
Abstract
The objective of this study was the development of a colon-targeted microspheres which were compressed into tablets containing the non-steroidal anti-inflammatory bumadizone calcium dihydrate. A 32 full factorial design was adopted for the evaluation of the prepared microspheres. The effect of two independent variables namely polymer type (Eudragit RS100, ethyl cellulose and cellulose acetate butyrate), and drug: polymer ratio (1:1, 9:1 and 18:1) was studied on the entrapment efficiency and in vitro drug release for 12 h. Colon targeting aims to minimize the release of the drug off target area (pH 1.2 and 6.8) and to maximize the release of the drug in target area (pH 7.4). Candidate formulae were compressed into core tablets and colon targeting was achieved using the enzyme-dependent polymer (pectin) as coat in three different concentrations 50, 75 and 90%. Candidate formula F15 (microspheres prepared using BDZ:CAB in a ratio of 18:1 and compressed into tablets using 50% pectin and 50% Avicel in the coat) was able to adequately modulate drug release avoiding drug release in the gastric ambient, and reaching the colonic targeting where 99.7% release was achieved within 12 h following zero-order model. In vivo studies showed that F15 achieved significant decrease in myeloperoxidase activity and inflammation with delayed Tmax (4 h) and lower Cmax (2700 ng/ml) when compared to marketed product.
Introduction
Inflammatory Bowel Diseases (IBD) are an immune-mediated chronic or relapsing disorders of the gastrointestinal (GI) tract. They are characterized by a chronic intestinal inflammatory process (Klein & Eliakim, Citation2010). They are gaining more and more attention. This is due to the fact that there is increased incidence of IBD in the past 50 years, up to 120–200/100 000 persons and 50–200/100 000 persons for ulcerative colitis (UC) and Crohn’s disease (CD) respectively (Cosnes et al., Citation2011). Also for the increased evidence, patients with IBD are at increased risk of developing colorectal cancer (Munkholm, Citation2003).
Site-specific delivery of drugs to the site of action has the potential to reduce side effects and to increase pharmacological response. One of the seemingly interesting areas to target drugs through oral route is the colon. Colon targeting denotes dosage forms that are able to sense the arrival to the colon and release the drug upon activation to achieve an optimum pharmacological action together with decreasing drug side effects (Vandamme & Lenourry, Citation2002).
Different mechanisms for colon targeting have been adopted; namely the use of prodrugs (Oz & Ebersole, Citation2008) and the use of pH-dependent polymers. These polymers have ability to withstand an environment ranging from low pH (∼1.2) to neutral pH (∼7.5) for several hours (Singh, Citation2007). A third mechanism in colon targeting involves the presence of time-dependent polymers. In these systems, the site of drug release is decided by the transit time of a formulation in the GI tract (Singh, Citation2007). The abundance of microflora in the colon area has also been used to achieve colon targeting (Sinha & Kumria, Citation2003). Hence, enzymatically controlled delivery systems are considered a convenient approach for site-specific drug delivery to the colon where no drug release can occur unless the system arrives to the colon. This helped the use of moieties that are susceptible to these enzymes including pectin and guar gum (El Gamal et al., Citation2013). Designing the previous systems using multiple-units, like microparticles and nanoparticles, is preferred for many reasons (Abdallah et al., Citation2009). They are less likely to be affected by food and demonstrate more consistent absorption compared to single unit systems (Amrutiya et al., Citation2009). They are well retained and they have greater potential to provide a uniform spreading of the drug particles to the inflamed parts of GI tract, which is advantageous for the topical therapy of inflamed bowel diseases. In addition, the residence time of other delivery systems like tablets or pellets is usually decreased by diarrhea, a common symptom of IBD (Meissner et al., Citation2006)
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used in the treatment of chronic inflammatory conditions. In addition, they showed that a promising activity for the prevention and treatment of colitis but with tendency to cause gastric bleeding and form ulceration in the gastric lining (El-kamel et al., Citation2008). Bumadizone calcium dihydrate (BDZ) is an acetic acid derivative NSAID. Formulation of BDZ into colon-targeted dosage forms will avoid these undesirable effects.
Literature search revealed that no previous studies were carried out to formulate colon-targeted dosage forms containing BDZ. The aim of this study was the development and evaluation of colon-targeted microspheres and compressing them into tablets containing BDZ to prevent drug release in the stomach, had an additional lag phase to retard drug release in the small intestine, and to deliver drug specifically to the colon to avoid gastrointestinal side effects of the drug and to increase the patient compliance. The promising dosage form was examined for its efficacy using acetic acid-induced rabbit colitis model in order to elucidate its usefulness as a specific drug delivery system for the treatment of ulcerative colitis. Finally, the pharmacokinetics of the drug was estimated following oral administration of the tablet to rabbits compared to marketed tablet.
Materials and methods
Materials
Bumadizone calcium dihydrate (BDZ) and Octomol® tablets (110 mg) were kindly provided by October Pharma, Egypt. Cellulose acetate butyrate (CAB) was obtained from Aldrich Chemical Company, Germany. Ethyl cellulose N100 (EC), pectin and acetonitrile (HPLC grade) and methanol (HPLC grade) were purchased from Sigma Chemical Co., St. Louis, (USA). Avicel pH 101 (microcrystalline cellulose) was obtained from Morgan Chemical Industrial Company, Egypt. Eudragit® RS 100 was kindly provided from Degussa, Rohm GmbH and Co. KG, Pharma Polymer, Germany. Mannitol was generously supplied from Roquette, France. Poly vinyl alcohol (PVA; (Mw 88 000) and triethyl citrate were obtained from Acros Organics, Belgium. Dichloro methane was purchased from El Nasr pharmaceutical Chemical Company, Egypt. Pectinex (3000 FDU/ml), was purchased from Fluka Chemicals, Switzerland.
Preparation of BDZ-loaded microspheres
A 32 full factorial design was adopted to study the effect of two independent variables: (i) polymer type and (ii) drug-to-polymer ratio. Three time-dependent polymers, namely, Eudragit® RS100, ethyl cellulose and CAB and three drug-to-polymer ratios, namely, 1:1, 9:1 and 18:1 were used. summarizes the independent variables along with their levels. Nine formulae were prepared as given in . PVA concentration, internal phase to external phase ratio and stirrer speed were kept constant and were selected based on preliminary studies (data not shown).
Table 1. The experimental plan of the 32 factorial design used for preparation of BDZ microspheres.
Table 2. Mean particle size (µm), production yield, % entrapment efficiency, % of total drug released (mg%) 12 h and desirability of the prepared BDZ-loaded microspheres (mean ± SD, n = 3).
Microspheres were prepared according to quasi-emulsion solvent diffusion method (Jain et al., Citation2011) using the different variables. Briefly the calculated amount of the drug, polymer and 400 mg triethyl citrate (plasticizer) were dissolved in 20 ml dichloromethane (internal phase). The resulting solution was poured onto a 200 ml of 0.5% PVA aqueous solution (external phase) saturated with 10% sodium chloride and 3% calcium chloride (to prevent the escape of the drug to the external phase) and stirred at 500 rpm. Stirring continued for 8 h until dichloromethane has evaporated completely. The formed microspheres were then separated by filtration, washed 4–5 times with 75 ml distilled water and left to dry at room temperature for 24 h.
Characterization of the microspheres
Scanning electron microscopy
For morphology and surface characteristics, candidate formula was taken and fixed on a scanning electron microscopy (SEM) holder with double sided adhesive tape and coated with gold–palladium under an argon atmosphere at room temperature and then the surface morphology of the microspheres was studied by scanning electron microscope (SEM) using a JEOL JXA 840A (Tokyo, Japan).
Particle size analysis
Particle size analysis was performed using laser diffraction (LD) technique. Samples were measured in distilled water; diameters and particle size distribution of the prepared microspheres were determined using Mastersizer Laser Diffractometer (Malvern Instruments, Malvern, Worcestershire, UK).
Determination of production yield and entrapment efficiency
The microspheres formed were collected and weighed. The percentage yield was determined using the following formula:
Fifteen micrograms of the prepared microspheres were crushed and stirred with 100 ml phosphate buffer (pH 7.4) for 24 h. The extract was filtered and the drug was assayed spectrophotometrically (UV–VIS spectrophotometer, Shimadzu UV 1601 PC, Kyoto, Japan) at λmax of 234 nm after doing the necessary dilution (Verma et al., Citation2010). To determine the entrapment efficiency the following formula was used:
Differential scanning calorimetry
Samples of the drug, blank microspheres and the candidate formula (F9) were heated in an aluminum pan at 5 °C/min in an atmosphere of nitrogen to 200 °C. The differential scanning calorimetry (DSC) thermograms were recorded on a differential scanning calorimeter (DSC-60, Shimadzu, Kyoto, Japan).
X-ray diffraction
X-ray diffraction (XRD) patterns of pure BDZ, blank CAB microspheres and candidate BDZ loaded microspheres were determined at a voltage of 40 kV and a current intensity of 30 mA at a scanning speed of 1 °C/min using a Philips PW 1729 X-ray diffractometer (Legroupe Interconnexion, Saint-Juire, Clubac, Canada).
In vitro drug release
Release of the drug from the microspheres was performed using USP dissolution tester type I (VK700 Dissolution Testing Station, Vankel Industries, Inc., NJ, USA). The baskets were covered by a piece of white muslin cloth (Lakshmana et al., Citation2009) to prevent the escape of the microspheres to the release media. Accurately weighed microspheres equivalent to 110 mg of the drug were used in the dissolution study. The basket was rotated at 100 rpm. In vitro release studies were carried out at 37 ± 1 °C in 900 ml of 0.1 N HCl containing 1% PEG 400 (to maintain sink conditions) for a period of 2 h followed by release in Sorensen's phosphate buffer pH 6.8 for 2 h then Sorensen's phosphate buffer pH 7.4 for 8 h to simulate the pHs pertaining to the stomach, proximal and middle small intestine (duodenum and jejunum), and distal small intestine (ileum), respectively (Saboji et al., Citation2012). Aliquots were withdrawn in a predetermined time intervals, filtered then assayed spectrophotometrically for the drug at λmax = 232.6 nm in pH 1.2 and λmax 234 nm (Ali et al., Citation2012) in pH 6.8 and 7.4. The withdrawn samples were replaced with equal volume of the release media. The cumulative drug percent released was plotted against time. The experiments were performed in triplicate for each sample.
Statistical analysis
The experimental results regarding production yield, entrapment efficiency and in vitro release after 12 h were analyzed using Design Expert® 7.0 software (Design-Expert trial version 7; State-Ease Inc., Minneapolis, MN).
Preparation of compression coated colon-targeted tablets
Based on the previously mentioned characterization, and the results of the main effects of the adopted factorial design two candidate formulae, namely, F6 (18:1 drug to EC) and F9 (18:1 drug to CAB) were chosen. Both formulae showed maximum yield and entrapment efficiency and the required drug release profile (minimum drug release in pH 1.2 and pH 6.8 and maximum drug release in pH 7.4). Microspheres of candidate formulae containing 110 mg drug was mixed with 15% Avicel, 2% sodium lauryl sulfate and mannitol as diluent to a total weight of 200 mg and were compressed with an applied force of 4000 kg using a single punch tablet machine equipped with concave punch 10 mm in diameter forming the core tablet (tablet single punch press machine, Royal Artist, Bombay, India). The core tablets were compression-coated with 300 mg different coat mixtures (). Different pectin: Avicel mixture was used as outer shell for compression coating where pectin is considered as an enzyme dependent polymer to achieve colon targeting. Fifty percent of coating material (150 mg) was placed in the die cavity, and the core tablet was placed in the center followed by addition of the remainder of the coating material. The coating material was compressed around the core tablet using round flat punches (12 mm) on the same tabletting machine. The mixture of powders prepared as core materials were evaluated for their flow properties using angle of repose method (Singh et al., Citation2012) and the prepared tablets were tested for weight variation, hardness, thickness and friability.
Table 3. Formulation of core and coat of different compression coated tablets for colon targeting.
In vitro drug release
Release of the drug from the compression-coated colon-targeted tablets was performed like that of the prepared microspheres. The experiments tests were performed in triplicates. The release of the formulae that showed the required drug release profile (minimum drug release before target area pH 1.2 and pH 6.8 and maximum drug release in target area pH 7.4) were repeated in the presence of pectinase enzyme to simulate the colonic in vivo conditions.
Kinetic modeling of the release profiles
The release profiles of the drug were fitted to zero-order, first-order, Higuchi model and Korsmeyer–Peppas model (Costa et al., Citation2001).
In vivo study
Study design
In vivo study was done to compare the anti-inflammatory activity on colonic mucosa and the pharmacokinetics of BDZ from the candidate formula F15 and the marketed product Octomotol® tablets using a non-blind, two treatments and randomized design. The protocol of the study was approved by the Research Ethics Committee in the Faculty of Pharmacy, Cairo University, Egypt. The study was conducted in accordance with EC Directive 86/609/EEC for animal experiments.
Twelve New Zealand white rabbits of either sex weighing 2–2.5 kg were randomly distributed between four groups of equal number. Group I: normal animals, group II: animals with acetic acid induced ulcerative colitis, group III: animals with induced ulcerative colitis and treated with marketed tablets and group IV: animals with induced ulcerative colitis and treated with F15.
Induction of colitis
Colitis was induced in rabbits following the method described by Hathaway et al. Citation(2012). Briefly, rabbits were fasted 24 h with free access to water before experimentation (Millar et al., 1996), a catheter was advanced rectally into the colon until its tip was 8 cm above the anus. The acetic acid/ethanol (1 ml of 5% acetic acid–30% ethanol) was retained in the colon for 30 s, after which the catheter was withdrawn.
Treatment with BDZ
The dose of the drug was adjusted using the equation developed by Reagan-Shaw et al. Citation(2008). Bumadizone calcium dihydrate formula (containing 15 mg) were administered, using per-oral intubation in rabbits of all groups except the control group, once daily till (not including) the day of scarification.
Sample collection
About 1 mL blood sample was withdrawn from the ear vein of the rabbits. The samples were collected into heparinized test tubes at time intervals of 0 (predose), 1, 2, 3, 4, 6, 8 and 12 h following oral administration of a treatment. Plasma was separated by centrifugation at 3000 rpm for 10 min, collected in polyethylene-capped tubes and stored at −20 °C until analysis.
Quantitation of BDZ in plasma
A simple HPLC method (Schroeder & Stevens, Citation2003) was developed for the determination of BDZ in plasma. A Shimadzu HPLC system with UV–Visible detector: SPD-10 A, liquid chromatogram: LC-10AD, integrator: C-R6A, injector (20 μL): 6E (Shimadzu, Japan). The analysis was carried out using Hypersil C18 column (Thermo Electron Corporation) with dimensions = 150 × 4.6 mm and particle size = 5 μm. The mobile phase consisted of acetonitrile:water (65:35) v/v, adjusted to pH 3.5 with phosphoric acid. The mobile phase solution was filtered and degassed. The flow rate was 0.8 mL/min. The eluent was monitored at 234 nm. Flufenamic acid was adopted as an internal standard. A standard calibration curve was constructed by plotting peak area ratio of BDZ and flufenamic acid against BDZ concentration in plasma. Validation parameters, namely, linearity, accuracy, recovery, interday and intraday precision were tested.
Pharmacokinetics analysis
The pharmacokinetic parameters of the two treatments were estimated for each subject these include: maximum drug concentration Cmax (ng/ml), the time to reach Cmax, Tmax, t1/2 and MRT.
Scarification of animals
Rabbits were sacrificed 5 days after the induction of colitis and a piece (5 cm) of colon was removed 8 cm from the rectum (El-kamel et al., Citation2008). The separated part was cleaned of fat and mesenteric blot, washed with 0.9% (w/v) saline solution and plotted on a filter paper.
Histopathological evaluation
Two tissue samples were excised from each colon and maintained in 10% (v/v) formalin in saline for histopathological evaluation. Sections of 5 μm were stained with hematoxylin and eosin (Kucharzik et al., Citation2001) and then examined using light microscope (Nikon, Tokyo, Japan) fitted with a Canon power shot G3 digital camera (Canon, Tokyo, Japan) and compared with control sample.
Measurement of myeloperoxidase activity
The activity of myeloperoxidase (MPO), which is found in neutrophils, can be used for evaluating the degree of inflammation in the intestine. The distal colon specimen (200 mg) was minced in a beaker (cooled on ice) containing 10 times its amount water for injection, transferred to a test tube and homogenized at 1000 rpm (Potter E1 Vejhem glass homogenizer, Poland) three times for 30 s each. The homogenate (0.5 mL) was mixed with 0.5 mL of HTAB buffer (0.5% HTAB in 50 mM phosphate buffer, pH 6.0) and the mixture was sonicated for 10 s and centrifuged at 10 000 rpm for15 min. The supernatant was assayed for MPO activity. MPO activity was measured spectrophotometrically (UV-1601 PC spectrophotometer, Shimadzu, Kyoto, Japan): 0.1 mL of supernatant was combined with 2.9 mL containing 0.167 mg/mL O-diansidine hydrochloride and 0.0005% hydrogen peroxide. The change in absorbance at 460 nm was measured every minute for 4 min. One unit of MPO activity is defined as the amount which degrades 1 mmol of the peroxide per minute at 25 °C (Cui et al., Citation1994; Kucharzik et al., Citation2001). The results were expressed as the mean ± standard deviation of the mean.
Results and discussion
Because of its simplicity and reproducibility, quasi-emulsion solvent diffusion method was used for the preparation of microspheres. Triethyl citrate was added as plasticizer and the external phase was saturated with 10% NaCl and 3% CaCl2 in order to favor the drug entrapment inside the microspheres. Nine BDZ microspheres formulations were successfully prepared and subjected to evaluation and the results were as follows.
Scanning electron microscopy
Scanning electron microscopy of the dried microspheres revealed that the produced microspheres were spherical, non-aggregated with rough and porous surface (). The pores might be induced by the diffusion of the solvent from the surface of the microspheres.
Figure 1. SEM of F9.

Particle size analysis
The mean particle size of the obtained microspheres ranged from 90.8 to 147.95 µm (). Both studied factors, namely, polymer type and drug:polymer ratio had non-significant effect on the mean particle size p > 0.05 (data not shown). This might be explained on the fact that particle size distribution of the microspheres was more dependent on the volume of dichloromethane and PVA concentration rather than on drug:polymer ratio (Lakshmana et al., Citation2009).
Production yield and entrapment efficiency
Results in revealed that the production yield of the prepared microspheres varied from 45.45 ± 5.23% (F7) to 80.2 ± 1.04% (F9) while their entrapment efficiency was in the range of 44 ± 10.36% (F1) to 99.77% ± 1.25% (F3).
The effect of polymer type
Polymer type had a significant effect (p < 0.05) on the percentage yield (). It was found that CAB had lower percentage yield when compared to the other two polymers used. Jelvehgari & Montazam (Citation2012) reported that CAB led to lower yield of theophylline microspheres when compared to Eudragit RS100. Similarly, polymer type had a significant effect (p < 0.05) on the entrapment efficiency. Microspheres prepared using CAB showed lower entrapment efficiency when compared to the other two polymers. This could be due to the high-permeability characteristics of CAB which would facilitate the diffusion of part of the entrapped drug to the surrounding medium during the preparation of the microspheres (Jelvehgari & Maghsoodi, Citation2010). On the other hand, microspheres prepared with ethyl cellulose showed the highest entrapment efficiency when compared to the other two polymers. This may be due to higher viscosity obtained in case of ethyl cellulose N100 when compared to Eudragit RS100 and CAB, decreasing drug diffusion to the outer phase and forcing it to stay in the microspheres formed resulting in higher entrapment. These findings are in accordance with the results reached by Swetha et al. (Citation2011) who formulated etodolac microspheres using ethyl cellulose and Eudragit RS100 as polymers.
Figure 2. Main effects of the different factors according to 32 factorial design on the yield and entrapment efficiency: polymer type (A) and drug:polymer ratio (B).
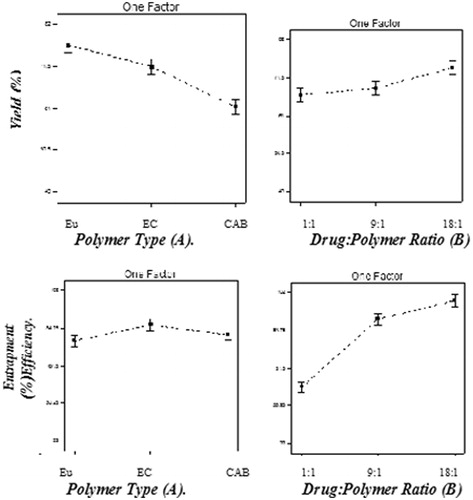
The effect of drug to polymer ratio
Increasing polymer to drug ratio led to a significant increase in production yield (p < 0.05) () This could be attributed to a slight increase in the inner phase viscosity providing more time for the droplet formation and improving yield also might lead to reduced diffusion rate of dichloromethane from concentrated solutions into aqueous phase. Our results are in accordance with Baykara (Citation2003) who found that increasing drug to polymer ratio with keeping amount of the polymer constant led to an increase in production yield. Similarly, the entrapment efficiency of the prepared microspheres was found to increase significantly with increasing drug:polymer ratio. The higher drug entrapment efficiencies were obtained at the higher drug: polymer ratios (F3, F6 and F9). This may be because increasing drug loading would increase the drug concentration difference between that in the microsphere and in the surrounding release medium which increase the driving drug release force, another reason could also be that the use of the higher amounts of BDZ when preparing microspheres caused slightly an increased viscosity of the dispersed phase. Increasing the viscosity of the internal media decreased the drug diffusion from the formed microspheres giving the drug enough time to be entrapped inside the particles formed. When dichloromethane diffused out, nearly all of the dispersed phase was converted to the form of solid microspheres and separated particles appear, furthermore, the highest drug entrapment efficiency of these formulations could be explained through the fact that the amount of drug in per unit polymer is greater than that in other formulations (Jelvehgari et al., Citation2006).
Differential scanning calorimetry
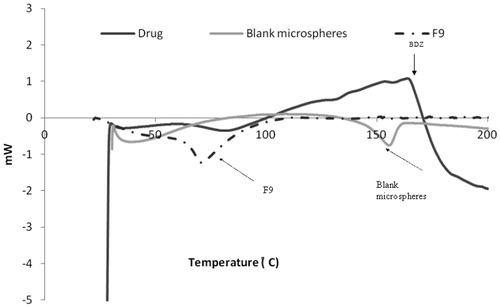
The DSC thermogram of pure BDZ showed that an exothermic peak associated with crystal melting at 158 °C (). On the other hand, the amorphous blank polymer did not show any fusion peak or phase transition, but showed a broad signal around 144–166 °C. The DSC thermogram of BDZ-loaded CAB microspheres (F9) showed a broad signal around 63.94–88.19 °C. The peak corresponding to the melting point of BDZ disappeared. This could be attributed to the possible change of the drug to an amorphous form. To confirm the previous suggestions, X-ray diffraction studies were conducted.
Figure 3. DSC thermograms of pure drug, blank microspheres and BDZ-loaded microspheres (F9).
X-ray diffraction
X-ray diffraction studies were done to investigate the crystalline status of the samples. The peak position (angle of diffraction) is an indicative of a crystal structure and peak heights are the measures of samples crystallinity in a diffractogram ().
Figure 4. X-ray diffractograms of (a) pure drug, (b) blank microspheres and (c) BDZ-loaded microspheres (F9).
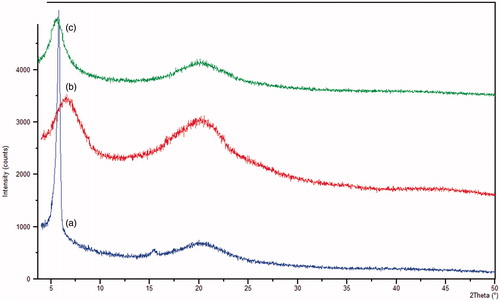
The diffractogram of BDZ shows three intense peaks at three different θ of 5.8°, 15.41° and 20.31°. The respective signal heights were 4080.31, 147.43 and 516.7, respectively. This was indicative of the crystallinity of the pure drug. While the diffractogram of plain microspheres shows three different peaks and three different angles namely 6.56°, 20.175° and 44.45°. The respective signal heights were 1564.98, 1435.08 and 102.88, respectively. The loaded microspheres (F9) had two different peaks and two different angles, namely, 5.584° and 20.29° with heights of 1121.37 and 545.49, respectively. These results showed that a decrease in intensity of the peaks of the pure drug which favored the entrapment of the drug in the formed microspheres and changing of the drug from crystalline to amorphous form.
In vitro release studies of BDZ from the microspheres formulations
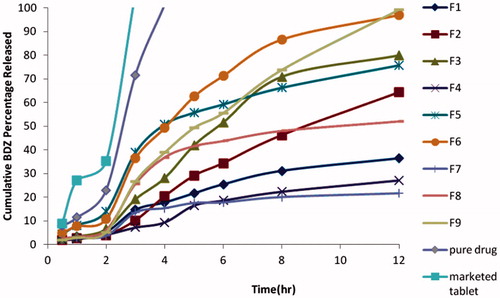
The in vitro release of the drug was done on a gradient pH basis so as to mimic the in vivo conditions (Saboji et al., Citation2012). Release profile of BDZ from the prepared microspheres are shown in (). The results revealed that the marketed bumadizone tablet and bumadizone powder showed fast dissolution when compared to the prepared microspheres as they showed 100% release after 3 and 4 h, respectively. The percentage of BDZ released from the prepared microspheres was found to be low for the initial 2 h in 0.1 N HCl, it was found to vary between 3.71% for F7 prepared using CAB in a drug to polymer ratio of (1:1) and 14.11% for F5 prepared using ethyl cellulose in a drug to polymer ratio of (9:1). Upon replacing 0.1 N HCl with phosphate buffer pH 6.8 for another 2 h, the percentage of BDZ released increased to 15.35% and to 50.82% for F7 and F5, respectively. On the other hand, the total BDZ released after 12 h from the prepared microspheres varied between 21.63% for F7 prepared with CAB in a drug:polymer ratio (1:1) and 99.1% for F9 prepared with CAB in a drug:polymer ratio of (18:1). To evaluate the effect of polymer type and drug:polymer ratio on the release profile, statistical analysis was done using design expert software. (Design- Expert trial version 7; State-Ease Inc., Minneapolis, MN, USA). Both factors had a significant effect on the total percentage released after 12 h (p < 0.05).
Figure 5. Release profile of BDZ from the prepared microspheres in gradient pH system (pH 1.2 for 2 h, pH 6.8 for 2 h and pH 7.4 till the end of 12 h).
Effect of drug:polymer ratio
Increasing drug:polymer ratio led to an increase in percentage of the drug released. This may due to that there was more drug than the polymer in the formed microspheres which led to an increase in entrapment efficiency and more homogenous drug distribution and consequently higher release. Also decreasing the polymer concentration in the formed microspheres, might facilitate drug diffusion to the release media. The increase in the rate and extent of drug release upon decreasing the polymer concentration might be due to the hydrophobic character of BDZ which hindered its delivery through the hydrophobic coat. Therefore, the diffusion of BDZ through the polymer wall became the controlling step of drug release. Delay in drug release at higher polymer concentration could be also attributed to the formation of thicker coating membranes, where 1:1 drug to polymer ratio contains 50% polymer, increasing drug concentration decreased polymer amount in formed microspheres as 9:1 ratio contains 90% drug and 10% polymer and 18:1 ratio contains 94.7% drug and 5.3% polymer.
Effect of polymer type
The used time-dependent polymers, namely, Eudragit® RS 100, Ethyl cellulose and CAB are known to be of hydrophobic nature, insoluble in water and buffers and are mainly used for sustainment of drug release (Rowe et al., Citation2006). In this study, ethyl cellulose and CAB showed faster release when compared to Eudragit® RS100. This could be attributed to the difference in permeability between the three polymers. Eudragit® RS 100 are copolymers of methacrylic acid and methyl methacrylate with known low permeability. Kotagale et al. (Citation2011) reported that ranitidine hydrochloride microspheres prepared using ethyl cellulose showed higher and faster release if compared to microspheres prepared using Eudragit® RS 100 or combination of both ethyl cellulose and Eudragit®. Another reason for the slow release of BDZ from Eudragit® RS100 when compared to ethyl cellulose and CAB could be a possible interaction between the positively charged Eudragit RS100 and the negative charge of the drug.
Although F3, F6 and F9 showed higher % of drug released after 12 h which was 79.81, 96.98 and 99.1%, respectively. Unfortunately, none of the developed microspheres showed the desired drug release profile which must be less than 20% before target area (pH 1.2 and 6.8) so further optimization was required.
The aim of optimization of pharmaceutical dosage formulation is generally to determine the levels of variables from which a robust product with high quality characteristics may be produced. Design Expert® software was used to determine the best desirability for further optimization. The criteria selected were maximizing the yield, entrapment efficiency and total drug released after 12 h and minimizing the percentage of BDZ released before target area (pH 1.2 and 6.8). It excluded particle size as it was statistically insignificant.
Formulae F6 and F9 were chosen (Table 2) with a desirability of 0.963 and 0.872, respectively, for further optimization (to be compressed into tablets in order to decrease the percentage of drug released in pH 1.2 and 6.8). It is well known that desirability increases as this number closes to 1.
Pre-compression parameters
The value of angle of repose of the powders (different core and coat blends) ranged from 19.91 ± 2.02° to 23.32 ± 1.02° indicating good flow properties. The angle of repose is considered as a characteristic of the internal friction or cohesion of the particles. Powders with angle of repose above 50° have unsatisfactory difficult flow properties, 25–40° reasonable flow potential, whereas minimum angles close to 25° correspond to very good flow properties (Jones, Citation1988).
Post-compression parameters
All prepared tablet formulations met the British Pharmacopeia (Ivy Leaf, 2007) requirements for drug content and weight variation. The average drug content of the prepared formulations was found to be 101.44% ± 1.25 while the average thickness ranged from 4.47 ± 0.85 to 4.79 ± 0.3 mm. None of the formulated tablets deviated from the average weight (500 mg) by more than 5%, indicating that all formulae fulfilled the pharmacopoeial specifications for weight variation.
According to the British Pharmacopeia hardness values between 4–6 and friability value <1% are considered acceptable. Hardness of the prepared tablets was within the acceptable range except F10 and F13 that had hardness values of 2.89 and 3.36 kg. The friability of the prepared tablets was within the compendial limits except F10 and F13 which had friability of 1.47 and 1.37%, respectively, that exceeded the pharmacopoeial limitation.
In vitro release of BDZ from compression coated tablets
The release profiles of BDZ from the compression coated tablets in pH gradient system are shown are shown in .
Figure 6. Release profile of BDZ from compression-coated colon-targeted tablets in gradient pH system (pH 1.2 for 2 h, pH 6.8 for 2 h and pH 7.4 till the end of 12 h).
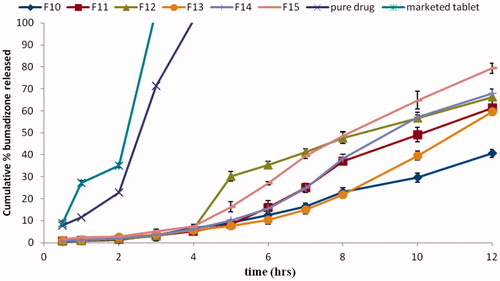
Compression of the microspheres into coated tablets led to a decrease in the percentage of drug released in pH 1.2 and 6.8 when compared to the drug released from the corresponding microspheres ().
All prepared tablets showed less than 20% of BDZ released in pH 1.2 and 6.8. F12, F14 and F15 showed 66.23, 67.9 and 79.42% of BDZ released after 12 h, respectively. This decrease in drug release might be attributed to the presence of pectin that acted as an additional barrier in front of the drug release before target area. Pectin may also have enabled drug targeting due to its bioactivity (Allena et al., Citation2012).
To evaluate the effect of core polymer type and pectin concentration in the coat on the total percentage of drug released, statistical analysis was done using Design Expert® software. Both factors had a significant effect (p < 0.05) on the total percentage released after 12 h.
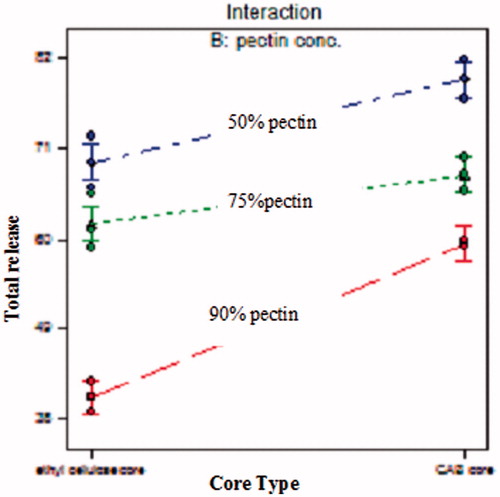
Drug released from the compression-coated tablets increased as the percentage of pectin in the coat decreased. This might be because polymer (pectin) becames hydrated and forms a viscous gel layer that slowed down further seeping-in of dissolution fluids towards the core tablets (Patel et al., Citation2010). The interaction line plots () show that the effect of the polymer type varied according to the pectin concentration used. At different pectin concentration, CAB showed higher drug release when compared to ethyl cellulose. This effect was prominent at 90% pectin but less clear at lower pectin concentrations. This could be attributed to higher permeability characteristics and lower viscosity of CAB when compared to ethyl cellulose. Ethyl cellulose is one of the most widely used water-insoluble polymers in film-coating to achieve extended drug release. In order to evaluate enzyme-triggered drug release of pectin compression-coated tablets, 7.4 to simulate the degradation of the polymers by the microflora (Turkoglu & Ugurlu, Citation2002).
Figure 7. Interaction line plots showing the effect of core type and pectin concentration on the total percentage of drug released after 12 h.
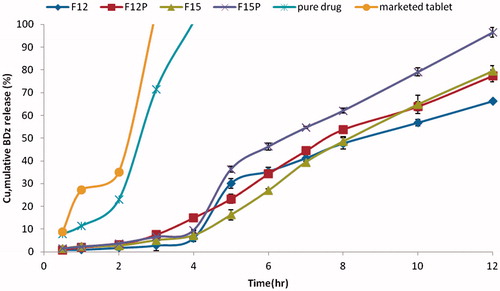
Using Design Expert software, F12 and F15 were selected and their release was repeated with the addition of 1 ml of pectinolytic enzyme to Sorensen's buffer (pH 7.4; F12P and F15P). Results are illustrated in (). The results revealed that there was an increase in release of the drug in target area pH 7.4 in the presence of pectinolytic enzyme that hydrolyzed pectin in the coat formulation. The presence of pectinolytic enzyme in pH 7.4 led to hydrolysis of the pectin of the coat, hence decreasing the barrier in front of the drug together with forming channels in the coat. This in return allowed the release media to reach the core, releasing the drug (Okafor et al., Citation2010). F15 achieved the lowest percentage of drug released before target area (pH 1.2 and 6.8) of 7.26% and maximum percentage of drug released in target area (pH = 7.4) of 72.16%. After the addition of 1 mL pectinase enzyme the percentage of BDZ released increased from 72.16% to 99.7% after 12 h.
Figure 8. In vitro release of BDZ from prepared formulations (F12 and F15) in the presence (F12P and F15P) and in the absence (F12 and F15) of pectinase enzyme.
Kinetic analysis of the release profiles of the compression-coated colon-targeted tablets are shown in Table . In all tested formulations, calculated n values were >0.89 indicating a super case II release. A super case II release mechanism usually indicates that polymer erosion is the technique involved in the release of the drug. It occurs when the erosion rate of the polymer is higher than its swelling rate. Regarding F12 P and F15P, the presence of pectinase enzyme aided to dissolve the pectin present in the coat blend (Arunachalam & Asha, Citation2010) and this is the main reason for the use of pectin in colon targeting (El-Gamal et al., Citation2013). It was previously reported that a super case II release is the expected mechanism when the polymer dissolves in the release media (Costa et al., Citation2001).
Table 4. Mathematical modeling and release kinetics of BDZ from the compression-coated colon-targeted tablets.
In vivo evaluation
Plasma-time curve
Plasma concentration–time curve was investigated to confirm the ability of the candidate formula to prevent or minimize drug absorption before target area and to sense the arrival of the dosage form to the colon where drug release should start and be maintained up to the 12th hour.
An HPLC method was adopted (Schroeder & Stevens Citation2003), a calibration curve was constructed and a linear response (r2 = 0.998) across the full range of concentrations from 100 to 1500 ng/ml was achieved, satisfactory validation parameters, namely, linearity, accuracy, recovery percentage, interday and intraday precision were obtained.
The pharmacokinetic parameters of bumadizone calcium dihydrate following oral administration of a single dose (15 mg) of (i) marketed product and (ii) F15 are shown in .
Table 5. Pharmacokinetic parameters of BDZ following oral administration of octomotol and the prepared colon targeted tablets (F15).
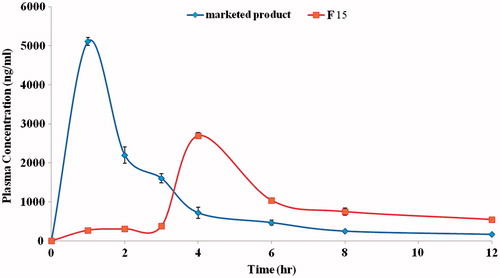
The drug plasma concentration–time profiles of both treatments are depicted in (). It is clear that remarkable differences between the two treatments were found. The peak plasma concentration (Cmax) of both treatments were found to be 5110.3 ± 103.21 (ng/ml) and 2700.2 ± 135.14 (ng/ml) for the marketed product Octomotol® tablets and the candidate formula (F15), respectively. Statistical analysis of Cmax revealed significant difference (p < 0.05) where candidate formula showed lower Cmax in comparison to the marketed product. Tmax was 1 and 4 h for marketed product and candidate formula, respectively (). These results confirmed that the candidate formula was able to delay the drug absorption (Tmax 4 h) till the 4th hour (minimum drug absorption at the upper parts of GIT) and to sense the arrival of the dosage form to the target area (colon) when compared to the marketed product that showed a lower Tmax of only 1 h. This could be furtherly confirmed by the increase of the MRT from 2.49 h of the marketed product to 5.91 h of the candidate formula. All the above-mentioned pharmacokinetic parameters (Cmax, Tmax and MRT) were statistically significant (p < 0.05) and correlate well with the in vitro release data.
Figure 9. Mean plasma concentrations of BDZ following administration of an oral dose of 15 mg of Octomotol® tablets and F15 compression-coated colon-targeted tablets to three rabbits.
Histopathological analysis
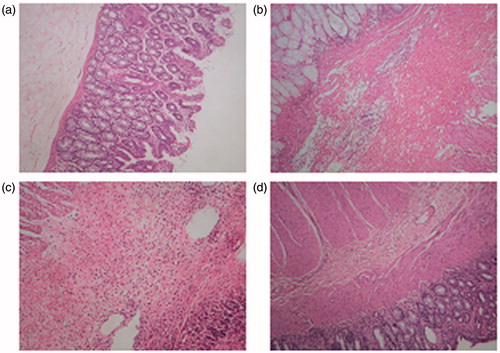
Histopathological examination of the obtained animal tissue revealed that acetic acid was able to induce a severe inflammation in the colon tissue. This resulted in a pan mural inflammation together with ulceration and gangrene if compared to normal tissue. Treatment with the marketed product did not show significant decrease in formed ulceration and inflammation. However, treatment with F15 showed a marked decrease in inflammation and neutrophil infiltration ().
Figure 10. Histopathological analysis of colon tissue. (a) Normal mucosa. (b) Acetic acid induced colitis:pan hemorrhagic infarction, marked inflammation. (c) Treatment with marketed product showing neutrophil infiltration. (d) Treatment with F15: marked decrease in inflammation and neutrophil infiltration.
MPO measurement
The acetic acid induced experimental rabbit colitis model was selected because the pharmacological responses of rabbit colonic smooth muscle to inflammatory mediators closely resemble those of the human colon (Percy et al., Citation1990). MPO activity, which is an important quantitative index for colonic inflammation, was determined in terms of units per gram tissue weight (Soad et al., Citation2009). MPO activity for normal control group was 2.724 ± 0.809 U/g tissue weight. However, MPO activity of the induced colitis group was found to be 15.363 ± 1.205 U/g tissue weight. This MPO was significantly decreased to 11.96 ± 1.30 and 5.7 ± 0.389 U/g tissue weight in the case of the group that received the marketed product and F15, respectively (). Statistical analysis was done using SPSS statistical package version 17 (SPSS Inc., Chicago, IL). Post-hoc test revealed that the groups are statistically significant (p < 0.05) concerning MPO activity.
Table 6. Mean myeloperoxidase activity of treated and untreated animals (mean ± SD, n = 3).
Conclusion
BDZ was successfully colon targeted using the techniques adopted. Candidate formula (F15) (microspheres prepared using BDZ:CAB in a ratio of 18:1 and compressed into tablets using 50% pectin and 50% Avicel in the coat) was able to adequately modulate drug release avoiding drug release in the gastric ambient, and reaching the colonic targeting where 99.7% release was achieved within 12 h following the zero-order model. Histopathological investigation showed a marked decrease in inflammation and colorimetric assay of inflammatory marker (Myelo peroxidase enzyme) revealed its efficacy when compared to the marketed product. Statistical analysis revealed significant increase in Tmax (4 h) and in MRT (5.91 h) with a decrease in Cmax (2700 ng/ml) when compared to marketed product. This indicates decreased and delayed absorption of the drug. These findings suggest that the microspheres compressed into tablets containing BDZ (F15) are likely to become one of choices for the treatment of ulcerative colitis.
Declaration of interest
No conflict of interest.
References
- Abdallah M, Yuichi T, Hirofumi T. (2009). pH-Sensitive nanospheres for colon-specific drug delivery in experimentally induced colitis rat model. Eur J Pharm Biopharm 72:1–8
- Allena RT, Gowda DV, Teja R. (2012). Formulation and evaluation of pectin-HPMC mesalamine tablets coated with Eudragit L 100 for ulcerative colitis. Der Pharm Lett 4:1093–102
- Ali NW, Zaazaa HA, Abdelkawy M, Magdy MA. (2012). Simultaneous HPTLC and RP-HPLC methods for determination of bumadizone in the presence of its alkaline-induced degradation product. Biomed Chromatogr 26:1143–9
- Amrutiya N, Bajaj A, Madan M. (2009). Development of microsponges for topical delivery of mupirocin. Mercury 10:402–9
- Arunachalam C, Asha S. (2010). Pectinolytic enzyme – a review of new studies. Adv Biotechnol 561:1–5
- Baykara T. (2003). Preparation and in vitro evaluation of modified release ketoprofen microsponges. Il Faramaco 58:101–6
- Cosnes J, Grower-Rousseau C, Seksik P, Cortot A. (2011). Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 140:1785–94
- Costa P, Manuel J, Lobo S. (2001). Modeling and comparison of dissolution profiles. Eur J Pharm Sci 13:123–33
- Cui N, Friend DR, Fedorak RN. (1994). A budesonide prodrug accelerates treatment of colitis in rats. Gut 35:1439–46
- El-kamel AH, Abdelaziz AM, Fatani AJ, El-subbagh HI. (2008). Oral colon targeted delivery systems for treatment of inflammatory bowel diseases: synthesis, in vitro and in vivo assessment. Int J Pharm 358:248–55
- El Gamal SS, Naggar VF, Sokar MS. (2013). Colon targeting of theophylline from enzyme-dependent release tablet formulations: in vitro evaluation. Am J Res Commun 1:116–33
- Hathaway CA, Appleyard CB, Percy WH, Williams JL. (2012). Experimental colitis increases blood-brain barrier permeability in rabbits experimental colitis increases blood-brain barrier permeability in rabbits. AJP Gastrointest Liver Physiol 276:1174–80
- Ivy Leaf. (2007). In British Pharmacopoeia, 1st edn. London: The Stationary Office, 111–12, 153
- Jain V, Jain D, Singh R. (2011). Factors effecting the morphology of Eudragit S-100 based microsponges bearing dicyclomine for colonic delivery. J Pharm Sci 100:1545–52
- Jelvehgari M, Siahi-Shadbad MR, Azarmi S, et al. (2006). The microsponge delivery system of benzoyl peroxide: preparation, characterization and release studies. Int J Pharm 308:124–32
- Jelvehgari M, Maghsoodi M. (2010). Development of theophylline floating microballoons using cellulose acetate butyrate and/or Eudragit RL 100 polymers with different permeability characteristics. Res Pharm Sci 5:29–39
- Jelvehgari M, Montazam SH. (2012). Comparison of microencapsulation by emulsion-solvent extraction/evaporation technique using derivatives cellulose and acrylate-methacrylate copolymer as carriers. Jundishapur J Nat Pharm Prod 7:144–52
- Jones BE. (1988). Pharmaceutics: the science of dosage form design. Edinburgh, Churchill Livingstone: Longman Group, 322
- Klein A, Eliakim R. (2010). Non-steroidal anti-inflammatory drugs and inflammatory bowel disease. Pharmaceuticals 3:1084–92
- Kotagale NR, Parkhe AP, Jumde AB, et al. (2011). Ranitidine hydrochloride-loaded ethyl cellulose and Eudragit RS 100 buoyant microspheres: effect of ph modifiers. Indian J Pharm Sci 73:626–33
- Kucharzik T, Walsh SV, Chen J, et al. (2001). Short communication neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol 159:2001–9
- Lakshmana Prabu S, Shirwaikar AA, Shirwaikar A, Kumar A. (2009). Formulation and evaluation of sustained release microspheres of rosin containing aceclofenac. ARS Pharmaceutica 50:51–62
- Meissner Y, Pellequer Y, Lamprechat A. (2006). Nanoparticles in inflammatory bowel disease: particle targeting versus pH-sensitive delivery. Int J Pharm 316:138–43
- Millar AD, Rampton DS, Chander CL, et al. (1996). Evaluating the antioxidant potential of new treatments for inflammatory bowel disease using a rat model of colitis. Gut 39:407–15
- Munkholm P. (2003). Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Alimentary Pharmacol Ther 18:1–5
- Okafor UA, Okochi VI, Chinedu SN, Onygemeokerenta BM. (2010). Pectinolytic activity of wild-type filamentous fungi fermented on agro-wastes. Afr J Microbiol Res 4:2729–34
- Oz HS, Ebersole JL. (2008). Application of prodrugs to inflammatory diseases of the gut. Molecules (Basel, Switzerland) 13:452–74
- Patel NV, Patel JK, Shah SH, Patel JJ. (2010). Design, development and in vitro evaluation of mesalamine tablets containing pectin and chitosan for colon-specific drug delivery. Int J Res Pharm Sci 1:94–102
- Percy WH, Burton MB, Fallick F, Burakoff R. (1990). A comparison in vitro of human and rabbit distal colonic muscle responses to inflammatory mediators. Gastroenterology 99:1324–32
- Reagan-Shaw S, Nihal M, Ahmad N. (2008). Dose translation from animal to human studies revisited. FASEB J 22:659–61
- Rowe RC, Sheskey PJ, Owen SC. (2006). Handbook of Pharmaceutical Excipients. London, UK: Pharmaceutical Press
- Saboji JK, Gadve RB, Patil SM. (2012). Development of oral colon specific pH dependent microcapsules of NSAID drug naproxen. J Appl Pharm Sci 2:202–11
- Schroeder B, Stevens J. (2003). An automated SPE procedure and analysis for the determination of ibuprofen and ketoprofen in plasma. GILSON 1–6
- Singh BN. (2007). Modified-release solid formulations for colonic delivery. Recent Pat Drug Deliv Formul 1:53–63
- Singh PK, Kumar S, Sharan G, Paradesh U. (2012). Formulation, devlopment and evaluation of colon targeted dosage form of Ibuprofen. Int J Pharm Sci Res 3:268–78
- Sinha VR, Kumria R. (2003). Microbially triggered drug delivery to the colon. Eur J Pharm Sci 18:3–18
- Soad AY, Ahmed HE, Ibrahim S, Ahmed HS. (2009). Optimization of budesonide compression-coated tablets for colonic delivery. AAPS PharmSciTech 10:147–57
- Swetha A, Gopal RM, Venkata RK, et al. (2011). Formulation and in-vitro evaluation of etodolac entrapped in mircosponge based drug delivery system. Int J Pharm 1:73–80
- Turkoglu M, Ugurlu T. (2002). In vitro evaluation of pectin–HPMC compression coated 5-aminosalicylic acid tablets for colonic delivery. Eur J Pharm Biopharm 531:65–73
- Vandamme T, Lenourry A. (2002). The use of polysaccharides to target drugs to the colon. Carbohydr Polym 48:219–31
- Verma A, Tripathi A, Saraf SA, Saraf S. (2010). Fabrication and evaluation of sustained release microspheres of ketorolac tromethamine. Int J Pharm Pharm Sci 2:44–8