
Abstract
This study is aimed to investigate the applicability of poloxamer 407 (P407) and 188 (P188)-based temperature-sensitive in situ hydrogel (TSHG) in sustained delivery of hydrophilic macromolecules following intramuscular administration. Polyethylene glycols (PEGs) with molecular weight of 5-, 20-, and 40-kDa were used as model drugs, which can represent the common size range of hydrophilic macromolecular drugs using TSHG. The correlation between the level of poloxamers and thermogelling transition temperatures (Tsol–gel) was established and two formulations “20% P407/10% P188” and “24% P407/10% P188” were chosen for further study. The results showed that the release kinetics of PEGs was close to zero order. Sustained in vivo behaviors were achieved by both of the two formulations for all the PEGs though variations were seen. Lower molecular weight PEG showed more remarkable pharmacokinetic improvements. No significant differences in pharmacokinetics were observed between the two formulations for the same PEG. This suggested that 20–24% P407/10% P188 formulations, with accordingly Tsol–gel in the range of 24.6 °C–31.7 °C, might be freely chosen to achieve comparable pharmacokinetics for hydrophilic macromolecular drugs after intramuscular injection.
Introduction
The rapid development of biotechnology, genetic engineering, and proteomic techniques provide a powerful weapon for human to systematically find a variety of macromolecular drug targets and macromolecular drugs. However, most of macromolecular drugs have short biological half-life, poor oral bioavailability, low delivery efficiency, and poor stability at room temperature. These properties not only confine their administration route to injection, but also necessitate frequent administration, which brings a lot of inconvenience and suffering to patients.
Currently, there are two main strategies to achieve the long acting administration of macromolecular drugs (Wright & Burgess, Citation2011): (i) loading the drug in a sustained- or controlled-release delivery vehicle, such as oily suspensions, microspheres, implants, and in situ gels, which would delay the drug release after administration; (ii) structural modification of the drug, such as hydrophobic modification and polymeric modification, which would achieve long-lasting efficacy by reducing drug in vivo elimination rate. Both of the two strategies have their own advantages and disadvantages. For example, compared to the latter, the former does not change the drug structure and the drug is administered in the form of prototype; however, drug can have rather longer plasma residence time as well as better stability and release behavior in the latter condition. In recent years, the temperature-sensitive in situ hydrogel (TSHG) is attracting more and more attention as a delivery system for proteins and other hydrophilic macromolecular drugs to prolong and optimize their therapeutic effects (Liu et al., Citation2007; Chung & Liu, Citation2010; Akash et al., Citation2012; Rouini et al., Citation2013). TSHG is made of water-soluble polymer(s) and water. It has broad applications in the field of biomedicine and pharmacy (Klouda & Mikos, Citation2008; Gong et al., Citation2013). It exists in the form of a sol prior to administration, and rapidly forms non-chemically cross linked gel after injection due to temperature-induced phase transition. It can wrap both hydrophilic and hydrophobic drugs. Hence it is an effective formulation way to enhance the efficacy of a single dose with a stabilizing capacity on macromolecular drugs. Moreover, the preparation method of drug-loaded TSHGs is simple, involving simple mixing only and thus avoiding extreme conditions such as heating, organic solvents, and chemical crosslinking agents.
Poloxamer 407 (P407), also known as Pluronic® F127, is a commonly used polymer to form TSHG for prolonged drug delivery due to its suitable reverse thermo-sensitive property. P407 is a commercially available member of the family of poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) (PEO-PPO-PEO) triblock copolymers, whose average molecule weight and oxyethylene weight percentage are 9840–14 600 Da and 71.5–74.9, respectively. P407 has been used as the controlled release vehicle for many macromolecular drugs with molecular weight ranging from several thousands to tens of thousands, such as interleukin-2 (Johnston et al., Citation1992), urease (Pec et al., Citation1992), recombinant hirudin (Liu et al., Citation2007), insulin (Barichello et al., Citation1999; Chung & Liu, Citation2010), recombinant human somatropin (Wei et al., Citation2006), and interleukin-1 receptor antagonist (Akash et al., Citation2012). However, the effect of molecular weight on release was missing and intramuscular injection was rarely studied.
The purpose of the current study is to illuminate and improve the applicability of poloxamer-based TSHG in controlled delivery of hydrophilic macromolecular drugs with different molecular weights following intramuscular administration. Ratio and gelation temperature were studied to screen P407 and poloxamer 188 (P188)-based TSHGs and then polyethylene glycols (PEGs) with molecular weights of 5-, 20-, and 40-kDa were used as model drugs to evaluate two representative formulations by in vitro and in vivo studies. The reasons for choosing PEGs as model macromolecular drugs include (i) both the release behavior of a hydrophilic drug from TSHGs and the following absorption mainly depend on its hydrodynamic volume rather than its chemical composition; (ii) PEGs with different and homogeneous molecular weight are easily available, which not only makes possible the simulation of a large range of macromolecular drugs with different hydrodynamic volumes but also is favorable to explain the effect of molecular weight; (iii) PEG is one of few hydrophilic polymers that were approved by the U.S. Food and Drug Administration (FDA) to be used in injections; and (iv) the release and absorption behaviors of PEG would accurately reflect those of its mono-PEGylated drug whose hydrodynamic volume attributes mostly to the PEG part.
Materials and methods
Materials and animals
Poloxamer 407 (Pluronic® F127) (P407) and Poloxamer 188 (Pluronic® F68) (P188) were purchased from BASF (China) company Co., Ltd. Linear amino-terminated poly (ethylene glycol) methyl ethers (mPEG-NH2) were purchased from Jenkem technology Co., Ltd. (Beijing, China). Fluorescein isothiocyanate (FITC) was purchased from Sigma (St. Louis, MO). Extra dry dimethyl sulfoxide (DMSO) was purchased from Acros Organics (Geel, Belgium). All other chemicals were of reagent grade and purchased from commercial sources. Male Sprague–Dawley rats, weighing 250 ± 25 g, were supplied by Lab Animal Center of Shanghai University of Traditional Chinese Medicine. The rats were kept in an environmentally controlled breeding room for 6–7 d before starting the experiments and fed with standard laboratory food and water ad libitum. All procedures of the animal experiments were approved by the Animal Ethical Experimentation Committee of Shanghai University of TCM according to the requirements of the National Act on the Use of Experimental Animals (PR China).
Preparation of FITC-labeled mPEG-NH2 (FITC-PEG)
Labeling of mPEG-NH2 using FITC was carried out according to a reported method with minor modification (Sun et al., Citation2012). Briefly, mPEG-NH2 (1 g) was dissolved in DMSO (10 ml) containing 0.5 ml of pyridine. FITC (0.1 g) was added, followed by 0.2 ml of a solution of dibutyltin dilaurate in DMSO (1/4, v/v). The mixture was well vortexed and then heated for 30 min at 95 °C. After reaction, the product was precipitated by nine volumes of a cold diethyl ether/ethanol (3:1, v/v) mixture and then filtered. The final product was washed with the aforementioned precipitation reagent at least nine times till free FITC and the other excess reagents were fully removed, and finally dried in vacuo. The amounts of FITC conjugated to mPEG-NH2s were determined by Hitachi F-4500 fluorescence spectrophotometer (Tokyo, Japan) and were calculated in terms of their fluorescence intensity.
Preparation of poloxamer-based gels
The “Cold method” was adopted for preparing poloxamer-based gels as described in a previous report (Matthew et al., Citation2002). The required amounts of P407 and P188 for each formulation were carefully weighed and placed in a flat-bottomed vial. After addition of the required amount of 0.9% NaCl solution, the vial was placed at 4 °C until P407 and P188 were dissolved completely and a clear solution was obtained. In the study, P407 and P188 concentrations in sols or gels were expressed as the weight percentage (w/w; wt.%). The PEG-contained formulations for in vitro and in vivo studies were prepared by mixing FITC-PEG (8 mg) with 4 ml of the cold poloxamer sol by vortex. Physiological saline solution of FITC-PEG was used as reference solution.
Measurement of the phase transition temperature
A glass vial containing a magnetic bar and 5 ml of blank poloxamer sol was placed in a cold water bath. A thermometer with accuracy of 0.1 °C was immersed in the glass vial. The sol was heated at a constant rate with constant stirring at a rate of 50 rpm (Temperature magnetic stirrer, Model 85-2B, Jiangsu Jintan medical Instrument Factory, China). The temperature, at which the magnetic bar completely stopped moving, was regarded as gelling temperature (Choi et al., Citation1998). The result was the mean of triplicates.
Measurement of gelling time and viscosity
To determine the viscosities of sols or gels, 1 ml of each sol was added to a cone/plate viscometer (Physica MCR-101, Anton Paar, Austria) that contained a cone with an angle of 1°, inverted on a stainless steel plate. The viscosities of each sol at 10 °C and gel at 37 °C were continuously measured at a constant shear rate (75 s−1) and data were automatically logged from 60 s to 400 s. The effect of temperature on the viscosity of each formulation was determined by making the aforementioned measurement while varying the temperature from 5 °C to approximately 37 °C. To calculate the gelling time, 5 ml of each sol (4 °C) and a magnet bar were transferred into a transparent vial, which was then immediately placed in water bath (37 °C). The stirring rate of the bar was set at 50 rpm. The time (seconds) at which the sol lost its fluidity and the magnet bar did not move further due to gelation was recorded and considered as the gelling time. The result was the mean of triplicates.
In vitro studies
The membrane-less model was used as described elsewhere (Wei et al., Citation2006). The vial containing approximately 0.5 ml FITC-PEG-loaded or blank sol was placed in a water bath (37 °C) until a clear gel formed. 0.3 ml of the release medium (PBS, pH 7.4) pre-equilibrated at 37 °C was carefully layered over the surface of the gel. Then the vial was placed in a water bath (37 °C). At predetermined time points, the release medium was totally replaced by fresh medium (0.3 ml) and the weight of the vial plus the rest blank gel was recorded to calculate the weight of gel dissolved. The concentration of FITC-PEG in the release medium was determined by Hitachi F-4500 fluorescence spectrophotometer (Tokyo, Japan). Samples were diluted with PBS 100 times before assay.
In vivo studies
Rats (250 ± 25 g, male) were divided into nine groups (three animals each) to be dosed different formulations as follows: (1) 5-kDa FITC-PEG physiological saline solution; (2) 5-kDa FITC-PEG-loaded 20% P407/10% P188 sol; (3) 5-kDa FITC-PEG-loaded 24% P407/10% P188 sol; (4) 20-kDa FITC-PEG physiological saline solution; (5) 20-kDa FITC-PEG-loaded 20% P407/10% P188 sol; (6) 20-kDa FITC-PEG-loaded 24% P407/10% P188 sol; (7) 40-kDa FITC-PEG physiological saline solution; (8) 40-kDa FITC-PEG-loaded 20% P407/10% P188 sol (9) 40-kDa FITC-PEG-loaded 24% P407/10% P188 sol. 0.5 ml of each formulation was intramuscularly injected to the thigh. At predetermined time points after administration, blood samples were taken from the orbit venous plexus and anti-coagulated with 1.0% heparin (10 μl). To a 100 μl portion of each plasma sample, 40 μl of 1 M perchloric acid was added to precipitate plasma proteins. After centrifugation at 10 000 r/min for 2 min, the supernatant of the sample was transferred to another clean tube into which 1 M NaOH (30 μl) were added. The mixture was diluted with PBS (pH 7.4) 40 times before determined spectrofluorimetrically at λex 495 nm and λem 515 nm.
Statistical analysis
The one-way analysis of variance (ANOVA) was used for comparison among groups. Differences between the mean values were evaluated by t-test. A p < 0.05 was considered significant.
Results and discussion
Screening of gel formulations
In most reported studies on poloxamer-based in situ gels, P407 was used alone as the gelling agent, which leads to two main shortcomings. First, the concentration range of P407 suitable for use is very narrow, approximately 16–20% (w/w). When the concentration is less than 15% (w/w), the preparation cannot form a gel regardless of the temperature. If above 20% (w/w), the critical temperature will be lower than 20 °C, causing inconvenience in use and storage. Second, the control of drug release from the gel formed is sometimes inadequate due to the lower consistency of the gel and its poor anti-dilutability. The latter means that the strength of the gel decreases sharply with body fluid penetration. The thermogelation of P407 aqueous solutions results from the dehydration of the hydrophobic PPO block of the copolymer (Rassing et al., Citation1984); therefore, the phase transition temperature of the formulation could be modulated by simply adding another poloxamer that has a different PEO to PPO block ratio. P188, with a higher PEO to PPO block ratio than P407 (weight percent oxyethylene: 79.9–83.7 versus 71.5–74.9), was used in combination with P407 in the study to (i) up-regulate the phase transition temperature of the formulation and, thus, the P407 upper limit of use, and (ii) improve both the mechanical strength and the in situ dilutability of the gel formed by increasing the total amount of poloxamers used in the preparation. Thermogelling transition temperatures (Tsol–gel) of different P407 and P188 combinations were determined and listed in . In order to examine the impact of poloxamer leves on Tsol–gel, the following equation was drawn from multiple regression analysis, and the p values for CP407, CP188, and were all 0.000.
(1)
Table 1. Thermogelling transition temperatures (Tsol–gel) of different poloxamer 407 (P407) and 188 (P188) combinations (means ± SD, n = 3).
It was clearly showed in that the influence of P407 level on Tsol–gel was more significant than P188 level and when P407 level was constant, the up-regulation effect of P188 on Tsol–gel reached a peak value of ∼10 °C at about 10%. Based on the above result and the characterizing results ( and ), two formulations “20% P407/10% P188” and “24% P407/10% P188” were chosen for further study as they represent a reasonable and applicable range for P407 and P188 combined formulations.
Figure 1. Effect of compositions on the phase transition temperature of poloxamer 407 (P407)- and 188 (P188)-based thermosetting gels.
Figure 2. The changing of viscosity of four poloxamer-based formulations (○ for 24% P407 alone, □ for 20% P407 alone, ⋆ for 24% P407/10% P188, ▵ for 20% P407/10% P188) with temperature increased by 2 °C per min.
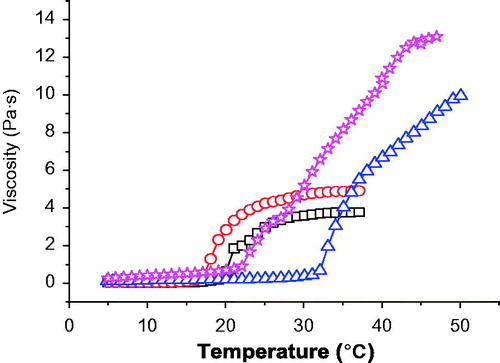
Table 2. Rheological parameters of poloxamer-based gel formulations (means ± SD, n = 3).
In vitro release
It was clearly showed in that the release kinetics of PEGs with different molecular weights from both of two gel formulations was close to zero order, which is consistent with the release behavior of many drugs from gels contained P407 alone, such as paclitaxel (Nie et al., Citation2011), chlorhexidine (Peppas, Citation1985), recombinant hirudin (Liu et al., Citation2007), ceftiofur (Zhang et al., Citation2002), insulin (Barichello et al., Citation1999), and IL-2 (Johnston et al., Citation1992). In addition, the release of PEGs with different molecular weights from the same gel formulation showed no significant differences (p > 0.05) in terms of the slopes of release profiles of three PEGs, which are close to 0.13 and 0.09 for 20% P408/10% P188 gel and 24% P407/10% P188 gel, respectively (). This indicates that hydrophilic macromolecules with different molecular weights may have the same release mechanism from P407/P188 gels. The release rate of the same PEG from the two gel formulations showed a significant difference (p < 0.05). Increasing the concentration of P407 from 20% to 24% led to a retarded PEG release from the gel. Poloxamer-based gels consist of a large amount of micelles and aqueous channels. Increasing the concentration of P407 could not only cause a greater tortuosity in the aqueous phase of the gel structure but also result in a shorter inter-micellar distance and thus a greater number of cross-links between neighboring micelles as well as a greater number of micelles per unit volume (Bhardwaj & Blanchard, Citation1996). All of these contribute to the drug release rate reduction. For hydrophilic drugs, drug diffusion and gel erosion occur simultaneously after intramuscular or subcutaneous administration of the in situ gel formulation with the markedly faster one being the main mechanism of drug release. shows a good linear correlation with the slope being almost 1.0 between the percentage of PEG released and the percentage of gel eroded, indicating a gel erosion-controlled release mechanism.
Figure 3. The in vitro release curves of 5-kDa PEG (left), 20-kDa PEG (middle), and 40-kDa PEG (right) from 24% P407/10% P188 gel (red open squares) and 20% P407/10% P188 gel (black open circles) measured by the membrane-less model at 37 °C. Data are expressed as means ± SD (n = 3)..
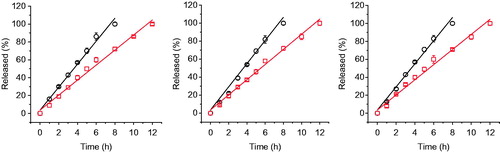
Figure 4. Correlation between cumulative % gel degradation and cumulative % 5-kDa PEG released from 20% P407/10% P188 gel (○) and 24% P407/10% P188 gel (□).
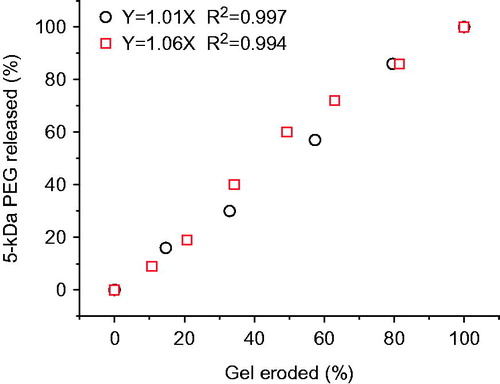
Table 3. The linear regression of release curves of PEGs with different molecular weights from two poloxamer-based gels.
In vivo studies
In general, sustained in vivo behaviors were achieved by gels for all the PEGs studied though variations were seen. Compared to solution formulations, their plasma level versus time profiles show a broader, lower, and delayed plasmatic peak after intramuscular administration of the gel formulations (), and their main pharmacokinetic parameters were significantly improved (). After intramuscular or subcutaneous administration, the drug entrapped in gels needs finish two steps in tandem to be absorbed, i.e. (i) release from the in situ formed gel and (ii) diffusion through the interstitial space and the blood and lymphatic vessel endothelium, with the markedly slower step as the rate limiting one. Both of the steps are drug hydrodynamic volume-dependant and the first one is also formulation-dependant. The in vitro study showed that, regardless of their molecular weight, PEGs released from the gels studied mainly by erosion of rather than diffusion through the gels. Therefore, with increasing the molecular weight of PEGs from 5 kDa to 40 kDa, the contribution of the second step to the whole absorption process increased while that of the first step reduced. This resulted in that more remarkable improvement in pharmacokinetics was achieved by gels for lower molecular weight PEG (). For example, compared to aqueous solution, the AUC(0–∞) values for 5-kDa PEG released from the two gel formulations studied increased 3.25- and 5.15-times, respectively; however, only comparative or slightly increased values were achieved for 20- and 40-kDa PEGs. Similar results were observed for the t1/2z value, which increased 6.16- and 12.3-times for 5-kDa PEG released from the two gel formulations but only increased slightly (1.04–1.59 times) for 20- and 40-kDa PEGs. On the other hand, the Cmax value decreased more significantly for 5-kDa PEG (∼55% decreased) than for 20- and 40-kDa PEGs (∼25% decreased) by gel formulations. As to Tmax, comparable results, i.e. two to four times increase, were observed for all of the three PEGs and both of the two gel formulations. Disagreed with the in vitro release results, which showed that the release rates of all of the three PEGs from 24% P407/10% P188 gel were significantly slower than from 20% P407/10% P188 gel (p < 0.05), no significant differences in plasma level-time profiles and most pharmacokinetic parameters were observed between the two gel formulations for the same PEG. This is also inconsistent with a previous report (Liu et al., Citation2007) that the recombinant hirudin pharmacokinetics showed significant differences between 20% P407 and 25% P407 formulations after subcutaneous injection in rats. The reasons for the above disagreement might include (i) the addition of 10% P188 reduced the influence of P407 level difference on the in vivo behavior of the gel formulation; (ii) intramuscular conditions are more vigorous than both the in vitro release test ones used in the study and subcutaneous ones, which would reduce the actual in vivo release difference between the two gel formulations studied; and (iii) as stated above, with increasing the molecular weight of drug, the contribution of drug release from the gel to the whole drug absorption process reduced. As a whole, the results observed in the study suggest that 20–24% P407/10% P188 gels, with accordingly Tsol–gel in the range of 31.7 °C–24.6 °C, might be freely chosen to achieve comparable pharmacokinetics for hydrophilic macromolecular drugs with hydrodynamic volumes between those of 5- and 40-kDa PEGs after intramuscular injection.
Figure 5. Plasma PEG (A: 5-kDa PEG; B: 20-kDa PEG; C: 40-kDa PEG) concentration–time profiles of PEG-contained aqueous solution (○), 20% P407/10% P188 gel (□), and 24% P407/10% P188 gel (⋄) following intramuscular administration in rats. The dose for a PEG is same. Data are represented as mean ± SD (n = 3).
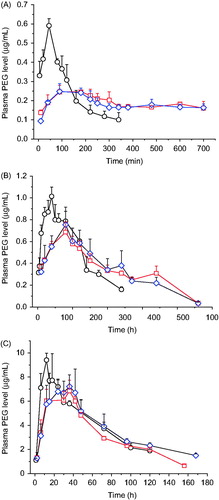
Table 4. Pharmacokinetic parameters of PEGs in aqueous solution, 20% P407/10% P188 gel (Gel 1), and 24% P407/10% P188 gel (Gel 2).
Table 5. Ratios of pharmacokinetic parameters of PEGs in 20% P407/10% P188 gel (Gel 1) and 24% P407/10% P188 gel (Gel 2) to those in aqueous solution (AS).
Some investigations, which used poloxamer-based gels to control the release of proteinic drugs, observed results similar to our findings. For example, consistent with the results for 5-kDa PEG in this study, the Tmax of recombinant hirudin (7 kDa) in rats were 1.1 and 5.2 h after subcutaneous injection of its solution and 25% P407-based gel, respectively, and the drug Cmax also reduced nearly half by the gel (Liu et al., Citation2007). In another report, the blood glucose levels of rats injected subcutaneously with insulin (5 kDa) solution and its 18% P407/15% P188 gel reached valley at about 1 and 2 h postdose, respectively, which indicates that the drug Tmax was doubled by the gel (Chung & Liu, Citation2010). The investigation on recombinant human somatropin (22 kDa) showed that compared to solution, the drug Tmax was delayed four times and AUC0–48 h increased 1.2 times by the 15% P407/15% P188 gel (Wei et al., Citation2006) after subcutaneous administration, which was consistent with those for 20-kDa PEG in the study. When using the results obtained in the study to predict the in vivo behavior of actual hydrophilic macromolecular drugs, it is necessary to take into consideration that macromolecules with the same molecular weight generally have different hydrodynamic volumes. As a rough guide, protein, polyvinylpyrrolidone, dextran, and PEG with molecular weights of 60–70, 25, 50, and 30 kDa, respectively, have a comparable hydrodynamic size of about 10 nm (Seymour, Citation1992; Davis et al., Citation2008). Sometimes, for better prediction, the absorption route, drug stability, and the macromolecular steric configuration, flexibility and deformability should also be considered. In general, the main absorption route for subcutaneously or intramuscularly administered drugs would change gradually from direct absorption into blood to indirect absorption via lymphatic system with increasing the molecular weight of drugs. For example, studies have shown that greater than 50% of a subcutaneous dose may be taken up by the peripheral lymphatics for macromolecules whose molecular mass exceeds approximately 20 kDa (Supersaxo et al., Citation1988, Citation1990; Charman et al., Citation2000). However, for some high-molecular-weight drugs, significant direct absorption can also happen attributed to their “snake-like” movement through hydrophilic pores in blood vessel endothelium.
Except for the results obtained in this study, there are some reports that also suggest that poloxamer-based in situ hydrogels could prolong drug release compared to solutions following injection, but the delivery period rarely exceeds a few days (Katakam et al., Citation1997; Veyries et al., Citation1999; Paavola et al., Citation2000; Wenzel et al., Citation2002; Zhang et al., Citation2002). This characteristic makes such systems interesting for short-term therapies like pain management (Paavola et al., Citation2000), infection treatment (Veyries et al., Citation1999; Zhang et al., Citation2002), and fertility control (Wenzel et al., Citation2002). Besides injection, other administration routes have also been evaluated, such as rectal, vaginal, transdermal, and ophthalmic (Ruel-Gariépy & Leroux, Citation2004). Similarly, poloxamer formulations, although with weak mechanical strength, high permeability, and rapid erosion (i.e. dissolution from the surface), generally increased drug residence time at application sites, resulting in improved bioavailability and efficacy. The results obtained in the study, when considered together with the physiological differences between different administration routes, might also provide some helpful information for these administration routes.
Poloxamers are generally regarded as nontoxic and nonirritant materials. They are used as emulsifying agents in intravenous fat emulsions. P188 has also been used as an emulsifying agent for fluorocarbons used as artificial blood substitutes. However, hyperlipidemia has been reported in rats, mice, and rabbits after injection of high doses of P407 (Wout et al., Citation1992; Palmer et al., Citation1998; Blonder et al., Citation1999). It was reported that the toxicity of poloxamer vehicles was proportional to their lipophilicity. The more lipophilic the poloxamer, the more severe the lesion produced following intramuscular injection and the greater the elevation in plasma CPK (Johnston & Miller, Citation1985). Therefore, the administered amount of P407 should be kept to a minimum, especially when repeated dosing is required. To further delay drug release from poloxamer-based in situ hydrogels, several strategies have been tried with some success, including (i) addition of thickening agents, such as methylcellulose and hydroxypropylcellulose, in gels; (ii) buffering the gels when drug solubility is pH-dependent; (iii) incorporation of drug-loaded nano-sized or microsized delivery systems, such as liposomes, microspheres, and polylactic-co-glycolic acid nanoparticles, rather than free drug in gels; and (iv) chemical modification of poloxamer to optimize gelling characteristics with lower polymer concentrations.
Conclusions
The addition of P188 to P407-based in situ gel formulations could not only up-regulate the phase transition temperature of the formulations and, thus, the P407 upper limit of use, but also significantly improve the mechanical strength of the formed gels. The release kinetics of 5-, 20-, and 40-kDa PEGs from two representative formulations, “20% P407/10% P188” and “24% P407/10% P188”, indicates that the main release mechanism was gel erosion-controlled. In general, sustained in vivo behaviors were achieved by both of the two formulations for all the three PEGs; however, more remarkable improvements in pharmacokinetics were achieved by the gels for the lower molecular weight PEG. No significant differences in pharmacokinetics between the two formulations for the same PEG suggest that 20–24% P407/10% P188 formulations, with accordingly Tsol–gel in the range of 24.6 °C–31.7 °C, might be freely chosen to achieve comparable pharmacokinetics for hydrophilic macromolecular drugs after intramuscular injection.
Declaration of interest
This work was supported by Program for New Century Excellent Talents in University (NCET-13-0906), the National Natural Science Foundation of China (81073065), the Key Discipline Project of Shanghai Education Committee (J50302), and the “085” Project (085ZY1219) and Xinglin Scholar Program of Shanghai University of TCM. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Akash MSH, Rehman K, Li N, et al. (2012). Sustained delivery of IL-1Ra from Pluronic F127-based thermosensitive gel prolongs its therapeutic potentials. Pharm Res 29:3475–85
- Barichello JM, Morishita M, Takayama K, Nagai T. (1999). Absorption of insulin from pluronic F-127 gels following subcutaneous administration in rats. Int J Pharm 184:189–98
- Bhardwaj R, Blanchard J. (1996). Controlled-release delivery system for the a-MSH analog melanotan-I using Poloxamer 407. J Pharm Sci 85:915–9
- Blonder JM, Baird L, Fulfs JC, Rosenthal GJ. (1999). Dose-dependent hyperlipidemia in rabbits following administration of poloxamer 407 gel. Life Sci 65:261–6
- Charman SA, Segrave AM, Edwards GA, Porter CJH. (2000). Systemic availability and lymphatic transport of human growth hormone administered by subcutaneous injection. J Pharm Sci 89:168–77
- Choi HG, Jung JH, Ryu JM, et al. (1998). Development of in situ-gelling and mucoadhesive acetaminophen liquid suppository. Int J Pharm 165:33–44
- Chung TW, Liu DZ. (2010). Effects of interpenetration of thermo-sensitive gels by crosslinking of chitosan on nasal delivery of insulin: in vitro characterization and in vivo study. Carbohydr Polym 82:316–22
- Davis ME, Chen ZG, Shin DM. (2008). Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov 7:771–82
- Gong C, Qi T, Wei X, et al. (2013). Thermosensitive polymeric hydrogels as drug delivery systems. Curr Med Chem 20:79–94
- Johnston TP, Miller IR. (1985). Toxicological evaluation of poloxamer vehicles for intramuscular use. J Parenter Sci Technol 39:83–9
- Johnston TP, Punjabi MA, Froelich CJ. (1992). Sustained delivery of interleukin-2 from a poloxamer 407 gel matrix following intraperitoneal injection in mice. Pharm Res 9:425–34
- Katakam M, Ravis WR, Banga AK. (1997). Controlled release of human growth hormone in rats following parenteral administration of poloxamer gels. J Contr Rel 49:21–6
- Klouda L, Mikos AG. (2008). Thermoresponsive hydrogels in biomedical applications. Eur J Pharm Biopharm 68:34–45
- Liu Y, Lu WL, Wang JC, et al. (2007). Controlled delivery of recombinant hirudin based on thermosensitive pluronic® F127 hydrogel for subcutaneous administration: in vitro and in vivo characterization. J Contr Rel 117:387–95
- Matthew JE, Nazario YL, Roberts SC. (2002). Effect of mammalian cell culture medium on the gelation properties of Pluronic PF127. Biomaterials 23:4615–9
- Nie S, Hsiao WW, Pan W, Yang Z. (2011). Thermosensitive pluronic® F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: in vitro drug release, cell cytotoxicity, and uptake studies. Int J Nanomed 6:151–61
- Paavola A, Kilpelainen I, Yliruusi J, Rosenberg P. (2000). Controlled release injectable liposomal gel of ibuprofen for epidural analgesia. Int J Pharm 199:85–93
- Palmer WK, Emeson EE, Johnston TP. (1998). Poloxamer 407-induced atherogenesis in the C57BL/6 mouse. Atherosclerosis 136:115–23
- Pec EA, Wout ZG, Johnston TP. (1992). Biological activity of urease formulated in poloxamer 407 after intraperitoneal injection in the rat. J Pharm Sci 81:626–30
- Peppas NA. (1985). Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv 60:110–11
- Rassing J, Mckenna W, Bandyopadhyay S, Eyring E. (1984). Ultrasonic and 13C-NMR studies on gel formation in aqueous solution of ABA block copolymeric Pluronic F-127. J Mol Liq 27:165–78
- Rouini MR, Atyabi F, Foroumadi A, et al. (2013). In vitro and in vivo evaluation of an in situ gel forming system for the delivery of PEGylated octreotide. Eur J Pharm Sci 48:87–96
- Ruel-Gariépy E, Leroux JC. (2004). In situ-forming hydrogels—review of temperature-sensitive systems. Eur J Pharm Biopharm 58:409–26
- Seymour LW. (1992). Passive tumor targeting of soluble macromolecules and drug conjugates. Crit Rev Ther Drug Carrier Syst 9:135–87
- Sun GL, Lin X, Hong YL, et al. (2012). PEGylation for drug delivery to ischemic myocardium: pharmacokinetics and cardiac distribution of poly(ethylene glycol)s in mice with normal and ischemic myocardium. Eur J Pharm Sci 46:545–52
- Supersaxo A, Hein W, Gallati H, Steffen H. (1988). Recombinant human interferon alpha-2a: delivery to lymphoid tissue by selected modes of application. Pharm Res 5:472–6
- Supersaxo A, Hein WR, Steffen H. (1990). Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm Res 7:167–9
- Veyries ML, Couarraze G, Geiger S, et al. (1999). Controlled release of vancomycin from poloxamer gels. Int J Pharm 192:183–93
- Wei G, Lu LF, Zhong GR, et al. (2006). A probe study on thermosensitive in situ gel as carrier of injectable sustained-release protein delivery system. Chin J Pharm 37:597–601
- Wenzel JGW, Balaji KSS, Koushik K, et al. (2002). Pluronic F127 gel formulations of Deslorelin and GnRH reduce drug degradation and sustain drug release and effect in cattle. J Contr Rel 85:51–9
- Wout ZGM, Pec EA, Maggiore JA, et al. (1992). Poloxamer 407-mediated changes in plasma cholesterol and triglycerides following intraperitoneal injection to rats. J Parent Sci Technol 46:192–200
- Wright JM, Burgess DJ, eds. (2011). Acting injections and implants, advances in delivery science and technology. Berlin, Germany: Springer
- Zhang L, Parsons DL, Navarre C, Kompella UB. (2002). Development and in-vitro evaluation of sustained release poloxamer 407 (P407) gel formulations of ceftiofur. J Contr Rel 85:73–81