with distilled water in the ratio of 1:100. The diluted samples were placed in a water bath and its temperature was increased gradually. Cloud point was spectrophotmetrically determined as the temperature at which there was a sudden appearance of cloudiness (Elnaggar et al., Citation2009).
Drug release studies
Dissolution studies were carried out for OPT formulation in triplicate, employing USP Apparatus 2 (M/s Labindia, DS A/S 8000, Mumbai, India) with 500 ml of simulated gastric fluid (SGF) containing 0.3% (w/v) sodium lauryl sulphate (SLS), stirred at 50 rpm at a temperature of 37 ± 0.5 °C (Elsheikh et al., Citation2012). At predetermined time intervals, an aliquot (1 ml each) of the sample was collected, filtered, and analyzed for the content of t-RVT by the HPLC method (Singh et al., Citation2013). An equivalent volume (1 ml) of fresh dissolution medium was added to compensate for the loss due to sampling. Dissolution study was also conducted on pure drug in an analogous manner.
In vivo pharmacokinetic studies in rat
The animal investigations were performed as per the requisite protocol approved by the Institutional Animal Ethics Committee [Letter no AACP/IAEC/Jun-2012-02]. The Committee is duly approved for the purpose of control and supervision of experiments on the animals by the Government of India.
The pharmacokinetic study involved two groups. Six rats (male Wistar, weighing 250–300 g) were randomly distributed among each group. Group I received OPT formulation and Group II received the pure drug in the form of 0.3% carboxymethyl cellulose suspension. All the animal groups received a dose equivalent to 20 mg of t-RVT per kg of body weight (Almeida et al., Citation2009; Singh et al., Citation2013). Following oral drug administration, the rats kept in the cages were allowed access to food and water ad libitum. Serial aliquots of the blood samples (100 μl each) were withdrawn from the retro-orbital plexus under mild ether anesthesia at 0, 0.33, 0.5, 1, 1.5, 2, 3, 4, 6, and 8 h post-dosing and placed in the heparinized microcentrifuge tubes (50 units heparin/ml of blood). Plasma was harvested by centrifugation at 15, 000×g for 15 min and stored at −20 °C until analyzed. Plasma samples were analyzed by HPLC (Singh et al., Citation2013). Non-compartmental pharmacokinetic parameters for extravascular input, i.e. Cmax, Tmax, AUC, t1/2, Ke, and MRT were computed by choosing Kinetica 5.0.11 version software (Thermo Fisher Scientific Inc. Waltham, MA).
In vitro/in vivo correlation
Attempts were made to correlate the in vivo plasma level data obtained for OPT formulation and pure drug with the corresponding in vitro drug release data and establish IVIVC. For establishing Level A IVIVC, percent drug absorbed data at various time points were obtained using the modified Wagner–Nelson method and correlated with percent drug release data.
In situ SPIP studies
In situ SPIP studies were performed in male Wistar rats. The animals were before made to abstain from solid food at least 24 h prior to the study. The SPIP studies were carried out on the OPT formulation and pure drug each in hexaplicate, employing an in-house fabricated assembly. The animals were anesthetized using thiopental sodium (50 mg/kg) intraperitoneally. Following mid-line incision of the abdomen, the proximal part of the jejunum, 2–5 cm below the Ligament of Trietz, was cannulated with a glass cannula and connected to a reservoir (Ho et al., Citation2008). The second incision was made 10–15 cm below the first incision and connected with an outflow glass cannula. The intestinal segments were perfused with Krebs–Ringer's solution until the perfusate was clear. The intestinal segments were consequently perfused with OPT formulation and pure drug maintained at 37 ± 1 °C at a perfusion rate of 0.25 ml/min. Steady state was achieved within 30 min, after which aliquots of samples (1 ml each) were periodically withdrawn at a regular intervals of 10 min each. The volume of sample for each time interval was 2 ml. Samples were stored at −20 °C until analysis. Samples were filtered and directly injected onto HPLC column and required no sample preparation prior to analysis.
Transmission electron microscopy (TEM)
The morphology of the OPT formulation was observed using TEM attached with a mega view II digital camera (H 7500, Hitachi, Tokyo, Japan). A drop of sample diluted with water was placed on a copper grid and the excess was drawn off with a filter paper. Samples were subsequently stained with 1% of uranyl acetate solution for 1 min. The image was magnified and focused on a layer of photographic film.
Stability studies
The OPT formulation was subjected to stability studies, carried out at 25 ± 2 °C/60% ± 5% RH, as per the ICH guidelines for the climatic zone IV. The formulation was kept in air-tight glass vials and assayed periodically, at the time points of 0, 1, 3, and 6 months, for globule size, dissolution performance, and emulsification time.
Results
Initial studies for screening of excipients
Solubility studies
Equilibrium solubility studies were carried out to investigate the maximum soluble fraction of t-RVT in different lipids and surfactants. The maximum solubility of t-RVT was observed in Lauroglycol FCC (0.37 mg/ml), while the minimum was found in olive oil (0.19 mg/ml). The dose of t-RVT (i.e. 100 mg), higher solubility of drug in any lipid, would augment its drug loading potential. Accordingly, on the basis of maximum solubilities, Lauroglycol FCC was chosen as the lipidic constituent(s) for SNEDDS formulation. Likewise among the surfactants, the maximum solubility of t-RVT was observed in Transcutol P (216.43 mg/ml) and minimum solubility was found in Labrasol (0.41 mg/ml).
Phase diagram study
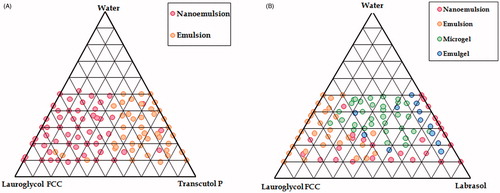
Construction of phase diagrams provides a picture of probable concentrations of components that might yield spontaneous emulsion and the nature of the resultant dispersions, such as separated dispersions, coarse emulsions, and self-nanoemulsifing systems. As indicated in , the nanoemulsion region for SNEDDS was prominent with Transcutol P, whereas it was minimal with Labrasol. Further, the ternary plot between Lauroglycol FCC and Transcutol P yielded only two regions (i.e. nanoemulsion and emulsion), where all the four regions were observed with Labrasol (). Hence, Lauroglycol FCC and Transcutol P were selected for the formulation of SNEDDS.
Figure 1. Ternary phase diagrams employing Lauroglycol FCC with surfactants namely Transcutol P (A) and Labrasol (B).
Response surface analyses
The response surface plots were constructed to facilitate the understanding of contribution of formulation variables and their interactions. The plot () shows that a sharp linear decrease in globule size was observed from lower to intermediate levels of lipid following which there was minuscule increase in the globule size with higher levels of lipid. With an increase in surfactant concentrations, however, a relatively little curvilinear descending trend was observed on the globule size.
Figure 2. Response surface plots showing the influence of Lauroglycol FCC with Transcutol P on Dnm, Perm45 min and %DE30 min.
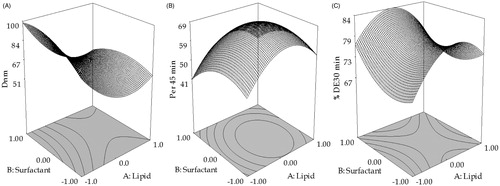
A curvilinear response surface plot was observed for Perm45 min. The plot () shows increase in Perm45 min till intermediate levels of lipid, where after a sharp decline was observed. With surfactant, Perm45 min showed marginal reduction from lower to higher levels at lower levels of lipid. A similar pattern of permeation was reported employing the LCT’s (Bandyopadhyay et al., Citation2012).
As illustrated in , %DE30 min increased till intermediate levels of lipid, where after a sharp decline was observed at lower levels of surfactant. However, at higher levels of surfactant, %DE30 min decreased till intermediate levels of lipid, after which an insignificant increase was observed at low levels of surfactant.
Determination of droplet size
The globule size of all the 13 SNEDDS formulations, prepared as per the experimental design, ranged between 48 and 100 nm.
In vitro drug release studies
All the formulations tended to release ∼80% of drug within initial 30 min, with relatively miniscule amount of drug release after 5 min.
DoE validation and selection of OPT formulation
The OPT formulation was selected by “trading off” various response variables while adopting the following criteria:
The OPT formulation (containing Lauroglycol FCC: 262.0 mg and Transcutol P: 225.0 mg) was found to fulfill maximal criteria for optimal performance. The said formulation exhibited Dnm of 56.73 nm, Perm45 min of 89.47%, and DE30min of 86.62%. Selection of the OPT formulation was based on “trading off” of various response variables, i.e. maximization of Perm45 min (i.e. indicating drug transport potential across GI tract) and %DE30 min (i.e. indicating adequate drug release), and minimization of Dnm (i.e. necessary for micellar absorption via lymphatic route).
Formulation characterization of OPT formulation
Determination of the emulsification time
The OPT formulation exhibited a rapid rate of emulsification (∼43 s).
Determination of globule size and zeta potential
The globule size and zeta potential of the OPT formulation were found to be 56.73 nm and −29.76 mV, respectively.
Thermodynamic stability studies
The OPT formulation did not show any signs of precipitation, creaming, or cracking thereby establishing the kinetic stability of the system.
Cloud point measurement
The cloud point for OPT formulation was observed to be 58–61 °C.
In vitro drug release comparison with pure drug
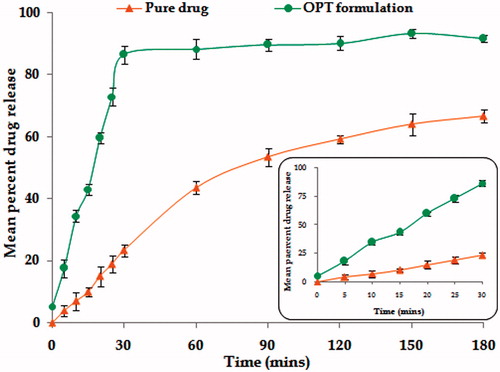
Marked improvement was observed in the release profiles of OPT formulation vis-à-vis pure drug (). Drug dissolution was nearly completed within 30 min in case of the OPT formulation, as compared with that of the pure drug, where drug release was only 66.56%.
Figure 3. Plot showing mean percent t-RVT release from OPT formulation and pure drug. The inset shows the mean percent drug release in 30 min. (n = 6 ± SD).
In vivo pharmacokinetic studies in rats
The mean plasma concentration–time profile () depicts significant higher plasma levels from OPT formulation with respect to pure drug (p < 0.001). Further, the lower magnitude of SEM of OPT formulation indicated that the pharmacokinetic profile was more reproducible than that of pure drug.
Figure 4. Plasma drug level profiles of OPT formulation and pure drug. Each point represents mean of six replicates and each cross bar indicates 1 SEM.
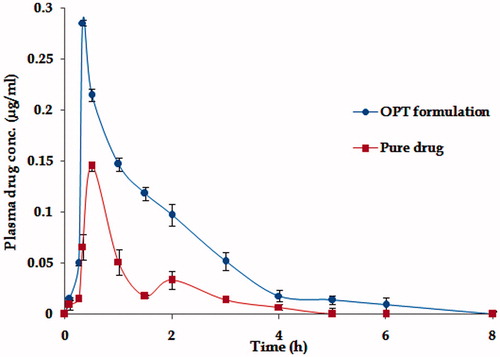
The absorption rate was markedly enhanced, as revealed by distinct improvement in various pharmacokinetic parameters, i.e. Tmax and AUC. Maximal change in the pharmacokinetic parameter was observed in case of absorption rate constant (Ka), which is known to reflect the first-order rate of drug absorption. Nearly, 3.29-fold augmentation in the magnitude of Ka was perceptible with the OPT formulation. On the whole, the values of Cmax and AUC rose considerably vis-à-vis pure drug to 2.34 and AUC 4.31-fold, respectively, in case of the OPT formulation ratifying distinct improvement in extent of bioavailability too (p < 0.001). Subsequent a one-way ANOVA carried out on various pharmacokinetic parameters namely AUC, Ka, and Tmax connoted highly statistically significant variation (p < 0.001) in the drug absorption potential of t-RVT from, i.e. OPT formulation vis-à-vis pure drug.
In vitro/in vivo correlation (IVIVC)
Level A correlation was investigated between the two variables.
In situ single-pass intestinal perfusion (SPIP) studies in rat
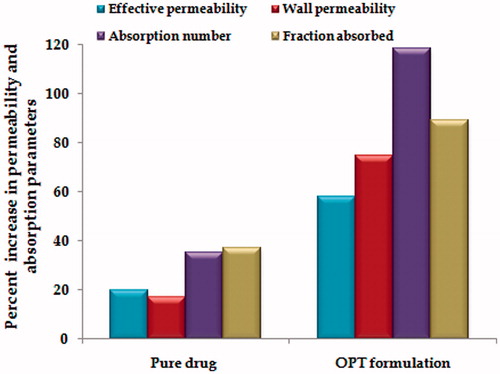
The OPT formulation showed nearly 2.94 - and 3.79-fold enhancement in effective and wall permeability vis-à-vis pure drug, respectively, as depicted in . There was tremendous augmentation in the values of absorption number and fraction absorbed with the OPT formulation as compared with pure drug. The increase in absorption number with the OPT formulation was 3.37-fold, while the corresponding increase in the fraction absorbed was 2.41-fold. Overall, the results indicated that the OPT formulation was remarkably superior to the pure drug in absorption and permeability.
Figure 5. Percent increase in permeability parameters and absorption parameters in OPT formulation vis-à-vis pure drug using in situ single pass intestinal perfusion technique.
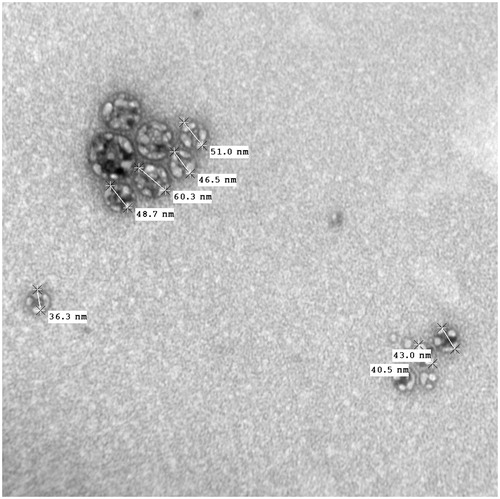
Transmission electron microscopy
shows the electron microscopic image, depicting the morphology of the reconstituted OPT formulation. As is evident from the figure, all the globules were of uniform shape, with globule size of most of them as less than 100 nm. The figure clearly illustrate that there are no signs of coalescence, indicating thereby the enhanced physical stability of the formulation.
Figure 6. TEM image showing particle shape and size of OPT formulation.
Discussion
The current work entails the development of OPT formulation of t-RVT and its subsequent pharmacokinetic evaluation for ratifying its biopharmaceutical superiority. SNEDDS spontaneously forms nanoemulsions when exposed to GIT fluids. The spontaneous formation of nanoemulsions advantageously presents the drug in a dissolved form. The resultant small droplet size provides a large interfacial surface area for drug release and absorption. In addition, the specific system components promote the intestinal lymphatic transport of drugs.
Equilibrium solubility studies were carried out in an attempt to find out the maximum soluble fraction of t-RVT in different lipids and surfactants. Based on the maximal drug solubility, Lauroglycol FCC was selected as the lipidic excipient for formulating SNEDDS formulation. Lauroglycol FCC is a known bioavailability enhancer and increases the oral bioavailability in associated with inhibition of the enterocytic drug-metabolizing enzyme CYP3A4. Oily phase is the most important component of SNEDDS formulations, as it can solubilize lipophilic drugs and increase the fraction of lipophilic drug transported via the intestinal lymphatic system, thereby increasing the absorption from the gastrointestinal tract, depending on the molecular nature of the triglycerides.
Non-ionic surfactants are generally considered less toxic than ionic surfactants. They are usually accepted for oral ingestion (Elnaggar et al., Citation2009). In addition, they can fabricate reversible changes in intestinal mucosa, thus leading to augment permeability absorption of drug (Swenson at al., Citation1994). Transcutol, which is a solubilizer and absorption enhancer, was found to be a very efficient solubilizer for t-RVT, and so was chosen as a surfactant in the development of SNEDDS formulation aiming to improve the drug-loading capabilities.
To ensure the spontaneity of SNEDDS to form the nanoemulsions within the GI conditions, the construction of ternary plots is considered to be a vital exercise (Bandyopadhyay et al., Citation2012). There are reports which clearly indicate that HLB value of surfactant facilitates in lowering the interfacial energy, leading to the formation of a stable nanoemulsion. The surfactant used in the present study, i.e. Transcutol, has HLB value of 4.2. Thus, the HLB values and ternary plots constructed clearly indicate the synergism of the excipients in reducing the interfacial tension as well as the formation of stable nanoemulsion region.
The CCD is documented as the most efficient design. The high values of r2 exhibited by the polynomial relationships vouched high statistical validity (p < 0.001) of polynomial models for fitting to the experimental data. The linearity of the correlation plots between predicted and observed responses (p < 0.001), and nearly uniform and random scatter of the corresponding residual plots indicate high prognostic ability of the postulated model.
All the formulations, prepared as per the experimental design, exhibited nano globule size, ranging between 48.777 and 100 nm, which is considered ideal for a low emulsification time and enhanced micellar absorption (Porter & Charman, Citation2001). Remarkably, small globule size was observed at the higher level of the lipid and lower level of the surfactant.
The OPT formulation exhibited a rapid rate of emulsification (∼43 s). It is obvious that rapid emulsification is correlated with lower content of oil and higher content of co-surfactant which result in lower viscosity of the system (Basalious et al., Citation2010).
The nano globule size is considered ideal to result in lower emulsification time, enhanced absorption through lymphatics and subsequent augmentation in the therapeutic efficacy of drugs. High values of zeta potential construe an increase in electrostatic repulsive forces, thus ruling out the plausibility of coalescence. As such, the results clearly show that phase separation did not occur which indicate the formation of stable SNEDDS (Gupta et al., Citation2011).
Thermodynamic stability indicates the kinetic stability of a formulation and is employed to study the chemical reaction occurring between the excipients of a formulation. The SNEDDS system undergoes spontaneous in situ solubilisation in the GI lumen to form a nanoemulsion system. As such, formulation should possess considerable stability in order to prevent precipitation, creaming, or cracking. The OPT formulation was subjected to centrifugation studies and, heating and cooling cycle exposure.
Determination of cloud point is considered an important factor in the SNEDDS formulation consisting of non-ionic surfactant. At temperatures higher than the cloud point, an irreversible phase separation occurs due to dehydration of its ingredients, which may affect drug absorption (Chen et al., Citation2000). Hence, to avoid this phenomenon, the cloud point for the SNEDDS formulation should be above body temperature (i.e. 37 °C). Much higher magnitude of the cloud point of SNEDDS formulation was found to indicate high stability of the SNEDDS formulation without any risk of phase separation.
Significant reduction in the values of dissolution time for OPT formulation is quite indicative of higher bioavailability potential of t-RVT, a BCS class II drug exhibiting dissolution-limited absorption.
SNEDDS formulation was designed to augment the oral bioavailability of t-RVT. To our knowledge, there was no comprehensive study reported in the literature demonstrating the in vivo performance of t-RVT SNEDDS. We believe that no single parameter can alone influence the fate of the formulations in vivo; rather it is a collective influence of many parameters. Blood levels after oral administration of pure drug were compared with OPT formulation are shown in .
Low values of AUC and Cmax after administration of adequate dose of pure drug (20 mg/kg) also point toward incomplete absorption of drug from rats. The values of Tmax, i.e. 0.5 h observed for pure drug in rats are quite in accordance with the literature (Singh et al., Citation2013). This low absorption rate potential can be attributed to slower and incomplete dissolution of this BCS class II drug in GI tract. Thus, besides poor aqueous solubility of t-RVT, decreased extent of bioavailability in rat may be due to a multitude of other reasons. The potential causes for decreased absorption include thicker GI barriers, higher hepatic first-pass metabolism, enterohepatic circulation, lower residence time in GI tract, and lower absorptive surface area.
The oral bioavailability of t-RVT is less than 1% owing to various factors which include partial solubility in water, extensive metabolism in the intestine, intense hepatic first-pass metabolism, colonic bacterial metabolism, and poor absorption. Further, following rapid and extensive metabolism, various metabolites of t-RVT are formed namely t-RVT glucuronides and t-RVT sulfates. Extremely rapid sulfate conjugation by the intestine/liver appears to be the rate-limiting step in t-RVT bioavailability. Second peak at 2 h indicates enterohepatic recirculation of the drug that has been well documented and enterohepatic recirculation is susceptible to circadian variation (Almeida et al., Citation2009) (as is evident from where two peaks of Cmax and Tmax were observed). A major portion of the bile acids secreted are reabsorbed from the intestines and returned via the portal circulation to the liver, thus completing the enterohepatic cycle (Marier et al., Citation2002). The parent compound and its conjugated metabolite undergo enterohepatic recirculation, resulting in multiple peaks in the plasma concentration–time profile.
The higher Cmax levels coupled with higher values of AUC observed in OPT formulation indisputably vouch distinct improvement in rate and extent of drug bioavailability. This can possibly be attributed to the lymphatic transport of t-RVT by such systems. This augmentation in bioavailability would eventually result in an escalation in the intensity of therapeutic effect of t-RVT. Exclusively higher values of Ka and lower values of Tmax for the OPT formulation vis-à-vis the pure drug indicate significantly quicker absorption of t-RVT from the self-nanoemulsifying systems.
Excellent Level A correlation was observed with the OPT formulation. Since t-RVT is a poorly water-soluble drug exhibiting nearly complete and dissolution limited absorption, it is anticipated to show good point-to-point correlation between in vitro and in vivo performance (i.e. Level A).
Significant improvement in the magnitudes of fraction absorbed and absorption number exhibited by the OPT formulation () indicated distinct improvement in the amount of drug transported across the intestine. This corroborated their high bioavailability improvement potential vis-à-vis pure drug. Much higher values of the absorptivity parameters from OPT formulation can be ascribed to the transport of drug via lymphatic pathways, in accordance with an earlier literature report (Sun et al., Citation2011). The existence of enterohepatic circulation and extensive first-pass metabolism by CYP3A4 in liver has been documented as the plausible cause for low oral bioavailability of t-RVT (Marier et al., Citation2002; Amri et al., Citation2012). Marked enhancement in the effective permeability, a direct measure of absorption across the intestine (Bandyopadhyay et al., Citation2012), of the SNEDDS clearly demonstrates that the OPT formulation was able to augment drug absorption in rat. In addition, bypassing the portal circulation also avoids further metabolism in liver and enterohepatic circulation. Earlier literature reports also document the role of SNEDDS in metabolism and enterohepatic circulation inhibition of drugs like raloxifene hydrochloride (Elsheikh et al., Citation2012).
Also, the improvement in permeability of OPT formulation may be attributed to the high concentration of surfactant present in the formulation. Analogous results of increased intestinal permeability on increasing the surfactant concentration have been reported in the literature (Gao et al., Citation2011). The OPT formulation seems to have marginal advantage in enhancing in situ drug permeability and absorptivity, probably owing to faster emulsification and dissolution. The close analogy of the in vivo results with that of the SPIP studies ratifies the usefulness of this in situ technique in the estimation the bioavailability and/or permeation parameters.
Drug-release parameters during the stability studies unambiguously show that the globule size, dissolution performance, and emulsification time of the OPT formulation were negligibly altered during 6 months storage under stability conditions, ratify high robustness of the formulations even under stability condition.
Conclusions
The novelty of the current work is the development of SNEDDS solely through the judicious selection of apt blends of high-melting lipids and surfactants, and optimizing their composition using systematic “DoE” methodology. Considerable augmentation in the rate and extent of oral drug absorption ratified the better performance of the SNEDDS in enhancing the bioavailability of t-RVT. Besides drug-release enhancement and nano globule size, significant improvement in oral bioavailability of t-RVT from SNEDDS may be attributed ostensibly to reduced metabolism by CYP3A4 in liver and overcome enterohepatic circulation.
Acknowledgements
The authors gratefully acknowledge financial support and granting research fellowship (45/38/2011/Nan-BMS) from ICMR (Indian Council of Medical Research, Govt of India, New Delhi). Authors are also grateful to Sami Labs, Bangalore, India for providing the gift sample of t-RVT.
Declaration of interest
The authors have no conflict of interest.
References
- Almeida L, Vaz-da-Silva M, Falcao A, et al. (2009). Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol Nutr Food Res 53: S7–15
- Amri A, Chaumeil JC, Sfar S, Charrueau C. (2012). Administration of resveratrol: what formulation solutions to bioavailability limitations. J Control Release 158:182–93
- Balakrishnan P, Lee BJ, OH DH. (2009). Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm 72:539–45
- Balakumara K, Raghavana CV, Selvana NT, et al. (2013). Self-nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf B Biointerfaces 112:337–43
- Bandyopadhyay S, Katare OP, Singh B. (2012). Optimized self-nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf B Biointerfaces 100:50–61
- Basalious EB, Shawky N, Badr-Eldin SM. (2010). SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: development and optimization. Int J Pharm 391:203–11
- Chen F, Wang Y, Zheng F, et al. (2000). Studies on cloud point of agrochemical microemulsions. Colloids Surf A: Physicochem Eng Aspects 175:257–62
- Date AA, Desai N, Dixit R, Nagarsenker M. (2010). Self-nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine 5:1595–616
- Elnaggar YSR, El-Massik MA, Abdallah OY. (2009). Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm 380:133–41
- Elsheikh MA, Elnaggar YSR, Gohar EY, Abdallah OY. (2012). Nanoemulsion liquid preconcentrates for raloxifene hydrochloride: optimization and in vivo appraisal. Int J Nanomedicine 7:3787–802
- Gao F, Zhang Z, Bu H. (2011). Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: performance and mechanism. J Control Release 149:168–74
- Gupta S, Chavhan S, Sawant KK. (2011). Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids Surf A: Physicochem Eng Aspects 392:145–55
- Ho YF, Lai MY, Yu HY, et al. (2008). Application of rat in situ single-pass intestinal perfusion in the evaluation of presystemic extraction of indinavir under different perfusion rates. J Formos Med Assoc 107:37–45
- Hung CF, Chen JK, Liao MH. (2006). Development and evaluation of emulsion-liposome blends for resveratrol delivery. J Nanosci Nanotechnol 6:2950–8
- Jeevana JB, Sreelakshmi K. (2011). Design and evaluation of self-nanoemulsifying drug delivery system of flutamide. J Young Pharm 3:4–8
- Lawrence MJ, Rees GD. (2000). Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev 45:89–121
- Marier JF, Vachon P, Gritsas A. (2002). Metabolism and disposition of resveratrol in rats: extent of absorption, glucuronidation, and enterohepatic recirculation evidenced by a linked-rat model. J Pharmacol Exp Ther 302:369–73
- Nam JB, Ryu JH, Kim JW, et al. (2005). Stabilization of resveratrol immobilized in monodisperse cyano-functionalized porous polymeric microspheres. Polymer 46:8956–63
- Nielsen FS, Petersen KB, Müllertz A. (2008). Bioavailability of probucol from lipid and surfactant based formulations in minipigs: influence of droplet size and dietary state. Eur J Pharm Biopharm 69:553–62
- Porter CJ, Charman WN. (2001). Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev 50:61–80
- Porter CJ, Trevaskis NL, Charman WN. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–48
- Porter CJ, Charman WN. (2001). Transport and absorption of drugs via the lymphatic system. Adv Drug Deliv Rev 50:1–2
- Singh G, Pai RS, Pandit V. (2013). In vivo pharmacokinetic applicability of a simple and validated HPLC method for orally administered trans-resveratrol loaded polymeric nanoparticles to rats. J Pharm Investigation. [Epub ahead of print]. doi:10.1007/s40005-013-0105-0
- Singh G, Pai RS, Devi VK. (2012a). Response surface methodology and process optimization of sustained release pellets using Taguchi orthogonal array design and central composite design. J Adv Pharm Tech Res 3:30–40
- Singh G, Pai RS, Devi VK. (2012b). Optimization of pellets containing solid dispersion prepared by extrusion/spheronization using central composite design and desirability function. J Young Pharm 4:146–56
- Singh G, Pai RS. (2013). Pharmacokinetics and in vivo biodistribution of optimized PLGA nanoparticulate drug delivery system for controlled release of emtricitabine. Drug Deliv. [Epub ahead of print]. doi:10.3109/10717544.2013.867382
- Sun M, Zhai X, Xue K, et al. (2011). Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci 43:132–40
- Swenson ES, Milisen WB, Curatolo W. (1994). Intestinal permeability enhancement: efficacy, acute local toxicity, and reversibility. Pharm Res 11:1132–42
- Troche-Pesqueira E, Perez-Juste I, Navarro-Vazquez A, Cid MM. (2013). A β-cyclodextrin–resveratrol inclusion complex and the role of geometrical and electronic effects on its electronic induced circular dichroism. RSC Adv 3:10242–50
- Wegiel LA, Mauer LJ, Edgar KJ, Taylor LS. (2013). Crystallization of amorphous solid dispersions of resveratrol during preparation and storage-impact of different polymers. J Pharm Sci 102:171–84