
Abstract
The objective of this study was to develop self-emulsifying drug delivery system (SEDDS) to improve solubility and enhance the oral absorption of the poorly water-soluble drug, nevirapine. This lipid-based formulation may help to target the drug to lymphoid organs where HIV-1 virus resides mainly. The influence of the oil, surfactant and co-surfactant types on the drug solubility and their ratios on forming efficient and stable SEDDS were investigated in detail. Two SEDDS (F1 and F2) were prepared and characterized by morphological observation, droplet size and zeta potential determination, cloud point measurement and in vitro diffusion study. The influence of droplet size on the absorption from formulations with varying concentration of oil and surfactant was also evaluated from two self-emulsifying formulations. Oral bioavailability of nevirapine SEDDS was checked by using rat model. Results of diffusion rate and oral bioavailability of nevirapine SEDDS were compared with marketed suspension. The absorption of nevirapine from F1 and F2 showed 1.92 and 1.98-fold increase (p < 0.05) in relative bioavailability, respectively, compared with that of the suspension. There was no statistical significant difference (p < 0.05) between F1 and F2 in their AUC and Cmax. This indicated that there was apparent poor correlation between the droplet size and in vivo absorption. However, nevirapine in SEDDS showed higher ex vivo stomach and intestinal permeability and in vivo absorption than the marketed suspension, suggesting that the SEDDS may be a useful delivery system for targeting nevirapine to lymphoid organs.
Introduction
Nevirapine (NVP) is an orally active antiretroviral drug approved by FDA that is currently used in the treatment of human immunodeficiency virus type 1 (HIV-1) infections (Devi & Pai, Citation2006; Waters et al., Citation2007), is particularly insoluble in water at physiological pH conditions and soluble only under extremely acidic media (Merck Index, Citation2006). NVP belongs to Biopharmaceutical Classification System (BCS) class II (low solubility/high permeability), poses a challenge in achievement of optimal dissolution kinetics from the dosage form (Kasim et al., Citation2004). NVP is a weak base (pKa = 2.8) with low intrinsic water solubility (0.06 mg/ml) which gives rise to difficulties in the formulation of dosage forms and leads to variable dissolution rates with a resultant decrease in bioavailability (Hawi & Bell, Citation1994; Lamson et al., Citation1995). A range of novel strategies are currently being developed for efficient delivery of antiretroviral drugs. Several delivery systems have been reported for the delivery of ARV drugs including bioadhesive coated matrix tablets (Betagiri et al., Citation2001; Govender et al., Citation2005), ceramic implants (Benghuzzi, Citation2000), liposomes (Desormeaux & Bergeron, Citation1998; Makabi-Panzu et al., Citation1998; Jin et al., Citation2005; Ramana et al., Citation2010), solid colloidal nanoparticles (Kuo & Su, Citation2007; Chattopadhyay et al., Citation2008; Kaur et al., Citation2008; Mainardes et al., Citation2009), microparticles by supercritical antisolvent method (Sanganwar et al., Citation2010), dendrimers (Dutta et al., Citation2007), micelles & microemulsion (Griffin & Driscoll, Citation2006), nanopowders (Erdenburgh et al., Citation2007) and suspensions/nanosuspension (Kinman et al., Citation2003; Shegokar & Singh, Citation2011; Shegokar et al., Citation2011; Yang et al., Citation2011). In this study we demonstrate the development of self-emulsifying drug delivery system (SEDDS) employing widely used and Generally Regarded As Safe (GRAS) lipid-based excipients. The present investigation also describes the detailed procedures for characterization of SEDDS with help of reported techniques.
A self-emulsifying formulation has an ability to form fine colloidal droplets with very high surface area. In many cases, this accelerates the digestion of the lipid formulation, improves absorption and reduces food effect and inter-subject variability (Constantinides, Citation1995; Humberstone & Charman, Citation1997; Pouton, Citation1997, Citation2000).
The self-emulsification process was shown to be dependent on the lipid/surfactant pair, the surfactant concentration, the ratio between lipid and surfactant (Pouton, Citation1985; Wakerly et al., Citation1987). However, only specific combinations can lead to an efficient self-emulsifying system (Charman et al., Citation1992; Shah et al., Citation1994). It is advantageous to use medium chain triglycerides (MCT) like ethyl oleate, caprylic/capric triglycerides derived from fractionated coconut oil or palm seed oil, due to their higher fluidity, better solubility properties and self-emulsification ability compared with long chain triglycerides (LCT) (Charman et al., Citation1992; Shah et al., Citation1994). Additionally, drug substances have better chemical stability in MCT due to the purity of the lipid and the lack of double bonds, which can catalyze oxidation. The two lipids are differently transported in the body, MCT is directly transported by the portal blood to the systemic circulation (Porter & Charman, Citation1997), whereas LCT is transported in the intestinal lymphatics. However, some of the medium-chain saturated fatty acids are resynthesized or re-esterified to triacylglycerols or neutral lipids, packaged within apoprotein surface layers and transported via the lymphatic system (Green & Glickman, Citation1981; Ikeda et al., Citation1991; Carvajal et al., Citation2000; Mu & Hoy, Citation2001; Holm et al., Citation2002). Recently, Sun et al. (Citation2011) have reported that, the lymphatic transport of sirolimus from SMEDDS formulation containing ≥25% MCT was a major contributor to its oral bioavailability.
HIV/AIDS patients often have severe fat malabsorption and thus their quality of life is greatly diminished. If they could ingest a diet containing a readily absorbable fat source, such as MCTs, their steatorrhea and stool nitrogen excretion could be diminished and their nutritional status may be improved. (Wanke et al., Citation1996; Craig et al., Citation1997). Craig et al. (Citation1997) have demonstrated that the MCT-containing formula to be better tolerated by AIDS patients, with fat malabsorption, compared to control liquid formulas (relative to solid food diet) and suggested as a source of dietary fat for such patients. The aim of the present study was to evaluate the potential of self-emulsifying drug delivery system to improve the absorption of dissolution-rate limited bioavailability of NVP, using traditional and pharmaceutically accepted lipid-based excipients. The objectives of the present study were: (1) to develop and optimize the self-emulsifying formulation of NVP; (2) to assess its properties in vitro; (3) to investigate the influence of droplet size of self-emulsifying formulation on drug absorption; and (4) to assess its oral bioavailability compared with a conventional dosage form using animal model.
Materials and methods
Materials
Nevirapine was a generous gift from Matrix Laboratories Ltd. (Hyderabad, India); Caprylic and Oleic acids were obtained from Soofi Traders (Mumbai, India); Polyoxyl 35 castor oil (Cremophor EL) was obtained from BASF Corp. (Mumbai, India). Diethylene glycol monoethyl ether (Transcutol P®), PEG-8 glycol caprylate (Labrasol®), Propylene glycol monocaprylate (Capryol 90®) were provided by Gattefosse, France; and Glycerol Monocaprylocaprate (Capmul MCM®) was provided by Abitec Corp., Janesville, WI. PEG-400, Tween 20, Tween 80, Propylene glycol and diethyl ether were purchased from S. D. Fine chemicals (Mumbai, India). Carbamazepine was purchased from HIMEDIA. All other chemicals and solvents used were of analytical grade.
Methods
Solubility studies
The solubility of NVP in various oils, surfactants and co-surfactants was determined. An excess of NVP was placed in 2 ml of various vehicles in a screw-capped glass vial and the mixture was heated at 60 °C in a water-bath (Equitron, Chennai, India) for 2 min to facilitate the solubilization using a cyclomixer (CM 101, Remi instruments Ltd., Ahmedabad, India). Mixtures were equilibrated at 25 °C for 48 h in a water bath and then centrifuged in a laboratory centrifuge (Biofuge, Thermo Electron Corp., Waltham, MA, USA) at 2000 rpm for 15 min to separate the undissolved drug. Aliquots of supernatant were diluted with methanol and analysed for the dissolved drug by HPLC (Jasco, Japan) using RP column (LCGC Qualisil BDS C18; 5 µm 250 mm × 4.6 mm, i.d) injecting 20 µl volume. The mobile phase consisted of a mixture of methanol: water (50:50) at a flow rate of 0.9 ml/min that led to retention time of 7.23 min when detection was carried out at 265 nm. The assay was linear (r2 = 0.999) in the concentration range of 2–20 µg/ml. The method was validated with respect to accuracy, and inter-day and intra-day precision as per ICH guidelines (Q2(B), 2003) and the relative standard deviation was less than 2% in both the cases.
Pseudoternary phase diagrams
Pseudoternary phase diagrams of oil, surfactant/co-surfactant (S/CoS) and water were developed using the water titration method (Djordjevic et al., Citation2004). The mixtures of oil and S/CoS at certain weight ratios (9:1 to 1:9 w/w) were diluted with water in a drop wise manner. For each phase diagram at a specific ratio of S/CoS (i.e. 2:1, 1:1 and 1:2 w/w), a transparent and homogenous mixture of oil and S/CoS was formed by vortexing for five minutes. Then each mixture was titrated with water and visually observed for phase clarity and flowability. During titration, the samples were agitated gently in order to reach equilibrium quickly. The phase boundary was determined by observing the changes in the sample appearance from transparent to turbid or from turbid to transparent. Phase diagrams were then constructed using Tri plot v1–4 software (David Graham &Nicholas Midgley, Loughborough, Leicestershire, UK). The following studies were carried out by constructing a ternary phase diagram: (1) the influence of the ratio of surfactant to co-surfactant on the formation of emulsion; (2) the effect of more than one surfactant in different ratios with Transcutol P as co-surfactant; (3) the influence of surfactants on emulsion formation with Transcutol P as co-surfactant.
Preparation of self-emulsifying drug delivery systems
The formulations consisted of nevirapine: Caprylic acid: (Labrasol: Tween 80, 1:3):Transcutol P (2.5:19.5:62.4:15.6 and 2.5:9.75:75.22:12.54%, w/w) (). The formulations were prepared by dissolving NVP in the mixture of Transcutol P and Caprylic acid at 50 °C in an isothermal water bath (Equitron, Chennai, India). Labrasol and Tween 80 were then added. This mixture was mixed by vortexing until a transparent preparation was obtained.
Table 1. Composition of NVP SEDDS (F1 and F2).
Characterization of self- emulsifying drug delivery systems
Drug content
The concentration of NVP was determined by HPLC method (described in solubility studies). SEDDS formulation equivalent to 25 mg of NVP was taken and diluted in methanol. Volume was made up to 25 ml with methanol (1 mg/ml). From the above stock solution, 0.1 ml (100 µg) was withdrawn and diluted up to 10 ml with methanol (10 µg/ml). Aliquots of 20 µl were injected onto the HPLC system, to analyze NVP at 265 nm.
Morphological characterization
The morphology of self-emulsifying formulation was observed by using a transmission electron microscope (TEM) (Phillips Tecnai 20, Holland) at an acceleration voltage of 200 kV and typically viewed at a magnification of 43 000×. The size of the colloidal structures was determined using Analysis® software (Soft Imaging Systems, Reutlingen, Germany).
Formulations were diluted with distilled water 1:25 and shaken. Carbon-coated copper grids were glow-discharged (Edwards E306A Vacuum Coater, Crawley, England) and 10 µl of samples were deposited on a holey film grid. After drying, the samples were photographed by transmission electron microscopy.
Determination of droplet size and zeta (ζ) potential
Droplet size and the zeta potential of the formed emulsion were determined by photon correlation spectroscopy that analyzes the fluctuations in light scattering due to Brownian motion of the particles, using a Zetasizer ZS 90 (Malvern Instruments, UK). Light scattering was monitored at 25 °C at a 90° angle. All the measurements were performed at room temperature (25 °C) in triplicate.
In vitro diffusion study
In vitro diffusion study of the NVP SEDDS was compared with a conventional marketed suspension (Nevimune®, Nevirapine Oral Suspension, CIPLA) using a dialysis technique (Kang et al., Citation2004; Patil et al., Citation2004; Dixit et al., Citation2010). The dialyzing medium was 0.1 N HCl. One end of dialysis tube (Dialysis membrane 70, HIMEDIA; MWCO 12 000–14 000 daltons; pore size 2.4 nm) was clamped and then the experimental formulation sample, equivalent to 100 mg drug, was placed in it. The other end of the tube was also secured with dialysis closure clip (HIMEDIA, Mumbai) and was placed in 900 ml of dialyzing medium and stirred at 100 rpm over a magnetic stirrer (Remi Instrument Ltd., Mumbai, India) at 37 °C. Aliquots of 1 ml were removed at 15, 30, 45, 60, 120, 180, 240 and 300 min time intervals and suitably diluted further. Moreover, each time the volume of aliquots was replaced with the fresh dialyzing medium. These samples were analyzed for NVP present in the dialyzing medium at corresponding time by UV-visible spectrophotometer (Shimadzu UV-2450, Japan) at 313 nm.
Ex vivo stomach permeability
Male Sprague–Dawley rats (250–300 g) were euthanized in a carbon-dioxide vacuum chamber. All experiments and protocols described in this animal study were approved by the Institutional Animal Ethics Committee of B. V. Patel PERD Centre, Ahmedabad, India and were in accordance with the guidelines of the committee for purpose of control and supervision of experiments on animals, Ministry of Social Justice and Empowerment, Government of India. To check the stomach permeability, the stomach part was isolated carefully and taken for the ex vivo permeation study. Then this tissue was washed with physiological acid solution containing 100 mM HCl and 54 mM NaCl to remove the mucous and gastric contents. The method employed was modified from experimental procedures well described in the literature (Gharzouli et al., Citation1995; Hung & Neu, Citation1997; Shah & Khan, Citation2004)
SEDDS formulations, 0.2 ml (equivalent to 5 mg of NVP) were injected into the stomach using a syringe, and the two sides of stomach tissue were tightly closed with the help of threads. In a similar way 0.5 ml (equivalent to 5 mg of NVP) market suspension formulation (Nevimune®-CIPLA) was also filled in the stomach tissue. The filled stomach tissue was placed in a beaker with constant stirring and temperature of 37 °C on a magnetic stirrer. The receiver compartment was filled with 30 ml of 100 mM HCl. Aliquots of 3 ml were withdrawn at 0.5, 1.0, 2.0 and 3.0 hours time intervals from the beaker and suitably diluted further. The absorbance was measured using a UV-Visible spectrophotometer at a wavelength of 313 nm, keeping the respective blank. The percent permeation of drug was calculated against time and plotted on a graph.
Ex-vivo intestinal permeability studies
This was carried out by the method well described in the literature (Araya et al., Citation2006; Ghosh et al., Citation2006). Male Sprague–Dawley rats (250–300 g) were euthanized in a carbon-dioxide vacuum chamber. All experiments and protocols described in this animal study were approved by the Institutional Animal Ethics Committee of B. V. Patel PERD Centre, Ahmedabad, India and were in accordance with the guidelines of the committee for purpose of control and supervision of experiments on animals, Ministry of Social Justice and Empowerment, Government of India. To check the intestinal permeability, small portion of small intestine was isolated and used for the ex vivo permeability study. The tissue was thoroughly washed with pH 6.8 phosphate-buffered saline (USP 36) (USP 36/NF31, 2014) to remove any mucous and lumen contents. The NVP SEDDS, 1.04 g (equivalent to 25 mg NVP) and conventional marketed suspension, 1 ml (equivalent to 10 mg NVP) were diluted up to 10 ml and 4 ml with distilled water, respectively and mixed over cyclomixer for one minute. The resultant samples (2.5 mg/ml) of both NVP SEDDS and conventional marketed suspension were injected separately into the lumen of the small intestine tissue using a syringe, and the two sides of the intestine were tightly closed with the help of a thread. The tissue was placed in a beaker filled with 30 ml of pH 6.8 phosphate-buffered saline containing 20% PEG-400 with constant stirring at 37 °C. The two ends of tissues were fixed horizontally on to a beaker with the help of a thread. Aliquots of 3 ml were withdrawn at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4 and 5 hours time intervals from the beaker and suitably diluted further. The absorbance was measured using a UV-visible spectrophotometer at a wavelength of 283 nm (Kabra et al., Citation2009; Mandloi et al., Citation2009). The amount of drug diffused (%) was calculated against time and plotted on a graph.
HPLC analysis
The concentration of nevirapine in the plasma samples was analyzed by modified reported methods (Hollanders et al., Citation2000; Kabra et al., Citation2009; Minzi & Ngaimisi, Citation2010). The HPLC apparatus consisted of Jasco PU-980 Intelligent HPLC pump (Jasco, Japan) equipped with a Jasco UV-975 Intelligent UV/VIS detector (Jasco, Japan), an autosampler Jasco AS-950–10 Intelligent sample (Jasco, Japan), a Jasco Borwin chromatography software (version 1.50) integrator software and a LCGC Qualisil BDS C18 (4.6 mm × 250 mm and 5 μm particle size) column. The mobile phase consisted of a mixture of 10 mM sodium phosphate buffer pH 5.0 and acetonitrile (70:30 v/v) at a flow rate of 1.0 ml/min that led to retention time of 6.75 min when detection was carried out at 245 nm. The assay was linear (r2 = 0.999) in the concentration range of 25–5000 ng/ml. The method was validated with respect to accuracy and inter and intra-day precision at three quality control samples (75, 500 and 3500 ng/ml) as per ICH guidelines (ICH Q2(B), 2003) Limit of detection (LOD) and limit of quantitation (LOQ) of the developed method were estimated on the basis of standard deviation and slope of the calibration curve as 3.3 δ/m and 10 δ/m, respectively. Here δ is the standard deviation of the response (chromatogram peak area) and m is the slope of the calibration curve. The LOD and LOQ were found to be 19.8 ng/ml and 60 ng/ml, respectively.
Bioavailability studies
Bioavailability studies were performed in male Sprague–Dawley rats weighing 250 to 300 g. All experiments and protocols described in this study were approved by the Institutional Animal Ethics Committee of Sri Dhanvantary Pharmaceutical Analysis and Research Centre, Surat, India and were in accordance with guidelines of the committee for purpose of control and supervision of experiments on animals, Ministry of Social Justice and Empowerment, Government of India (Ministry of Environment & Forests, Govt. of India, Citation2007). Three groups were made for the study, and four rats were kept in each group. The animals were kept under standard laboratory conditions, temperature at 25 ± 2 °C and relative humidity (55 ± 5%). The animals were housed in polypropylene cages, four per cage, with free access to standard laboratory diet (Lipton feed, Mumbai, India) and water ad libitum. The animals were fasted overnight prior to the experiment but had free access to water. The formulations (SEDDS (F1 and F2) and marketed suspension (Nevimune®)) were given orally using feeding snode. Dose for the rats was selected as reported (Rao & Shinde, Citation2009) and calculated based on the weight of the rats (20 mg/kg body weight) according to the surface area ratio (Paget & Barnes, Citation1964; Akhila et al., Citation2007). The developed SEDDS and marketed suspension (Nevimune®) were administered by oral snode in an equivalent dose of 20 mg/kg of NVP. The animals were anesthetized using ether and blood samples (approximately 500 μl) were collected from the retro-orbital vein using a heparinized needle (18–20 size) at 0, 1, 2, 4, 8, 12, 16 and 24 hours after oral administration. The blood samples were collected into a heparinized microcentrifuge tubes (13 × 75 mm, 2 ml, Accuvac, BD, NJ), subjected to centrifugation on a laboratory centrifuge and supernatant plasma was collected and kept at −20 °C (Thermo Scientific, USA) until analysis.
Plasma analysis
Frozen plasma samples were thawed just prior to extraction. A 100 µl of plasma sample was transferred in 2 ml centrifuge tube (Tarson, Kolkata, India). To that 100 µl of carbamazepine (1 µg/ml in methanol) was added as an internal standard (Minizi & Ngaimisi, Citation2010) and vibrated on cyclomixer for 10 s. Then, 25 µl of 0.1 M sodium hydroxide was added, vortex-mixed for 20 s and to it 1 ml of ethyl acetate was added and vortexed for 2 min. The tube was centrifuged at 15 000 rpm for 20 min at 4 °C; the organic layer was collected (top layer) and was transferred to a clean tube and dried under nitrogen stream at ambient temperature. The residue was reconstituted with a 100 µl aliquot of mobile phase, consisted of 10 mM Sodium phosphate pH 5.0 and acetonitrile (70:30 v/v) and 50 µl was injected directly onto the HPLC column at a flow rate of 1.0 ml/min.
Plasma concentration versus time data of NVP for rats was analyzed using standard non-compartment analysis. The area under the plasma concentration-time curve (AUC0→t) from zero to 24 hours was estimated by the linear trapezoidal method (Han & Lee, Citation1999; Lee & Ku, Citation1999). The relative bioavailability (F) of NVP in its SEDDS formulations (NVP SEDDS) to the suspensions was calculated using the following equation:
Statistical analysis
Statistical analysis for the determination of differences in diffusion, permeability and in vivo absorption profiles of NVP SEDDS and the marketed preparation was assessed by the use of Student’s t-test. The pharmacokinetic data between different formulations were compared for statistical significance by Student’s t-test. Statistical probability (p) values less than 0.05 were considered significantly different.
Stability studies
Chemical and physical stability of nevirapine SEDDS (F1 and F2) were assessed under various storage conditions namely room temperature (RT), 30 ± 2 °C/65 ± 5% RH and 40 ± 2 °C/75 ± 5% RH as per ICH guidelines (ICH Q1A(R2)) in ICH certified stability chambers (Humidity Chamber, EIE Instruments Ltd., Ahmedabad, India).
Nevirapine SEDDS equivalent to 25 mg was filled in a glass vial with a rubber closure and aluminium-crimped tops. Eight such glass vials of each F1 and F2 were filled and stored at various aforementioned storage conditions up to three months. Samples were removed at 0, 1, 2 and 3 months of interval and checked for NVP content (by HPLC), droplet size, polydispersity index and zeta potential after diluting formulation to 100 times with three different media viz. distilled water (DW), 0.1 N HCl and phosphate buffer saline pH 6.8 (PBS) maintained at 37 °C.
Results and discussion
Screening of oils and surfactants
The results of solubility of NVP in few vehicles are shown in . Solubility studies were carried out to identify potential ingredients for the formation of microemulsion. Amongst the various oils that were screened, Caprylic acid exhibited highest solubilizing potential for NVP and was selected as an oily phase for further studies (). Amongst various surfactants, Labrasol exhibited good solubilizing potential for NVP. Preliminary studies indicated that a mixture of Labrasol and Tween 80 showed reasonable solubilizing potential for NVP and also better emulsifying properties when evaluated by method reported by Date & Nagarsenkar (Citation2007). The components and their concentration ranges can be obtained by construction of a pseudo-ternary phase diagram with constant drug level fixed at 2.5% (w/w). The drug loading capability is the main factor when screening the oil phase.
Figure 1. Solubility of NVP in various vehicles at 25 °C (n = 3).
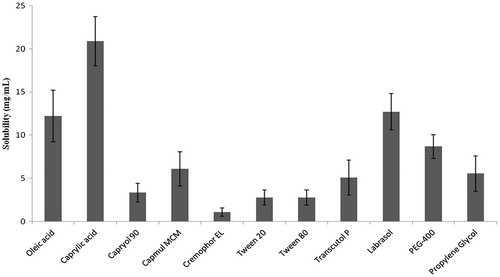
As to the selection of co-surfactant, Transcutol P showed better solubility in the presence of Labrasol than PEG-400 and propylene glycol. Therefore, it was reasonable to select Transcutol P as the co-surfactant.
Visual observation and percent transmittance data proved that Labrasol when used alone as a surfactant is unable to incorporate large amount of water compared to Tween 80. But the combination of these two surfactants showed enhanced water uptake capacity. Among the 1:1, 1:2 and 1:3 ratios of Labrasol: Tween 80; 1:3 showed better solubility and dilution potential. Hence the mixture of Labrasol and Tween 80 in the ratio of 1:3 was selected as a surfactant.
Phase diagrams of the systems containing Caprylic acid as an oil phase, mixture of Labrasol and Tween 80 as a surfactant and Transcutol P as a co-surfactant (CoS) were constructed at the surfactant (Labrasol: Tween 80; 1:3 (w/w))/co-surfactant ratio of 2:1, 1:1 and 1:2 (w/w) and shown in . Maximum microemulsifying region was found to be at the surfactant/co-surfactant in the ratio of 1:2 followed by 2:1 having surfactant mixture ratio of 1:3 (Labrasol: Tween 80).
Figure 2. Pseudo-ternary phase diagrams of the formulations composed of oil, surfactants and co-surfactant dispersed with distilled water at 37 °C. Surfactants = Labrasol: Tween 80, (1:3); Co-surfactant = Transcutol P. Surfactant to co-surfactant ratios were as follows: (a) 2:1, (b) 1:1, (c) 1:2. The shadow area represents microemulsion region.
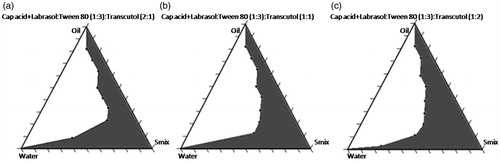
Usually Transcutol P is used as co-surfactant in less concentration compared to surfactants. Keeping this in mind, surfactant/co-surfactant in the ratios of 6:1, 4:1 and 2:1 with surfactant mixture 1:3 (Labrasol: Tween 80) were tried for formulation preparations in the ratios of oil:Smix of 1:4, 3:7 and 1:9 and were characterized for percent transmittance and globule size after 100 times dilution. The percent transmittance was in the range of 91.69–100.50 when Oil: Smix ratio was 1:9. The droplet size increases as the percent transmittance falls. The percent transmittance improved in the order of S/CoS ratio 6:1 > 4:1 > 2:1 > 1:2. There was decrease in the percent transmittance and increase in the average droplet size in the order of Oil: Smix ratio 1:9 < 1:4 < 3:7. The influence of different ratios of surfactant [Labrasol: Tween 80(1:3)] to co-surfactant (Transcutol P) on droplet size was also investigated.
Based on our results, two SEDDS formulations, F1 and F2 were established: (a) F1 comprised of 20% of Caprylic acid as oil, 64% mixture of Labrasol and Tween 80 (1:3) as surfactant and 16% Transcutol P as co-surfactant. (b) F2 comprised of 10% Caprylic acid as oil, 77.14% mixture of Labrasol and Tween 80 (1:3) as surfactant and 12.86% Transcutol P as co-surfactant.
SEDDS characterization
Morphological characterization
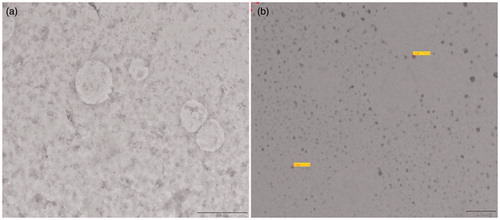
The NVP SEDDS turned into transparent emulsion when diluted with distilled water. The TEM pictures are shown in . The formed emulsion droplets were observed to be spherical.
Figure 3. TEM images of nevirapine microemulsion (×43 000) (Scale: 500 nm). TEM of (a) SEDDS F1 and (b) SEDDS F2.
Determination of droplet size and ζ-Potential
The average droplet size of emulsion, dispersed from the NVP SEDDS after dilution was within 260 nm and 100 nm when Oil: Smix ratio was 1:4 and 1:9, respectively and showed Gaussian distribution.
The effect of medium on droplet size was also investigated in the present study. The droplet size, polydispersity index and zeta potential of SEDDS formulation after 100 times dilution in different media is shown in . On diluting NVP SEDDS in distilled water, 0.1 N HCl and pH 6.8 phosphate buffer, the resulted droplet size was 210.8 ± 0.56 nm, 223.34 ± 0.48 nm and 163.1 ± 0.66 nm, respectively for Oil: Smix 1:4 and S/CoS 4:1 (F1). When Oil:Smix ratio was 1:9 and S/CoS 6:1 (F2), the resulted droplet size after dispersing in distilled water, 0.1 N HCl and pH 6.8 phosphate buffer was 14.63 ± 0.39 nm, 14.25 ± 0.35 nm and 13.17 ± 0.39 nm, respectively.
Table 2. Characterization of NVP SEDDS (F1 and F2).
Generally, an increase of electrostatic repulsive forces between microemulsion droplets prevents the coalescence of microemulsion droplets. On the contrary, a decrease of electrostatic repulsive forces may cause coalescence and phase separation. NVP SEDDS were diluted with distilled water, 0.1 N HCl and pH 6.8 phosphate buffer saline, and the resulted zeta potential was −5.2 ± 0.35 mV, 2.74 ± 0.17 mV and −2.73 ± 0.18 mV, respectively for F1 and −6.02 ± 0.54 mV, 15.2 ± 0.49 mV, −4.27 ± 0.21 mV, respectively for F2.
In vitro diffusion study
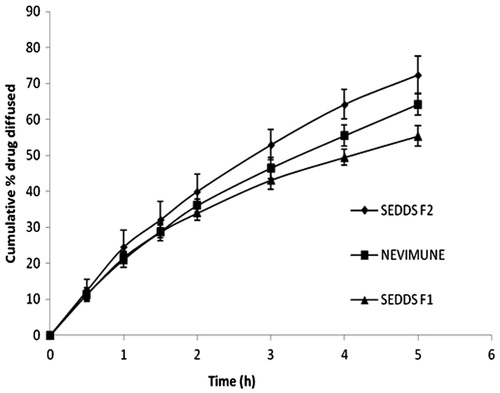
In vitro diffusion data are shown in . The rate of drug diffusion was in the order of F2 > marketed suspension > F1. The total percentage diffusion was higher for the F2 SEDDS than for the suspension dosage form. After five hours of diffusion, more than 70% of the drug was diffused from the self-emulsifying system, as compared with 64% diffused from the marketed suspension. The solubility of NVP increases as pH drops, therefore, the initial release profile of all the three formulations looks alike in 0.1 N HCl.
Figure 4. In vitro diffusion profiles of NVP SEDDS and marketed suspension in 0.1 N HCl (n = 6).
Ex-vivo stomach and intestinal permeability study
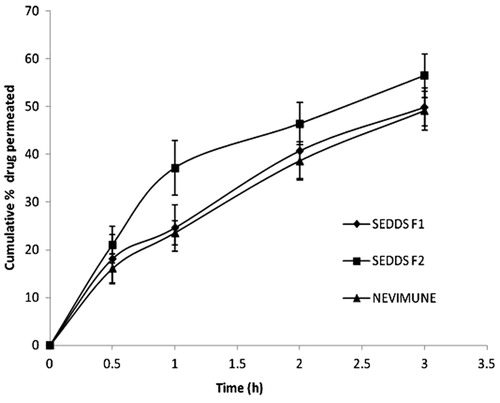
The profile of drug permeation across stomach is shown in . At the end of three hours, 49.86% ( ± 3.986) and 56.42% ( ± 4.582) of drug was permeated across the stomach from developed NVP SEDDS, F1 and F2 respectively, compared to 49.066 % ( ± 4.042) from marketed suspension. The total percentage permeation from rat stomach tissue was significantly higher (p < 0.05) for the NVP SEDDS F2 than for the marketed suspension. On the other hand, SEDDS F1 is composed of about 20% of oil and it needs some time to breakdown the oil globules before releasing the drug. SEDDS F2 which contains 10% oil, has higher proportion of surfactant compared to F1 and may be responsible for better release of NVP in an acidic medium.
Figure 5. Ex vivo stomach permeability of NVP SEDDS and marketed suspension in 100 mM HCl (n = 3).
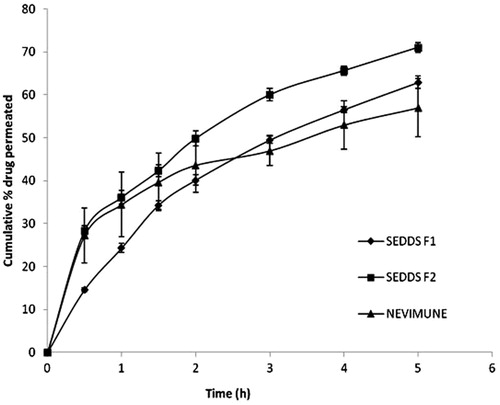
The profile of intestinal drug permeation is shown in . At the end of five hours, the percentage of NVP permeated across rat intestinal tissue was 62.88 ± 1.43, 71.03 ± 1.08 and 57.00 ± 6.77 for F1, F2 and markets suspension, respectively. More amount of oil needs to be broken down to release the drug from the oil globule as seen in F1. The initial similar permeation profile of marketed suspension to that of developed SEDDS may be due to dissolved and smaller drug particles of suspension, and also the availability of the fresh medium. The drug permeation (both stomach and intestinal permeability) from the NVP SEDDS formulation F2 was found to be significantly higher (p < 0.05) as compared to that of the marketed suspension and F1. There was no significant difference in the permeation of NVP SEDDS formulation F1 and marketed suspension at p < 0.05.
Figure 6. Ex vivo intestinal permeability of NVP SEDDS and marketed suspension in pH 6.8 PBS (n = 3).
Bioavailability study
To investigate the oral bioavailability of NVP SEDDS, the pharmacokinetic parameters of NVP were determined after oral administration of NVP SEDDS and marketed conventional suspension to male Sprague–Dawley rats. A representative chromatogram showing the elution of nevirapine and internal standard (carbamazepine) is shown in . The in vivo pharmacokinetic behaviour of NVP with SEDDS and suspension were investigated. shows the plasma profiles of NVP after oral administration of the marketed suspension and developed SEDDS (F1 and F2).
Figure 7. Typical chromatogram showing the elution of nevirapine (Rt = 6.76 min) and internal standard (carbamazepine) (Rt = 16.27 min) at their respective retention time.
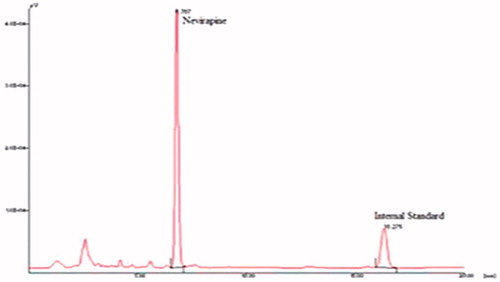
Figure 8. Plasma concentration profile of NVP after oral administration of SEDDS and marketed suspension in rats (n = 4 and 20 mg/kg).
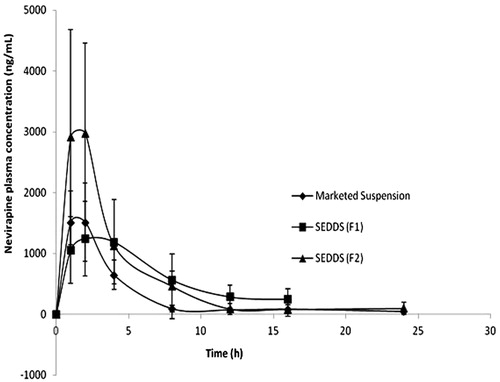
The pharmacokinetic parameters are given in . The AUC0→t and AUC0→∞ of the SEDDS were significantly higher than those of the suspension. However, there was not much difference with respect to Cmax for F1 and suspension, in fact the suspension showed slightly higher Cmax than that of F1. The SEDDS F1, having about 20% of oil has somewhat delay in action which may be due to the fact that drug is enclosed in the core and takes time to get released and absorbed into the intestinal linings. Suspension, initially, having drug particles with smaller diameter and some amount in dissolved form may be responsible for the initial rise in plasma concentration as seen in the plasma profile and same can be an explanation for ex vivo intestinal permeation (). Cmax of F2 was highest among all the three formulations evaluated. The role of particle size in the performance of the formulation in vivo is generally less important than formulators have assumed (Perlman et al., Citation2008). The main reason for this is that as soon as the dispersed formulation leaves the stomach it encounters the formidable digestive power of the small intestine (Charman et al., Citation1997). The fate of the drug after the formulation has been digested is a great deal more important than the initial particle size. Esters will be rapidly hydrolysed in the presence of pancreatic lipase and even the most commonly used surfactants (ethoxylated esters) are often rapidly hydrolysed (Dahan & Hoffman, Citation2008). The physical state of the degradation products will be changed significantly, by contact with the mixed bile salt micelles and the drug will partition between the various phases in the gut lumen, or could precipitate out if the total solvent capacity is reduced as a consequence of lipolysis (Pouton, Citation2006).
Table 3. Relative bioavailability and pharmacokinetic parameters of NVP after oral administration of NVP SEDDS and marketed suspension (MKT) to the rats (n = 4).
The relative bioavailabilities of NVP from SEDDS were approximately 1.92-fold and 1.98-fold for F1 and F2 formulations, respectively, when compared with the marketed suspension. There was no statistical significant difference between F1 and F2 for Cmax, AUC0→t and AUC0→∞ at p < 0.05.
Stability studies
Chemical and physical stability of the nevirapine SEDDS were assessed at various storage conditions as per ICH guidelines. The developed formulations were stored in glass vials with rubber closure and aluminium-crimped tops. No change in the physical parameters such as homogeneity and clarity was observed during the stability studies. Droplet size, polydispersity index and zeta potential remained unchanged even after keeping the formulations at different environmental storage conditions for three months, indicating the physical stability of developed formulations (data not shown), when compared to initial characterization as shown in . No decline in the drug content was observed at the end of three months indicating that drug remained chemically stable in SEDDS.
Conclusion
A SEDDS containing poorly water-soluble drug, NVP, was formulated for oral application. The components and their ratio ranges for the formulation of SEDDS were obtained by solubility study, pseudo-ternary phase diagram construction and droplet size analysis. The optimum formulations of the SEDDS consisted of 20% Caprylic acid, 64% mixture of Labrasol and Tween 80 (1:3) and 16% Transcutol P (F1) and 10% Caprylic acid, 77.14% mixture of Labrasol and Tween 80 (1:3) and 12.86% Transcutol P (F2). Both the SEDDS formulations had sufficient drug loading, rapid self-emulsification in aqueous media and forming droplet not more than 260 nm. The in vivo study showed that there is no significant difference among the developed formulations, F1 and F2, but showed greater extent of absorption than the marketed suspension. It is desirable to have less amount of surfactants and more amount of oil and therefore, the formulation F1 with 20% of Caprylic acid as oil, 64% mixture of Labrasol and Tween 80 (1:3) as surfactant and 16% Transcutol P as co-surfactant is acceptable. The relative bio-availabilities of SEDDS to suspension were 192.22 % and 198.45 % for F1 and F2, respectively. The data overall showed little apparent correlation between emulsion droplet size and oral absorption in rats, but differences in exposures were generally not large enough to be statistically significant. Both formulations did show significant differences in exposures, and these results were not consistent with a strong dependence on emulsion droplet size. However, there was an increase in the absorption of NVP with an increase in the concentration of surfactant in SEDDS. The increase in oral bioavailability could be a consequence of delivering the drug in a solubilized and rapidly dispersed manner. The results obtained illustrate the magnitude of bioavailability enhancement that can be achieved through rational design of lipid-based formulations.
Acknowledgements
The authors are thankful to the generosity of M/s Matrix Laboratories Ltd. (Hyderabad, India) for providing gift sample of nevirapine, and Soofi Traders, Mumbai; BASF Corp., Germany; Gattefosse, France; and Abitec Corp., Janesville WI for providing the gift samples of excipients. The authors are grateful to Dr Aravind Badiger, Director; Sri Dhanvantary Pharmaceutical Analysis and Research Centre, Surat, India for support and providing in vivo study facilities.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Akhila JS, Deepa S, Alwar MC. (2007). Acute toxicity studies and determination of median lethal dose. Curr Sci 93:917–20
- Araya H, Tomita M, Hayashi M. (2006). The novel formulation design of self-emulsifying drug delivery systems (SEDDS) type o/w microemulsion III: the permeation mechanism of a poorly water soluble drug entrapped o/w microemulsion in rat isolated intestinal membrane by the Ussing chamber method. Drug Metab Pharmacokinet 21:45–53
- Benghuzzi H. (2000). Long term sustained delivery of 3'-azido-2',3'-dideoxy thymidine in vivo by means of HA and TCP delivery devices. Biomed Sci Instrum 36:343–8
- Betagiri GV, Deshmukh DV, Gupta RB. (2001). Oral sustained- release bioadhesive tablet formulation of didanosine. Drug Dev Ind Pharm 27:129–36
- Carvajal O, Nakayama M, Kishi T, et al. (2000). Effect of medium-chain fatty acid positional distribution in dietary triazcylglycerol on lymphatic lipid transport and chylomicron composition in rats. Lipid 35:1345–51
- Charman SA, Charman WN, Rogge MC, et al. (1992). Self-emulsifying drug delivery systems: formulation and biopharmaceutics evaluation of an investigational lipophilic co mpound. Pharm Res 9:87–93
- Charman WN, Porter CJH, Mithani S, Dressman JB. (1997). Physicochemical and physiological mechanism for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci 86:269–82
- Chattopadhyay N, Zastre J, Wong HL, et al. (2008). Solid lipid nanoparticle enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm Res 25:2262–71
- Constantinides PP. (1995). Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res 12:1561–72
- Craig CB, Darnell BE, Weinsier RL, et al. (1997). Decreased fat and nitrogen losses in patients with AIDS receiving medium-chain-triglyceride-enriched formula versus those receiving long-chain-triglyceride-containing formula. J Am Diet Assoc 97:605–11
- Dahan A, Hoffman A. (2008). Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release 129:1–10
- Date AA, Nagarsenker MS. (2007). Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm 329:166–72
- Desormeaux A, Bergeron MG. (1998). Liposome as drug delivery system: a strategic approach for the treatment of HIV infection. J Drug target 6:1–15
- Devi KV, Pai RS. (2006). Antiretrovirals: need for an effective drug delivery. Indian J Pharm Sci 68:1–6
- Dixit AR, Rajput SJ, Patel SG. (2010). Preparation and bioavailability assessment of SMEDDS containing Valsartan. AAPS Pharm Sci Tech 11:314–21
- Djordjevic L, Primorac M, Stupar M, Krajisnik D. (2004). Characterization of caprylocaproyl macrogolglycerides based microemulsion drug delivery vehicles for an amphiphilic drug. Int J Pharm 271:11–19
- Dutta T, Agashe HB, Garg M, et al. (2007). Poly(propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocyte/macrophage, in vitro. J Drug Target 15:89–98
- Eerdenburgh BV, Froyen L, Martens JA, et al. (2007). Characterization of physiochemical properties and pharmaceuticals performance of sucrose co-freeze dried solid nanoparticulate powder of the anti-HIV agent loviride prepared by media milling. Int J Pharm 338:198–206
- Gharzouli K, Senator A, Gharzouli A, et al. (1995). Acid secretion and fluoride absorption by the ligated stomach of the rat-mutual influences of fluoride and luminal acidity. Fluor 28:3–9
- Ghosh PK, Majithiya RJ, Umrethia ML, Murthy RSR. (2006). Design and development of microemulsion drug delivery system of Acyclovir for improvement of oral bioavailability. AAPS PharmSciTech 7(3):E1--E6. Article 77. DOI: 10.1208/pt070377
- Govender S, Pillay V, Chetty DJ, et al. (2005). Optimisation and characterization of bioadhesive controlled release tetracycline microspheres. Int J Pharm 306:24–40
- Green PHR, Glickman RM. (1981). Intestinal lipoprotein metabolism. J Lipid Res 22:1153–73
- Griffin BT, Driscoll CMO. (2006). A comparison of intestinal lymphatic transport and systemic bioavailability of saquinavir from three lipid based formulation in the anaesthetized rat model. J Pharm Pharmacol 58:917–25
- Han KS, Lee MG. (1999). Effect of a new chemoprotective agent, 2-(allylthio)pyrazine, on the pharmacokinetics of intravenous theophylline in rats. Int J Pharm 184:237–42
- Hawi A, Bell G. (1994). Preformulation studies of nevirapine, a reverse transcriptase inhibitor. Pharm Res 11(Suppl):S236
- Hollanders RMF, Van Ewijk-Beneken Kolmer EWJ, et al. (2000). Determination of nevirapine, an HIV-1 non-nuceloside reverse transcriptase inhibitor, in human plasma by reversed-phase high-performance liquid chromatography. J Chromatogr B 744:65–71
- Holm R, Porter CJH, Mullertz A, et al. (2002). Structured triglyceride vehicles for oral delivery of halofantrine: examination of intestinal lymphatic transport and bioavailability in conscious rats. Pharm Res 19:1354–61
- Humberstone AJ, Charman WN. (1997). Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev 25:103–28
- Hung CR, Neu SL. (1997). Acid-induced gastric damage in rats is aggravated by starvation and prevented by several nutrients. J Nutr 127:630–6
- ICH (Q1A (R2) harmonized tripartite guideline, stability testing of new drug substances and products Q1A (R2). Available from: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html [last accessed 12 Oct 2013]
- ICH Q2(B) (2003). Guidelines: validation of analytical procedure: methodology Q2(B), 2003. Available from: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html [last accessed 12 Oct 2013]
- Ikeda I, Tomari Y, Sugano M, et al. (1991). Lymphatic absorption of structured glycerolipids containing medium-chain fatty acids and linoleic acid, and their effect on cholesterol absorption in rats. Lipid 26:369–73
- Jin SX, Wang DZJ, Wang YZ, et al. (2005). Pharmacokinetic and tissue distribution of zidovudine in rats following intravenous administration of zidovudine myristate loaded liposomes. Pharmazie 60:840–3
- Kabra V, Agrahari V, Karthikeyan C, Trivedi P. (2009). Simulataneous quantitative determination of zidovudine and nevirapine in human plasma using isocratic, reverse phase high performance liquid chromatography. Trop J Pharm Res 8:79–86
- Kang BK, Lee JS, Chon SK, et al. (2004). Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm 274:65–73
- Kasim NA, Whitehouse M, Ramachandran C, et al. (2004). Molecular properties of WHO essential drugs and provisional. Mol Pharm 1:85–96
- Kaur A, Jain S, Tiwary AK. (2008). Mannan-coated gelatine nanoparticles for sustained and targeted delivery of didanosine for site specific delivery. Acta Pharma 58:61–74
- Kinman L, Brodie SJ, Tsai CC, et al. (2003). Lipid drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissue and viral load reduction: a proof of concept study in HIV-22870-infected macaques. J Acquir Imm Def Synd 34:387–97
- Kuo YC, Su SL. (2007). Transport of stavudine on polybutylcyanoacrylate and methyacrylate-sulfopropylmethacrylate and solid lipid nanoparticle. Int J Pharm 340:143–52
- Lamson MJ, Sabo JP, MacGregor TR, et al. (1995). Single dose pharmacokinetics and bioavailability of nevirapine in healthy volunteers. Biopharm Drug Dispos 20:285–91
- Lee YH, Ku YS. (1999). Effects of cyclosporine on the pharmacokinetics of proparanolol after intravenous and oral administration to control rats and to rats with uranyl nitrate-induced acute renal failure. J Pharm Pharmacol 51:1149–54
- Mainardes RM, Gremiao GP, Brunetti IL, et al. (2009). Zidovudine loaded PLA and PLA-PEG blend nanoparticle: influence of polymer type on phagocytic uptake by polymorphonuclear cells. J Pharm Sci 98:257–67
- Makabi-Panzu B, Gourde P, Desormeaux A, Bergeron MG. (1998). Intracellular and serum stability of liposomal 2, 3'-deoxycytidine, effect of lipid composition. Cell Mol Bio 44:277–84
- Mandloi DK, Rai VK, Dey S, et al. (2009). Method development and validation of RP-HPLC method in the detection of nevirapine in bulk drug and tablet formulation and application in in vitro dissolution study. J Global Pharm Tech 1:54–61
- Merck Index. (2006). An encyclopedia of chemicals, drugs and biological. In: Budavari S, O'Neal MJ, Smith A, et al. eds. 14th ed. USA: Merck & Co. Inc., Monograph Number 6514. pp. 1123
- Ministry of Environment & Forests (Animal Welfare Division), Government of India. (2007). Guidelines on the regulation of scientific experiments on animals: 9–141
- Minizi OMS, Ngaimisi E. (2010). Bioanalytical method for determination of nevirapine in-vivo in resource constrained laboratories. J Chem Pharm Res 2:431–9
- Mu H, Hoy CE. (2001). Intestinal absorption of specific structured triacylglycerols. J Lipid Res 42:792–8
- Paget GE, Barnes JM. (1964). Evaluation of drug activities. In: Pharmacometrics, Vol. I., Laurence DR, Bacharach AL, eds. London and New York: Academic Press, 50
- Patil P, Joshi P, Paradkar A. (2004). Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS Pharm Sci Tech 5:1–8
- Perlman ME, Murdande SB, Gumkowski MJ, et al. (2008). Development of a self-emulsifying formulation that reduces the food effect for torcetrapib. Int J Pharm Sci 351:15–22
- Porter CJ, Charman WN. (1997). Uptake of drugs into the intestinal lymphatics after oral administration. Adv Drug Deliv Rev 25:71–89
- Pouton CW. (1985). Self-emulsifying drug delivery systems: assessment of the efficiency of emulsification. Int J Pharm 27:335–48
- Pouton CW. (1997). Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev 25:47–58
- Pouton CW. (2000). Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and self-microemulsifying drug delivery systems. Eur J Pharm Sci 11:93–8
- Pouton CW. (2006). Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Phar Sci 29:278–87
- Ramana LN, Sethuraman S, Ranga U, Krishnan UM. (2010). Development of a liposomal nanodelivery system for nevirapine. J Biomed Sci 17:57. DOI: 10.1186/1423-0127-17-57
- Rao RN, Shinde DD. (2009). Two-dimensional LC-MS/MS determination of antiretroviral drugs in rat serum and urine. J Pharm Biomed Anal 50:994–9
- Sanganwar GP, Sathigari S, Babu RJ, Gupta RB. (2010). Simulataneous production and co-mixing of microparticles of nevirapine with excipients by supercritical antisolvent method for dissolution enhancement. Eur J Pharm Sci 39:164–74
- Shah NH, Carvajal MT, Patel CI, et al. (1994). Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm 106:15–23
- Shah RB, Khan MA. (2004). Regional permeability of Salmon calcitonin in isolated rat gastrointestinal tracts: transport mechanism using Caco-2 cell monolayer. APPS J 34:1–5
- Shegokar R, Jansch M, Singh KK, Muller RH. (2011). In vitro protein adsorption studies on nevirapine nanosuspensions for HIV/AIDS chemotherapy. Nanomed 7:333–40
- Shegokar R, Singh KK. (2011). Nevirapine nanosuspensions for HIV reservoir targeting. Pharmaz 66:408–15
- Sun M, Zhai X, Xue K, et al. (2011). Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci 43:132–40
- United States Pharmacopoeia, 36/NF31. (2014). US Pharmacopoeial Convention, Rockville, MD, 1:807
- Wakerly MG, Pouton CW, Meakin BJ. (1987). Evaluation of the self emulsifying performance of a non-ionic surfactant-vegetable oil mixture. J Pharm Pharmacol 39:6
- Wanke CA, Pleskow D, Degirolami PC, et al. (1996). A medium chain triglyceride-based diet in patients with HIV and chronic diarrhoea reduces diarrhoea and malabsorption: a prospective, controlled trial. Nutri 12:766–71
- Waters L, John L, Nelson M. (2007). Non-nucleoside reverse transcriptase inhibitors: a review. Int J Clin Pract 61:105–18
- Yang J-T, Kuo Y-C, Chung J-F. (2011). Human serum albumin-grafted cationic solid lipid nanoparticles for delivery of nevirapine in vivo. Int Conf Biosci Biochem Bioinfo 11:66–70