
Abstract
Context: The sustained release implants can be directly implanted in tumor site by surgery and are promising for cancer treatment.
Objective: RGD-modified PEGylated polyamidoamine (PAMAM) dendrimer with doxorubicin (DOX) conjugated by acid-sensitive linkage (RGD-PPCD) was a potential conjugate for tumor-targeted therapy. In order to enhance tumor retention ability and long-term effect of drug, we developed the DOX and its conjugate implants using poly(dl-lactic-co-glycolic acid) (PLGA), poly(dl-lactic acid) (PLA) and polyethylene glycol (PEG) as carrier materials.
Methods: The implants were prepared by a simple solvent evaporation method. Different formulations with varying ratios of three polymers were designed, prepared and evaluated on the basis of viscosity, in vitro release and drying time. Furthermore, in vivo biodistribution and antitumor activity of the implants were studied in mice with subcutaneous C6 xenografts.
Results: The optimized formulation was obtained with the 3:1 ratio of PLGA/PLA (w/w) and 1% PEG (wt.%). The drug release behavior of DOX, PPCD and RGD-PPCD implants prepared by the optimized formulation was similar according to the assessment of similarity factor f2, and the release curves were fell into three phases, including a lag-period, then the second phase which was consistent with zero-order model followed by a plateau. Data of total DOX remained in implants indicated the release were faster in vivo than in vitro. Moreover, intratumoral drug amount of RGD-PPCD implants was the highest 45 days after implantation. Correspondingly, the RGD-PPCD implants exhibited the strongest antitumor activity compared with PPCD and free DOX implants.
Discussion and conclusion: This paper presents an exploratory research on macromolecule-drug conjugates, including RGD-PPCD and PPCD, which have the potential to be developed into long-term effect implants for tumor therapy with high efficiency and low systematic toxicity.
Introduction
Cancer is one of the most deadly diseases and causes death of millions of people around the world every year (Siegel et al., Citation2012). Despite aggressive treatment using surgery, radiotherapy, photodynamic therapy and chemotherapy, cancer has a poor prognosis (Stupp et al., Citation2006). Tumor-targeted delivery is a great challenge for drug delivery systems. Traditional chemotherapy has limited or even no specificity for cancer cells, normally accompanied by low regional concentration in tumor, and thus resulting in inefficacy (Arias, Citation2011). Another challenge is that the therapeutic agents cannot only accumulate in a lesion without diffusing into normal tissues, which is the cause of harmful side effects (Li et al., Citation2012). Targeting drug delivery systems for cancer treatment was promised to substantially improve the therapeutic outcomes and its capacity to enhance tumor accumulation of drugs or even decrease side effects was confined (Wang et al., Citation2008; Bragagni et al., Citation2012; Jiang et al., Citation2012; Puvanakrishnan et al., Citation2012; Vannucci et al., Citation2012). Based on polyamidoamine (PAMAM) dendrimers, our group has worked successfully to develop the DOX-polymer conjugates, in which acid-sensitive cis-aconityl linkage was used to conjugate DOX to PEG–PAMAM to achieve PPCD conjugate and further modified by the cyclic pentapeptide RGDyC to obtain the RGD-PPCD conjugate (Zhu et al., Citation2010a,Citationb, Citation2011). We demonstrated that the DOX release from the acid-sensitive conjugates guaranteed a higher concentration of free DOX in tumor and the RGD modification significantly improved the agent therapeutic effect against murine B16 melanoma and C6 glioma. However, it was also found that the accumulation of the conjugates in spleen and liver following intravenous administration was extremely higher than that in intracranial glioma, especially RGD-PPCD.
In order to overcome these obstacles, controlling drug released from surgically implanted polymers seems to be a promising way in treating tumors with several possible advantages over systemic administration (Wang et al., Citation1999). Implanting a biodegradable devices loaded with antineoplastic agent into tumor site can not only promote local drug concentration but also minimize the systemic side effects of the chemotherapy. The carmustine implant (GLIADEL® wafer) is incorporated into a biodegradable copolymer matrix consisting of 1,3-bis(p-carboxyphenoxy)propane (CPP) and sebacic acid (SA) in a 20–80 molar ratio (p(CPP:SA, 20:80)). GLIADEL® is the first implant preparation approved by the USFDA in 1996 as the post-surgical local chemotherapy against recurrent malignant glioma (Anonymous, Citation1996). Since then, many investigators have documented that local chemotherapeutics polymeric implants injection can exhibit controlled release and obtain effective drug concentrations within tumor (Haaga et al., Citation2005; Weinberg et al., Citation2007; Shikanov et al., Citation2008; Sivak et al., Citation2008; Ranganath et al., Citation2010; Aqil et al., Citation2012). The chemotherapeutics (doxorubicin, paclitaxel, 5-fluorouracil, camptothecin, carboplatin, and Withaferin A) delivering implants were fabricated by biocompatible and biodegradable polymers, mainly including PCPP-SA, poly(dl-lactic-co-glycolic acid) (PLGA), poly(dl-lactic acid) (PLA), poly(ester anhydride), polyurethane and polycaprolactone. Although the implants have shown great potential and provided significant effect in inhibiting tumor growth, there are still some problems. First, because of the weak penetration of the drugs, such as carmustine, doxorubicin, 5-fluorouracil and methotrexate, its concentration was far from enough to eliminate all tumor cells (Weinberg et al., Citation2007; Ranganath et al., Citation2010). Second, upon release, side effects in normal cells is inevitable due to the poor specificity to cancer cells of chemotherapeutics. Apart from that, a possible challenge is that the small molecule behaves rapid elimination from the adjacent capillaries to the circulation system.
All those above entailed us toward developing and investigating a novel delivering implants loading the macromolecular conjugates. The design ideas of the study through the use of DOX conjugates instead of free DOX loaded in the implants was to enhance tumor retention ability based on the EPR effect (the lack of lymphatic return results in the retention effect of macromolecule in tumor tissue). Furthermore, we used the RGD-modified PPCD as the loaded drug in the implants to further increase the tumor retention ability and anti-tumor effect, because RGD has high affinity with angiogenic vessels and tumor cells, which is highly expressed cell adhesion molecule integrin αvβ3 (Cai et al., Citation2008). In our study, PLGA and PLA were chosen as main polymer carriers, the implants were fabricated, respectively, by uniformly distributing DOX, PPCD and RGD-PPCD in solid polymer disks. After their in vitro release characteristics were investigated, we then use mice model-bearing subcutaneous C6 xenografts, studied the in vivo biodistribution and antitumor efficacy of the intratumoral implanted wafers.
Materials and methods
Materials and animals
PAMAM dendrimer generation 4 with an ethylenediamine core in methanol (10%, w/v) was obtained from Dendritech, Inc. (Midland, MI). Methoxy PEG succinimidyl carboxymethyl ester to synthesize of PPCD (MeO–PEG–SCM, MW 5187) and N-hydroxysulfo-succinimide-polyoxyethylene-maleimide to synthesize RGD-PPCD (NHS–PEG–MAL, MW 5147) were purchased from JenKem Technology Co., Ltd. (Beijing, China). PEG4000 used as one of the carrier materials of implants was acquired from Pudong Gaonan Co., Ltd. (Shanghai, China). RGD (Cyclo (RGD–DTyr–C)) was produced by ChinaPeptide Co. (Shanghai, China). 2-Mercaptpethanol was purchased from Aladdin Reagent, Inc. (Shanghai, China). Cis-aconitic anhydride was obtained from Alfa Aesar (Lancashire, UK). Anhydrous DMF, p-dioxane, was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). 1-Ethyl-3-(3-dimeth-ylaminopropyl) carbodiimide hydrochloride (EDC) was purchased from Shanghai Medpep Co., Ltd. (Shanghai, China). Doxorubicin hydrochloride (DOX) was acquired from Zhejiang Hisun Pharmaceutical Co., Ltd. (Zhejiang, China). PLGA lactide/glycolide ratio is 75:25, Mw = 10 000–25 000) and PLA Mw = 10 000–25 000) were obtained from Sigma Chemical Co. (St. Louis, MO, USA).
The C6 cell line was obtained from Institute of Biochemistry and Cell Biology (IBCB), Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
Male ICR mice (6–8 weeks old), ranging from 18 to 22 g, were provided by Sino-British Sippr/BK Laboratory Animal Co., Ltd. (Shanghai, China). All care and handling of animals were performed in accordance with the guidelines of the animal ethics committee of Fudan University (Shanghai, China).
Preparation of DOX and DOX-polymer conjugate implants
The implants were prepared in a perfectly clean environment after high-temperature sterilization of all the instrument. The PEG–PAMAM–DOX conjugates (PPCD) and RGD–PEG–PAMAM–DOX conjugates (RGD-PPCD) were first synthesized according to our previous reports (Zhu et al., Citation2010a,Citationb, Citation2011), respectively. About 20 molecules of PEG and 13 molecules of DOX were attached to one molecule of PAMAM dendrimer and the number of RGD per RGD-PPCD conjugate was around 10. After that the preparation of implants was performed as follows. PLGA and PLA were dissolved in methylene chloride at 20% (w/v) concentration. Drug (DOX or DOX conjugates) and PEG4000 in 30% methanol-70% water solution (by volume) were added to methylene chloride solution. The mixture was stirred at room temperature to evaporate most of the methylene chloride. After 3–4 h, the implants were formed by self-made die, followed by dried at room temperature for 2–3 weeks until residual organic solvent removed. The formulations with different compositions were designed, including PLGA/PLA in the ratios of 1/0, 3/1, 1/1 and 1/3 (w/w), and the PEG percentage content of 0%, 1% and 5% (wt.%) in carriers. The DOX-loaded implants with different formulations were prepared according to the above process. Viscosity, in vitro release and drying time were assessed in order to obtain an optimal formulation.
The HPLC method was used to determine DOX content in implants. Briefly, the implants were dissolved in methylene chloride/methanol (1:1, v/v) by vortex mixing for 5 min. The mixture was stirred at room temperature to evaporate most of the methylene chloride and then add 40% methanol to 10 ml. After centrifugation (10 000 rpm) for 5 min, 0.2 ml of the supernatant was processed acid hydrolysis to release doxorubicinone from conjugates, then determination of total DOX was achieved by HPLC separation according to our previous reports (Zhu et al., 2010a; Zhang et al., Citation2011).
In vitro drug release from the implants
In vitro release study was conducted in a 100 rpm shaking water bath at 37 °C, after immersing implants into 2 mL PBS buffer (pH 5.5 and 7.4) in 10 ml centrifuge tubes. At each specified time point, all buffer in tubes was withdrawn from tubes after centrifugation (10 000 rpm, 5 min) and 2 ml of fresh PBS buffer was added back to approximate infinite sink conditions. The total and free DOX amounts in release media were detected, respectively, by HPLC according to our previous methods (Zhu et al., 2010a). For the DOX, PPCD and RGD-PPCD implants prepared by the optimal formulation, the release curves were drawn, and the similarity of curves was examined by calculating similarity factor (f2) according to the following equation:
where n is the number of time points (Rt − Tt) is the distances of the released percentage between the two curves at time t. Two release curves are considered similar when the f2 value is ≥50. Furthermore, release data werefitted to zero-order, first-order, Higuchi’s and Korsmeyer–Peppas equations (Wang et al., Citation2008; Mesnukul et al., Citation2010; Koocheki et al., Citation2011).
Zero-order equation (cumulative amount of drug released versus time),
where Qt is the fraction released at time t and K0 is the zero-order rate constant.
First-order equation (log cumulative percentage of drug remaining versus time),
where Qt is the amount released at time t, Q0 is the initial amount of drug in solution and K1 is the first-order rate constant.
Higuchi’s equation (cumulative percentage of drug released versus square root of time),
where Qt is the fraction dissolved at time t and KH is the Higuchi dissolution constant.
The Korsmeyer–Peppas equation (log cumulative percentage of drug released versus log time), or its modified form which also known as the Power Law, Qt/Q∞ = a · tn, or Q(t-l)/Q∞ = a · (t − l)n, where Qt is the amount of drug released at time t, Q∞ is the amount of drug released after infinite time, l is the lag time, a is the Korsmeyer–Peppas dissolution rate constant and n is the diffusional exponent.
Cell culture and mice tumor model
C6 cells were maintained in F12 supplemented with 2.5% Fetal bovine serum, 15% horse serum, 100 U/ml penicillin and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. Then, 2 × 106 C6 cells (in 100 μl cell culture medium) in logarithmic growth phase were selected and inoculated into the subcutaneous tissue of the armpit of right anterior limb. The tumor was allowed to grow for approximately 10 days when the tumor volume reached to 0.3 cm3 before initiation of the studies.
Implant insertion
Administration of the implants was carried out in a sterile environment when the tumor volume reached to 0.3 cm3. All mice received either drug-loaded or blank implants. Subjects were anesthetized with 10% chloral hydrate (400 mg/kg), skin on the tumor site was shaved prior to cleaning and sterilized with iodine tincture and alcohol. The operative process is showed in . An incision as large as the implant was made carefully in the central of tumor. Then, the implants were placed in the incision and the wound was closed by suture.
Figure 1. The operative process of implantation. (A) C6-bearing mice model, (B, C) administration of DOX implants, (D, E) administration of blank implants, (F) the wound of mice was closed by suture.
In vivo biodistribution
Male ICR mice-bearing C6 tumor were randomly divided into three groups: DOX implants, PPCD implants and RGD-PPCD implants with 15 animals each. The implants at a dose of 0.2 mg DOX-equiv. per implant were given to the mice through the incision of the tumor. Five animals per group were sacrificed individually by cervical dislocation at 7, 30 and 45 days post-implantation. The plasma was collected and major organs were excised, washed with cold saline, dried over filter paper, weighed and frozen at −20 °C until analysis. Determination of total DOX in plasma and major organs was performed after acid hydrolysis to release doxorubicinone from polymer-bound DOX followed by HPLC separation according to our previous reports (Zhu et al., 2010a; Zhang et al., Citation2011). Additionally, the reliquus implants were carefully separated from tumor tissues and the total content of DOX was detected according to the method described above.
To visualize the drug distribution, the mice were anesthetized intraperitoneal (i.p.) with 10% chloral hydrate (400 mg/kg) on the day 0, day 7, day 30 and day 45 after implantation, the hair around the location of tumor was removed by surgery scissors and hair removal creams. Then, fluorescent imaging was performed via a Maestro in vivo imaging system (CRI, MA). The blue excitation light (wavelength: 500–720 nm, exposure time: 300 ms) was used to determine the fluorescence of DOX.
In vivo antitumor activity
The mice-bearing subcutaneous C6 tumor were randomly divided into 6 groups with 10 animals each, including group 1: the blank implants, group 2: the negative control without treatment, group 3–6: DOX saline solution, DOX implants, PPCD implants and RGD-PPCD implants at a dose of 0.2 mg DOX-equiv. per implant, respectively. Mice were then checked for survival every day post-implantation, tumor size was measured with calipers in two perpendicular diameters every five days and expressed as tumor volume V = a × b2/2 (a-longer diameter and b-shorter diameter). The inhibitory rate of tumor (IRT %) was calculated according to the equation: IRT (%) = (1 − D/S) × 100, where D and S were tumor volume of treatment and negative control group).
Data analysis
Data are expressed as mean ± standard deviations. Analysis of variance was used for comparisons among experimental data groups. p Value of 0.05 or less was considered to be statistically significant.
Results
Preparation and formula optimization of implants
As described above, the implants were given to mice through the incision of the tumor when the tumor volume reached to 0.3 cm3. On this account, the implants were prepared as the same size as the incision of the tumor. Thus, the implants were formed into wafer shape with a weight of 40 ± 0.58 mg, diameter of 5.59 ± 0.02 mm, and thickness of 1.83 ± 0.02 mm (). Additionally, based on clinical data and the effects of chronic DOX administration in rats (Voulgaris et al., Citation2002; Lesniak et al., Citation2005), DOX doses of 0.2 mg equivalent per implant were chosen.
Figure 2. Implants shape of DOX and DOX conjugates.
In order to obtain an optimal formulation, the different implants with varying ratios of carrier materials were designed and prepared by loading free DOX. On the basis of certain preparation process, the composition and the proportion of polymer carriers were main influence factors on viscosity, drug release and drying time. First, we only used PLGA as the carrier materials, but the viscosity of the mixture was too high to take shape. Then, PLA was added to modify viscosity and the different ratios of PLGA/PLA were investigated. When the ratio of PLGA/PLA enhanced, the DOX released from the implants was increased (). Thus, we choose PLGA/PLA with the ratio of 3:1.
Figure 3. In vitro release profiles of DOX from implants with different formulation compositions in pH 7.4 PBS, (A) different ratios of PLGA to PLA, (B) different percentage contents of PEG in the prescription of implants (PLGA/PLA, 3/1), n = 3.
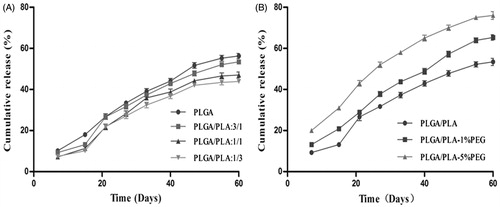
PEG also plays an important role in accelerating drug release from the carrier materials. While the PEG percentage was increased from 0% to 5%, the DOX release from implants was significantly accelerated (). The accumulative release within 60 days was more than 60% for the mixture with PEG (1% or 5%), which would ensure the lowest effective dose for the anti-tumor treatment. However, the mean drying time was extended from 14 days to 30 days with the increasing PEG percentage from 1% to 5%. The long drying time was unfavorable for preparation process of the implants. So PEG percentage of 1% was more suitable for the optimal formulation. Based on comprehensive consideration of sustained release, appropriate viscosity and shorter drying time, the optimal formulation was finally determined with the 3:1 ratio of PLGA/PLA (w/w) and 1% PEG4000 (wt.%).
DOX, PPCD and RGD-PPCD implants were prepared based on the optimum formulae and the determination of DOX content in implants is displayed in . The results indicated that the DOX content in each wafer was uniform. In addition, only about 2% of the active DOX was lost after the preparation of the implants.
Table 1. Contents (DOX-equiv.) and labeled amount of DOX and DOX conjugates implants, n = 5.
In vitro drug release from the implants
In vitro release was determined and release profile of total DOX from implants in pH 5.5 and 7.4 buffer was depicted in . All of the curves in both pH media displayed consistent features which fell into three phases, including a lag-period, linear pattern and then plateau. The mean cumulative release percentage was only 2.5% in the first 6 days showing a lag time. From day 6, the release curve began to display a nearly linear uptrend at a relatively faster speed until day 43 with an average of 55.3% of the drug released. So a plateau was presented from day 45 to the end, only a mean of 5.6% of the drug was released during this period. As a whole, the cumulative release percentage reached an average of 63.4% in 60 days, indicating that the implants had obviously sustained release characteristics. Comparability of the release curves was evaluated by calculating the similarity factor (f2). As presented in , all f2 values were over 50 between any two curves, so it could be confirmed that the release behaviors of three implants were similar, even in different pH conditions.
Figure 4. In vitro accumulation releasing curves of total DOX from implants in pH 5.5 and pH 7.4 PBS, n = 3.
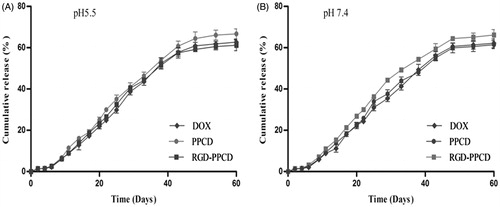
Table 2. Values of similarity factor between two different curves.
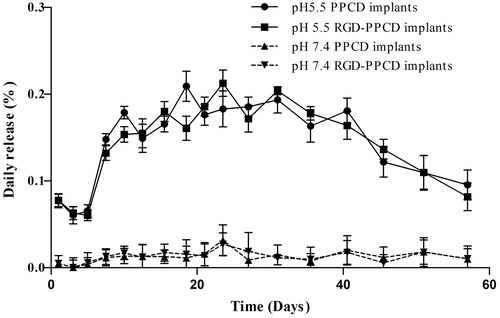
To confirm the acid-sensitive character of PPCD and RGD-PPCD released from implants, the amount of free DOX in different release media was directly detected by HPLC without acid hydrolysis. As shown in , when the DOX-conjugate implants were under physiological conditions (pH 7.4), free DOX release was extremely low with an 0.13‰ of average daily release and about 0.9% of the cumulative release within 60 days. In contrast, under the acidic condition (pH 5.5), the DOX daily release as well as the cumulative ones was 10-fold higher than in pH 7.4 media. Thus, the results demonstrated that the DOX-polymer conjugates in implants kept stable and still had the property of acid-sensitive drug release.
Figure 5. In vitro daily release profiles of free DOX from conjugate implants in pH 5.5 PBS and pH 7.4 PBS, n = 3.
Furthermore, the whole release process of total DOX follows a mixed order kinetics. The second phase was the longest segment of the curve, then the kinetics of the second phase was evaluated by zero-order, first-order and Higuchi’s models. The correlation coefficients were determined from regression plots of Qt versus t, log (Q0 − Qt) versus t and Qt versus t1/2, respectively. As shown in , all of the R2 values were close to 1 and have little difference between different models despite the slightly high R2 for zero-order equation. To understand the mechanism of drug diffusion from carrier materials, the release data of the second phase was also fitted to the Korsmeyer–Peppas model and its modified form which also known as the Power Law (). Obviously, the curves were fitted better to the Power Law due to greater R2 values than that of the Korsmeyer–Peppas equation. Based on the modified equation, the exponent “n” values for all formulas were much closer to 1, suggesting that the release mechanism of the second phase was zero-order release.
Table 3. In vitro kinetics analysis of DOX and its conjugates released from implantsa.
Table 4. The mechanism analysis of DOX and its conjugates released from implantsa.
In vivo biodistribution
The results of in vivo biodistribution are presented in . For all three groups, the DOX amounts in normal tissues and blood were at low levels and showed no significant difference at each time point. In contrast, the drug amounts of three groups in tumor tissue always maintained the highest levels when compared with other tissues within 45 days. Moreover, DOX amounts increased with the prolonging of implantation time. The concentration of intratumoral drug at day 30 and day 45 was significantly higher than that of day 7. Additionally, the obvious differences among three implants groups were observed on the day 30 and day 45 in which the intratumoral drug amount of RGD-PPCD and PPCD was higher than that of free DOX. Particularly, the concentration of RGD-PPCD in tumor was significant higher than that of PPCD at day 45 (p < 0.05).
Figure 6. In vivo disposition after inserting the implants into the middle of tumor (over 45 days), (a) – (c) were the biodistribution of total drug for DOX implants, PPCD implants and RGD-PPCD implants at a dose of 0.2 mg DOX-equiv. each mouse (n = 5), (d) and (e) were the total DOX concentration of three administration groups in the tumor and plasma, (f) was the total DOX remaining in reliquus implants *p < 0.05; #p < 0.01, as compared with DOX implant at the same time point and all the implanted matrix became gelatinous state resulting that could not be able to withdraw at day 45.
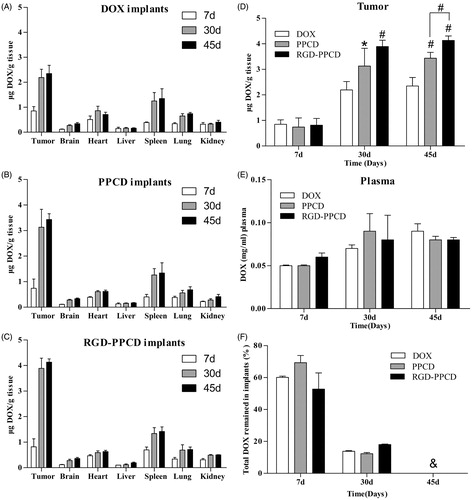
Moreover, it was clearly visible that the implanted matrix was gradually gelatinized and degraded from surface to interior over time. As shown in , the remaining DOX of 30 days after implantation significantly reduced as compared with that of 7 days. However, on the day 45, the residua had become viscous and gelatinous state, thus adhered to tumor tissue, and resulted in difficultly separating from tissues.
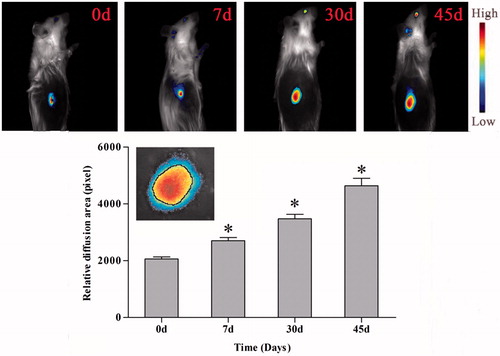
Furthermore, fluorescent imaging of tumor indicated the existence state of intratumoral implants. The penetration area of DOX, represented by red fluorescence in tumor location, expanded gradually over time. A black circle was defined between the area of yellow and blue in order to quantity the penetration area of intratumoral drug, as showed in inset. The area of the circle was measured by software ImageJ (NIH, Bethesda, MD, USA). It was found that the values expressing penetration area at d 7, d 30 and d 45 was 1.31-, 1.69- and 2.26-fold higher than that of d 0, respectively, indicating that drug did release from the implants and penetrated in tumor tissues gradually.
Figure 7. In vivo imaging and diffusion area of drug penetration in tumor after implantation. Upper figures are in vivo imaging of existence state of intratumoral implants, lower figure is relative diffusion area of DOX implants in tumor after implantation, *p < 0.01, inset: Screenshot of imaging of implants penetration in tumor (3×).
In vivo antitumor activity
The antitumor activity of the implants was reflected by the tumor growth rate in terms of mean tumor volume (cm3) and IRT (%). As presented in , the tumor growth of the blank implants group as well as the negative control group was fastest and had no significant difference with each other, indicating that the polymer carriers of the implants did not have any inhibitory effect on tumor growth. The conjugate implants were more effective in preventing tumor growth compared with free DOX implants group. In addition, on the day 35 after implantation, RGD-PPCD implants began to exhibit stronger antitumor activity than PPCD implants and showed significant difference till day 55 (p < 0.01). Correspondingly, IRT of RGD-PPCD implants group was the greatest followed by that of PPCD implants, with that of free DOX implant being the least (). The research also found that the tumor growth for the C6-bearing subcutaneous mice model began to slow down after 50 days. The decreased tumor volume in untreated groups resulted in lower IRT values at day 55 than that at day 45 ().
Figure 8. Tumor growth curve of in C6 tumor-bearing mice after the local application of DOX and DOX conjugate implants at the doses of 0.2 mg DOX-equiv. each mouse (n = 4). *p < 0.01, as PPCD implants compared with RGD-PPCD implants at the same time point.
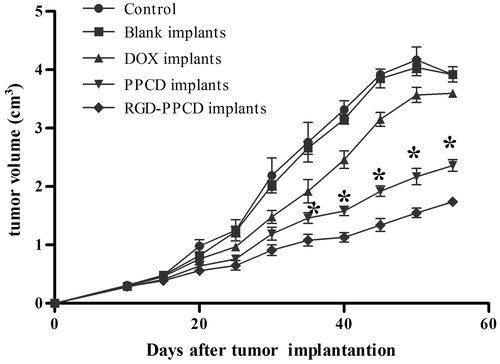
Table 5. The inhibitory rate of tumor (IRT %) after the local application of DOX and DOX conjugate implants at the doses of 0.2 mg DOX-equiv. each mouse, as compared with blank implants.
Discussions
Our previous work had already demonstrated that RGD-PPCD showed higher in vitro cytotoxicity and significant treatment efficacy via systemic administration, as compared with PPCD. In the present study, PLGA/PLA implants loading DOX-polymer conjugates were prepared for the first time to evaluate the potential of local therapy.
As is reported, the degradation rate of PLGA depended largely on the lactide/glycolide ratio of the polymers (Fu et al., Citation2002). Thus, the ratio of PLGA would influence on the drug release. Our previous study found that the release rate of drug from PLGA carrier speeded up with the increasing of the content of glycolic acid. Lactic acid is more hydrophobic than glycolic acid and hence lactide-rich PLGA are less hydrophilic, absorb less water and subsequently degrade more slowly (Jain, Citation2000). So PLGA 75:25, a common type used as the carrier of sustained or controlled release preparations, was selected in the present study. On the other hand, the high viscosity of the PLGA methylene chloride solution was weighted against the formability of implants. From this angle, PLA used as another carrier could decrease viscosity of PLGA and then improved the formability of implants. Consequently, the implants obtained with the 3:1 ratio of PLGA/PLA (w/w) had preferable formability and sustained release effect. In addition, it was found that uniform mixing of drug methanol-water solution and the PLGA-PLA methylene chloride solution was difficult to achieve, while adding PEG could not only heightened suspending effect resulting in a better drug-carrier mixing but also improved the formability of the carriers and regulated the release rate as well. Finally, the optimized formulation was confirmed with the 3:1 ratio of PLGA/PLA (w/w) and 1% PEG (wt.%).
It was reported that extracellular fluid in tumor tissue was slightly acidic and the lowest pH could be 6.2 (Ojugo et al., Citation1999). Within tumor cells, lysosomes and endosomes which performed uptake of drug-polymer conjugates were more acidic with the pH range from 4 to 6 (Liu & Shapiro, Citation2003). In order to imitate the release behave of the implants under the faintly acid condition of tumor interstitial fluid and inside of the tumor cells, the buffer of pH 5.5 was chosen as the release media. Meanwhile, the buffer of pH 7.4 was used to imitate the physiological condition.
With the same formulation and preparation procedure, three implants loaded free DOX and its conjugates showed similar release feature, illustrating that the release kinetics were a function of the polymer matrix rather than the chemotherapeutic agents. Unlike other compressed implantable tablets with burst release (Onishi et al., Citation2005; Santovena et al., Citation2009), our study fabricated the implants by a solvent evaporation method, successfully avoid occurring of burst release. The release curves were divided into three phases with different release rates. The first six days with negligible release showed a lag-period. This is due to the fact that, after the implants immersed in the release media, PEG hydrates and swells, then forms a hydrogel layer on the surface of the implants that slows down the rate of water intake and prolongs the drug release (Paun et al., Citation2011). After six days, according to the Power Law, the implants began to display a steady zero-order release rate about 30 μg/d, lasting near 40 days. The possible reason was that with the degradation of PLGA and PLA, drug gradually dissolved until saturation level in hydrated gel layer, thus a constant concentration gradient was attained between gel layer and release media resulting in a constant release rate. Afterward, the release rate obviously decelerated with only 5.6% of the cumulative release from day 43 to day 60, because of the concentration gradient decreasing with the most of drugs released.
Furthermore, there was a positive correlation between the release rate of drug from the wafer and the degradation of carrier material. The degradation of PLGA and PLA took place by hydrolysis and was determined by the rate of water diffusion into the wafer and the hydrolysis reaction rate. In our study and the other report (Herrmann et al., Citation2007), PEG significantly increased drug release from implants by promoting water diffusion into the wafer. Besides, water-solubility of the loaded drug also accelerated the rate of water diffusion (Siegel et al., Citation2006). Although the size of DOX conjugates was much larger than free DOX, DOX conjugates even had a better water solubility than free DOX which could improve diffusion of the macromolecular conjugates from the wafer. It was reported that the degradation index (including mass, molecular weight and morphology) of the PLGA foams at pH 5.0, 6.4 and 7.4 was similar until week 16 (Holy et al., Citation1999). In summary, compared with pH of the media, PEG and water-solubility of loaded drug had a stronger effect on drug release. Therefore, the total drug release was dependent on neither the loaded drugs (DOX or DOX conjugates) nor the pH of the release media in this study.
The sustained release character of the implants had been further testified by in vivo experiments. The results of biodistribution indicated that local administration could ensure the drug long-term residence in tumor and the lasting release from the sustained release carrier. The behavior was similar as multiple low-dose dosing. The intratumoral drug amount of DOX conjugate implants group was significant higher than that of DOX implants group. Though there were no significantly difference between PPCD implants group and RGD-PPCD implants group at day 30, growing trend did exist. The cumulative concentration of drug in tumor of different groups was ranked in ascending order of DOX implants <PPCD implants <RGD-PPCD implants at day 45. This would mainly depend on two limits: drug release rate from the implants into tumor tissue and transport rate from tumor into the circulation system. Although the release rate of three implants was consistent with each other, once released, the transporting of free drug to the circulatory system was easier than its conjugates due to the much smaller molecular weight. Furthermore, the retention ability of RGD-modified PPCD in tumor was higher than that of non-modified PPCD. It might be the reason that RGD have high affinity with angiogenic vessels and tumor cells which were highly expressed cell adhesion molecule integrin αvβ3 (Temming et al., Citation2005; Cai et al., Citation2008; Wang et al., Citation2011; Zhu et al., Citation2011; Zhang et al., Citation2012). As compared with DOX conjugates, despite of more free drug transporting into systemic circulation, a much more quick plasma clearance of free drug would balance it out and results in no difference in plasma drug concentration between DOX and its conjugates which further led to uniformity of the drug biodistribution in other normal tissues.
In vivo drug release behavior from the polymer carrier was evaluated by measuring the total drug amount in the residua. It was found that the amount of drug in the residua reduced gradually and the size of residue in vivo was significantly smaller than that in release medium in vitro at the same time point. In particular, the residue was difficult to separate from tumor tissues at day 45 after implantation. But in vitro release experiments revealed that the fragments of the implants could be collected even at day 60. Based on the measured data and observed phenomena, it can be inferred that the in vivo release of DOX and its conjugates from polymer matrix is faster in speed and shorter in duration than what our in vitro data suggest. As reported previously (Negrin et al., Citation2004; Takahashi et al., Citation2004; Onishi et al., Citation2005), the initial release in vivo might be stimulated by change in implant shape such as mechanical breakdown at the administration site. Besides, the in vivo environment might accelerate biodegradation rate of polymer due to the existence of various biological components involve enzymes.
The study of in vivo antitumor activity showed that RGD-PPCD implants exhibited stronger antitumor activity than PPCD implants. It might be explained by the higher tumor accumulation of RGD-PPCD and its stronger ability to induce apoptosis of tumor neovascular endothelial cells (Zhu et al., Citation2011). In addition, the phenomenon of the decreasing in tumor volume after 50 days might be related with the immunogenicity of C6 cells. It has been reported that the intensive immune responses against C6 xenografts can have a significant impact on tumor growth as well as obscuring therapeutic effects (Beutler et al., Citation1999; Badie & Schartner, Citation2000).
Conclusions
DOX and its conjugate implants were prepared by using the biodegradable polymer as matrix materials. The release curves were fell into three phases, including a lag-period, then the second phase which was consistent with the zero-order model followed by a plateau. After locally implanted in the tumor, the disposition of implants and the anti-tumor efficacy were evaluated in tumor-bearing animals. Results proved that RGD-PPCD implants showed the most retention dose in tumor and the most efficacy of inhibiting tumor growth among all the implants. Further work must be done to confirm the advantages of local RGD-PPCD implants therapy over systemic administration in orthotopic gliomas animal models.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. This study was funded by National Key Basic Research Program of China (2013CB932502), National Natural Science Foundation of China (81172185).
References
- Anonymous. (1996). Gliadel wafer recommended for approval as treatment for brain cancer by FDA’S Oncologic Drugs Advisory Committee. Drug News Perspect 9:292
- Aqil F, Jeyabalan J, Kausar H, et al. (2012). Multi-layer polymeric implants for sustained release of chemopreventives. Cancer Lett 326:33–40
- Arias JL. (2011). Drug targeting strategies in cancer treatment: an overview. Mini Rev Med Chem 11:1–17
- Badie B, Schartner JM. (2000). Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 46:957–61
- Beutler AS, Banck MS, Wedekind D, et al. (1999). Tumor gene therapy made easy: allogeneic major histocompatibility complex in the C6 rat glioma model. Hum Gene Ther 10:95–101
- Bragagni M, Mennini N, Ghelardini C, et al. (2012). Development and characterization of niosomal formulations of doxorubicin aimed at brain targeting. Pharm Pharm Sci 15:184–96
- Cai W, Niu G, Chen X. (2008). Imaging of integrins as biomarkers for tumor angiogenesis. Curr Pharm Des 14:2943–73
- Fu YJ, Shyu SS, Su FH, et al. (2002). Development of biodegradable co-poly(d,l-lactic/glycolic acid) microspheres for the controlled release of 5-FU by the spray drying method. Colloid Surface B 25:269–79
- Haaga JR, Exner AA, Wang Y, et al. (2005). Combined tumor therapy by using radiofrequency ablation and 5-FU-laden polymer implants: evaluation in rats and rabbits. Radiology 237:911–18
- Herrmann S, Winter G, Mohl S, et al. (2007). Mechanisms controlling protein release from lipidic implants: effects of PEG addition. J Control Release 118:161–8
- Holy CE, Dang SM, Davies JE, et al. (1999). In vitro degradation of a novel poly(lactide-co-glycolide) 75/25 foam. Biomaterials 20:1177–85
- Jain RA. (2000). The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 21:2475–90
- Jiang Z, Sun C, Yin Z, et al. (2012). Comparison of two kinds of nanomedicine for targeted gene therapy: premodified or postmodified gene delivery systems. Int J Nanomed 7:2019–31
- Koocheki S, Madaeni SS, Niroomandi P. (2011). Development of an enhanced formulation for delivering sustained release of buprenorphine hydrochloride. Saudi Pharm J 19:255–62
- Lesniak MS, Upadhyay U, Goodwin R, et al. (2005). Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res 25:3825–31
- Li Y, He H, Jia X, et al. (2012). A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials 33:3899–908
- Liu J, Shapiro JI. (2003). Endocytosis and signal transduction: basic science update. Biol Res Nurs 5:117–28
- Mesnukul A, Yodkhum K, Mahadlek J, et al. (2010). Characterization of indomethacin release from polyethylene glycol tablet fabricated with mold technique. Indian J Pharm Sci 72:92–100
- Negrin CM, Delgado A, Llabres M, et al. (2004). Methadone implants for methadone maintenance treatment. In vitro and in vivo animal studies. J Control Release 95:413–21
- Onishi H, Takahashi M, Machida Y. (2005). PLGA implant tablet of ketoprofen: comparison of in vitro and in vivo releases. Biol Pharm Bull 28:2011–15
- Ojugo A, McSheehy P, McIntyre D, et al. (1999). Measurement of the extracellular pH of solid tumors in mice by magnetic resonance spectroscopy: a comparison of exogenous F-19 and P-31 probes. Nmr Biomed 12:495–504
- Paun IA, Moldovan A, Luculescu CR, et al. (2011). Biocompatible polymeric implants for controlled drug delivery produced by MAPLE. Appl Surf Sci 257:10780–8
- Puvanakrishnan P, Park J, Chatterjee D, et al. (2012). In vivo tumor targeting of gold nanoparticles: effect of particle type and dosing strategy. Int J Nanomed 7:1251–8
- Ranganath SH, Fu Y, Arifin DY, et al. (2010). The use of submicron/nanoscale PLGA implants to deliver paclitaxel with enhanced pharmacokinetics and therapeutic efficacy in intracranial glioblastoma in mice. Biomaterials 31:5199–207
- Santovena A, Alvarez-Lorenzo C, Llabres M, et al. (2009). hGH release from directly compressed hGH-PLGA biodegradable implantable tablets: influence of physicomechanical factors. Eur Polym J 45:2830–8
- Shikanov A, Shikanov S, Vaisman B, et al. (2008). Paclitaxel tumor biodistribution and efficacy after intratumoral injection of a biodegradable extended release implant. Int J Pharm 358:114–20
- Siegel R, Naishadham D, Jemal A. (2012). Cancer statistics. Ca-Cancer J Clin 62:10–29
- Siegel SJ, Kahn JB, Metzger K, et al. (2006). Effect of drug type on the degradation rate of PLGA matrices. Eur J Pharm Biopharm 64:287–93
- Sivak WN, Pollack IF, Petoud S, et al. (2008). LDI-glycerol polyurethane implants exhibit controlled release of DB-67 and anti-tumor activity in vitro against malignant gliomas. Acta Biomater 4:852–62
- Stupp R, Hegi ME, van den Bent MJ, et al. (2006). Changing paradigms – an update on the multidisciplinary management of malignant glioma. Oncologist 11:165–80
- Temming K, Schiffelers RM, Molema G, et al. (2005). RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist Updat 8:381–402
- Takahashi M, Onishi H, Machida Y. (2004). Development of implant tablet for a week-long sustained release. J Control Release 100:63–74
- Vannucci L, Falvo E, Fornara M, et al. (2012). Selective targeting of melanoma by PEG-masked protein-based multifunctional nanoparticles. Int J Nanomed 7:1489–509
- Voulgaris S, Partheni M, Karamouzis M, et al. (2002). Intratumoral doxorubicin in patients with malignant brain gliomas. Am J Clin Oncol-Canc 25:60–4
- Wang CC, Li J, Teo CS, et al. (1999). The delivery of BCNU to brain tumors. J Control Release 61:21–41
- Wang JX, Wang ZH, Chen JF, et al. (2008). Direct encapsulation of water-soluble drug into silica microcapsules for sustained release applications. Mater Res Bull 43:3374–81
- Wang Q, Han Y, Jiang T, et al. (2011). MR Imaging of activated hepatic stellate cells in liver injured by CCl4 of rats with integrin-targeted ultrasmall superparamagnetic iron oxide. Eur Radiol 21:1016–25
- Weinberg BD, Ai H, Blanco E, et al. (2007). Antitumor efficacy and local distribution of doxorubicin via intratumoral delivery from polymer millirods. J Biomed Mater Res A 81:161–70
- Zhang C, Xie X, Liang S, et al. (2012). Mono-dispersed high magnetic resonance sensitive magnetite nanocluster probe for detection of nascent tumors by magnetic resonance molecular imaging. Nanomed-Nanotechnol Biol Med 8:996–1006
- Zhang L, Zhu S, Qian L, et al. (2011). RGD-modified PEG-PAMAM-DOX conjugates: in vitro and in vivo studies for glioma. J Eur J Pharm Biopharm 79:232–40
- Zhu S, Hong M, Tang G, et al. (2010a). Partly PEGylated polyamidoamine dendrimer for tumor-selective targeting of doxorubicin: the effects of PEGylation degree and drug conjugation style. Biomaterials 31:1360–71
- Zhu S, Hong M, Zhang L, et al. (2010b). PEGylated PAMAM dendrimer-doxorubicin conjugates: in vitro evaluation and in vivo tumor accumulation. Pharm Res 27:161–74
- Zhu S, Qian L, Hong M, et al. (2011). RGD-modified PEG-PAMAM-DOX conjugate: in vitro and in vivo targeting to both tumor neovascular endothelial cells and tumor cells. Adv Mater 23:H84–9