
Abstract
Self-microemulsifying drug delivery system (SMEDDS) has emerged as a vital strategy to formulate poor water soluble compounds for bioavailability enhancement. However, certain limitations are associated with SMEDDS formulations which include in vivo drug precipitation, formulation handling issues, limited lymphatic uptake, lack of predictive in vitro tests and oxidation of unsaturated fatty acids. These limitations restrict their potential usage. Inclusion of polymers or precipitation inhibitors within lipid based formulations helps to maintain drug supersaturation after dispersion. This, thereby, improves the bioavailability and reduces the variability on exposure. Also, formulating solid SMEDDS helps to overcome liquid handling and stability problems. Usage of medium chain triglycerides (MCT) and suitable antioxidants to minimize oxidation of unsaturated fatty acids are few of the steps to overcome the limitations associated with SMEDDS. The review discussed here, in detail, the limitations of SMEDDS and suitable measures that can be taken to overcome them.
Introduction
Majority of the new drug candidates (almost 40%) being developed are water insoluble. Such active pharmaceutical ingredients (API) poses several problems while developing their formulations (Tang et al., Citation2008). Popular formulation techniques used for delivering a poor water soluble drug includes: (a) micronization of crystalline solids (b) amorphous formulation or solid dispersions and (c) lipid based formulations. Among these approaches, lipid-microemulsion formulations, with a particular emphasis on self-emulsifying drug delivery system (SEDDS or SMEDDS), have gained great importance as a promising approach for poorly soluble drugs as well as for natural compounds (Yao & Li, Citation2010; Amri et al., Citation2013; Pund et al., Citation2013, Citation2014). You et al. (Citation2005) have improved the oral bioavailability of Oleum Curcumae wenchowensis by 57.7% using SEDDS.
Lipid based formulations mainly comprises of macroemulsion (coarse emulsion), microemulsion, self-microemulsifying drug delivery system (SMEDDS), solid-lipid nanoparticle (SLN), liposomes and lipoplexes. In recent years, much attention has been focused on lipid based formulation, with particular emphasis on SMEDDS (Pouton, Citation2006). Lipid based formulations, like SMEDDS, are emulsions having reduced globule size and high thermodynamic stability (Hauss, Citation2007; Nanjwade 2011; Müllertz et al., Citation2010).
Typically emulsions are dispersion of macroscopic droplets of one liquid in another liquid, with a droplet size of 1–10 µm consisting of water, oil and emulsifier. They are semi-transparent to cloudy, viscous liquid having tough interfacial film and are thermodynamically unstable. Emulsions can be oil-in-water (o/w), water-in-oil (w/o) or multiple emulsions (Eccleston, Citation2007). A dispersion of oil-in-water, of most interest for pharmaceutical applications, is referred to as an oil-in-water (o/w) emulsion, requires the emulsifying agent to be typically more soluble in the aqueous phase. The reverse emulsion, water-in-oil (w/o) is stabilized by surfactants that are stable in the oil phase. On the other hand, double emulsions are the ternary systems having either a w/o/w or o/w/o type, wherein the dispersed droplets contain smaller droplets of a different phase.
Here, it is essential to understand the differences between microemulsion and nanoemulsion as SMEDDS finally forms microemulsion after oral administration (Anton & Vandamme, Citation2010). Microemulsions are isotropic, thermodynamically stable systems composed of oil, water, surfactant (S) and co-surfactants (CoS) or co-solvents. The main driving force for microemulsion formation is the ultra low interfacial tension, which is usually achieved by the use of two or more emulsifiers (S/CoS). Out of the two emulsifiers used in microemulsion formation, one is predominantly water soluble (surfactant) while the other is predominantly oil soluble (co-surfactant). Co-surfactants reduce the interfacial tension to an ultra low value, required for microemulsion formation (Parmar et al., Citation2011; Rahman et al., Citation2012). On the other hand, nanoemulsions are the emulsions that have a globule diameter in the nanometer range (below 100 nm). The formation of nanoemulsion requires an input of energy. This energy can be supplied by either a mechanical equipment or the chemical potential inherent within the components (Lawrence & Warisnoicharoen, Citation2006). Unlike microemulsions, nanoemulsions are thermodynamically unstable (however, kinetically stable) systems. Like conventional emulsions, both emulsions and nanoemulsions are in thermodynamic non-equilibrium state, however the kinetics of destabilization of nanoemulsion is very slow (∼months) and hence they are considered as kinetically stable (Solans et al., Citation2005; Anton & Vandamme, Citation2010; Rao & McClements, Citation2012). As a result of this, nanoemulsion droplets remain stable in conditions of stress like temperature changes, dilutions. On the other hand, microemulsions are strongly affected by temperature changes and/or dilutions and are even broken-up by these alterations. Nanoemulsions are formed only if surfactants are first mixed with the oily phase, however, if surfactant is first mixed with water before adding the oily phase, then only a “macroscopic” emulsion is generated. On the other hand, formation of microemulsion is not affected by the order of mixing. This is a critical point and constitutes a preliminary test for characterizing the nature of the dispersion obtained. Dynamic light scattering (DLS), used for estimating the size distribution of the dispersions, often requires dilution of sample prior to measurement. Dilution, in case of microemulsion, results in a modification of the size of the swollen micelles, which simply invalidates the characterization using this technique and can even lead to the destruction of the micelles. While in the case of nanoemulsion, such dilutions do not have any influence on the droplet size and size distribution (Solans et al., Citation2005).
A common requirement for all the types of lipid formulation is that they should be able to keep the drug in the solubilized form in the gastrointestinal tract (GIT). Precipitation of the drug from the system nullifies the advantage offered by the lipid-based formulation system.
SEDDS/pre-microemulsion concentrate
SEDDS are isotropic mixtures of oils, hydrophobic surfactants and sometimes, containing co-solvents that rapidly produces fine o/w emulsions upon gentle agitation or digestive motility that would be encountered in the gastrointestinal tract (Boyd et al. 2009; Singh et al., Citation2008, Citation2009a). SEDDS is a broad term, typically producing emulsions with a droplet size ranging from 100 to 250 nm in case of SMEDDS and less than 100 nm in case of self-nanoemulsifying drug delivery Systems (SNEDDS) (Xi et al., Citation2009; Wei et al., Citation2012). Both SMEDDS and SNEDDS are isotropic mixture of oil, hydrophilic surfactant and co-solvents that rapidly form o/w microemulsion upon gentle agitation followed by dilution in an aqueous media that will be experienced in the gastrointestinal tract (Yang et al., Citation2004; Kohli et al., Citation2010; Shahba et al., Citation2012). It is worth noting that this method of producing a fine o/w emulsion using SMEEDS/SNEDDS is identical to the low-energy emulsification method for producing o/w nanoemulsions. The rate-limiting step for dissolution of drug substance from SMEDDS is the diffusion from oil globule to dissolution media, which was influenced by the concentration of surfactant and co-surfactant in the system (Zidan et al., Citation2007; Singh et al., Citation2009b; Wang et al., Citation2010). The basic difference between SEDDS and SMEDDS is that SEDDS typically produce opaque emulsions with a droplet size above 300 nm while SMEDDS form transparent microemulsions with a droplet size of less than 250 nm (Pouton 2000). In addition, concentration of oil in SMEDDS is less than 20% as compared to 40–80% in SEDDS. SEDDS are usually prepared using surfactants of HLB < 12 while SMEDDS and SNEDDS are prepared with surfactants of HLB > 12 (Mohsin 2012; Wadhwa et al., Citation2012). The microemulsion globule formed from SMEDDS/SNEDDS tends to be of larger size when compared to same drug administered as formulated microemulsion (Zidan et al., Citation2007; Xi et al., Citation2009). The major differences between SEDDDS, SMEDDS and SNEDDS are enlisted in .
Table 1 Major differences between SEDDS, SMEDDS and SNEDDS formulation.
Composition of SMEDDS formulation
Typically, a SMEDDS formulation comprises of drug, oil, surfactant and co-surfactant
Drug
Lipophilicity and dose of the drug are the main criteria to be considered before development of SMEDDS formulation. Ideally, drug should have low dose, log P ≥ 2 and should not possess extensive first pass metabolism. The drug should show substantial solubility in pharmaceutically accepted lipids, surfactants and co-solvents (Gao 2011; Gao & Morozowich, Citation2009).
Oils
Medium chain triglycerides (MCT) having carbon atoms between 6 and 12 are directly transported by portal blood to the systemic circulation. Whereas, long chain triglycerides (LCT) having carbon atoms greater than 12 are transported via intestinal lymphatics. As MCT have higher solvent capacity and are also not subjected to oxidation, they are widely used in lipid based formulations (Land et al., Citation2005; Hauss, Citation2007; Rahman et al., Citation2012).
Surfactants
Surfactants form the interfacial film and lower the interfacial tension to a small value which facilitates dispersion process. HLB value and concentration of surfactant is essential to be considered while selecting a surfactant. For attaining high emulsifying performance, the emulsifier involved in the formulation of SMEDDS should have high HLB greater than 12 which assists in formation of small o/w droplets and rapid spreading of formulation in aqueous media. Generally, non-ionic surfactants with HLB > 12 are suggested for design of self-dispersing systems as these are less toxic than ionic surfactants (Azaz et al., Citation1973; Hauss, Citation2007).
Co-surfactants
Co-surfactants ensures flexibility of the interfacial layer, i.e. it reduces the interfacial tension to a negative value. Co-surfactants form a flexible interfacial film in order to acquire different curvatures required to form microemulsion over a wide range of composition. Medium chain length alcohols (C3–C8) are commonly employed as co-surfactants (Humberstone & Charman, Citation1997; Parmar et al., Citation2011).
Advantages of SMEDDS over other emulsions
Storage: SMEDDS has the same advantage as emulsions, of facilitating the solubility of hydrophobic drugs. Macroemulsions undergo creaming over a period of time, whereas SMEDDS being thermodynamically stable can be stored easily (Khan et al 2012; Eccleston, Citation2007).
Stability: In contrast to micro/nanoemulsions, SMEDDS do not contain water and hence, they have improved physical and/or chemical stability on long-term storage. Self-nanoemulsifying tablets of carvedilol showed successful incorporation of carvedilol within the SNEDDS. This resulted in improvement of the stability of carvedilol on dilution with aqueous media in the presence of cellulosic polymers (Mahmoud et al., Citation2009).
Compliance: Most of the SMEDDS formulations are in capsule or tablet dosage forms, thus occupying smaller volume, easy to administer and hence improved patient compliance (Gao et al., Citation2003; Gao & Morozowich, Citation2006; Nazzal & Khan, Citation2006).
Palatability: SMEDDS formulation can be easily filled into capsules resolving the palatability issues associated with lipid formulations (Huang et al., Citation2012).
Effect of food: Absorption of drug from SMEDDS formulation is not affected by food. The lipophilic contents of fatty diet aid, aids in absorption of drug from these systems. It was observed that food had a marked effect on the absorption of itraconazole from the marketed formulation (Sporanox capsule), whereas the influence was less pronounced for the self-emulsifying formulation of itraconazole (ITRA-GSMP capsule) in human volunteers (Woo et al., Citation2008).
Quick onset of action: SMEDDS have the ability to facilitate rapid oral absorption of the drug, which results in quick onset of action. It was found that the tmax of vitamin A was reduced and bioavailability was increased when administered as SNEDDS capsule and SNEDDS tablet as compared to vitamin A oily solution-filled capsules without any additives (Taha et al., Citation2007).
Ease of manufacture and scale-up: SMEDDS can be easily manufactured at large scale as it requires simple and economical manufacturing facilities, such as simple mixer with an agitator and volumetric liquid filling equipment (Sander & Holm, Citation2009).
Limitations of SMEDDS
Although SMEDDS formulation has several advantages, there are certain limitations associated with this system represented in .
Figure 1 Challenges in SMEDDS formulation.
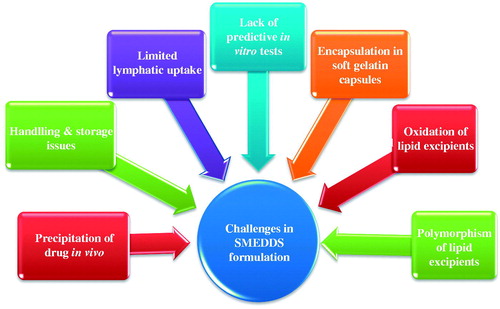
Drug precipitation on dilution: Diluted SMEDDS undergo precipitation of drug in gastrointestinal fluid. A common requirement for the lipid formulations is that they should be able to keep the drug in the solubilized form in the gastrointestinal tract (GIT). Precipitation of the drug from the system nullifies the advantage offered by the lipid-based formulation system.
The precipitation tendency of the drug on dilution is higher due to the dilution effect of the hydrophilic solvent. It thereby requires incorporation of polymers to minimize drug precipitation in vivo (Porter et al., Citation2007; Chen et al., Citation2012).
Encapsulation in soft gelatin capsules: Most of the marketed SMEDDS formulations are available as soft gelatin capsule. However, gelatin capsules are associated with few drawbacks. Manufacturing cost, transmissible spongiform encephalopathy (TSE) and consumer preference/religion are the few issues associated with animal gelatin (Bowtle, Citation2007). Volatile co-solvents in self-microemulsifying formulations are known to migrate into the shells of soft or hard gelatin capsules, resulting in the precipitation of the lipophilic drugs. These problems drive the market requirement to find substitute for soft gelatin capsules (Rahman et al., Citation2012). The current alternative material of choice, to animal gelatin capsules are those prepared from HPMC. The HPMC capsule shell has been explored as an alternative approach for encapsulating supersaturable SMEDDS formulation (Ku et al., Citation2010).
Storage and handling: Liquid SMEDDS exhibit problems in handling, storage and stability. Thus, formulating solid SMEDDS seems to be a logical solution to address these problems (Tang et al., Citation2008).
Limited targeting to lymphatics: Targeting lymphatics confers two primary advantages over conventional absorption via the portal blood. First, transport through the intestinal lymph avoids pre-systemic hepatic metabolism and thereby enhances the concentration of orally administered drugs reaching the systemic circulation. Second, site-specific drug delivery to lymphatic organs could be achieved. Normally high triglyceride solubility and high log P is required for lymphatic transport (Caliph et al., Citation2000). However, the amount of drug transported into lymphatics is variable from drug to drug. Hence, lipophilicity and triglyceride solubility of drug in correlation with lymphatic transport needs to be completely understood and a more adequate predictive model is required (Trevaskis et al., Citation2008).
Lack of good in vitro models: Another obstacle in the development of SMEDDS and other lipid-based formulations is the lack of good predicative in vitro models for the assessment of the formulations (Tang et al., Citation2008; Zhang et al., Citation2008). Traditional dissolution methods do not work, as these formulations potentially are dependent on digestion of lipid in the gut, prior to release of the drug. Although, to mimic this, an in vitro model simulating the digestive processes of the duodenum has been developed (Dahan & Hoffman, Citation2008). This in vitro model needs further refinement and validation before its strength can be evaluated. Further, development can be based on in vitro–in vivo correlations and therefore, different prototype lipid based formulations need to be developed and evaluated in vivo in a suitable animal model (Patil et al., Citation2007; Tang et al., Citation2007).
Oxidation and polymorphism of the lipids used in formulating SEDDS/SMEDDS: Lipid excipients containing unsaturated fatty acids and its derivatives are prone to lipid oxidation (Wasylaschuk et al., Citation2007). This requires inclusion of Lipid soluble antioxidant in capsule formulation (Bowtle, Citation2007). Polymorphism associated with thermo-softening lipid excipients requires specific process control in their application, in order to minimize polymorphic changes of the excipient matrix (Sato, Citation2001).
Precipitation of drug
Drug precipitation in vivo is an undesirable outcome after administration of SMEDDS formulation. It is a process in which a drug solute precipitates in vivo when the solubilization capacity of the formulation for the drug has decreased. Drugs may precipitate in vivo due to sharp pH change, dilution of formulation with body fluids or digestion of solubilizing excipients in formulations (Porter et al., Citation2007; Dai, Citation2010). Drug precipitation often reduces drug concentration in aqueous phase required for immediate action, leading to a delayed or reduced efficacy (Hoener & Benet, Citation2002). In SMEDDS formulation, drug must remain partitioned within the oil/water emulsion droplets; following dilution of SMEDDS formulation with aqueous media in the gastrointestinal tract. Aqueous dispersion of SEDDS resulted in turbid emulsions, subsequently followed by very slow precipitation of 3–7% of the dose of fenofibrate (Mohsin et al., Citation2008). Precipitation of drug is a complex process based on essentially three steps: supersaturation, nucleation and crystal growth.
A. Supersaturation: Supersaturation implies that the concentration of the solubilized drug is above the saturation solubility. When the solubility of drug in solvent exceeds the saturation solubility, the solution then becomes supersaturated and excess drug may precipitate or crystallize. In a supersaturated system, the drug solution is in thermodynamically unstable system and tends to return to the equilibrium state by drug precipitation. This essentially implies that precipitation process is a stabilization step. The degree of supersaturation: SS = S/Seq, where S represents the actual concentration and Seq is the equilibrium solubility. After supersaturation has reached, the process continues with nucleation (Gao & Shi, Citation2012).
B. Nucleation: Nucleation refers to the birth of very small bodies of a new phase within a homogenous supersaturated liquid phase. Nucleation is a consequence of rapid flocculations at the molecular level, when molecules/ions/atoms are in random motion in small volume. During nucleation, solute molecules initially gather onto the surface of an impurity within the solution. These nuclei reach a stable state by exceeding a critical size. Different types of nucleation exist. If a system does not contain any crystal, the process is named primary nucleation. Primary nucleation is subdivided into homogeneous and heterogeneous nucleation, depending if nucleation occurs spontaneously (primary nucleation) or if it is induced by foreign particles, the process is referred to as secondary nucleation. Further addition of molecules leads to crystal growth (Mullin, Citation2001).
C. Crystal growth: Crystal growth is a diffusional process and a surface phenomenon. Once a critical sized solute cluster is formed in solution, subsequent growth of the nuclei can occur. From solution, solute molecules or ions reach the faces of crystal by diffusion. However, growth does not occur in isolation and nucleation continues, with either process dominating depending on the physical conditions. When crystal growth is dominant, few large crystals are formed. Whereas significant ongoing nucleation leads to the generation of large number of small crystals (Bevernage et al., Citation2013).
Factors affecting drug precipitation
The precipitation of drug from supersaturated solutions is a complex function of both nucleation and crystal growth, which in general, is effected by the following factors.
A. Degree of supersaturation: Increasing the degree of supersaturation of SMEDDS formulation favors drug precipitation by increasing nucleation rate. Supersaturation can occur through (a) evaporation of solvent from the solution, (b) cooling of the solution, if solute has positive heat of solution, (c) formation of a new solute as a result of chemical reaction, (d) addition of a substance, which has higher solubility in solvent than the solid to be crystallized and (e) addition of solvent that lowers the solubility of the solute (Warren et al., Citation2010). Since in a supersaturated system, the drug solution is in thermodynamically unstable state, and thus it tends to return to the equilibrium state via drug precipitation (Gao & Shi, Citation2012).
B. Solubility: At constant supersaturation, increasing the solubility of SMEDDS increases the probability of intermolecular collision which thereby increases nucleation rate (Avdeef, Citation2003).
C. Impurities: Presence of impurities in solution stimulates nucleation process. The presence of impurities decreases the energy barrier for the formation of nuclei which ultimately lead to crystal formation (Rodriguez-Hornedo & Murphy, Citation1999). Therefore, the critical cluster size is significantly smaller than homogenous nucleation processes (Kashchiev & van Rosmalen, Citation2003).
D. Temperature: Binding between drug and polymer is decreased at higher temperatures, due to weakening of intermolecular interactions between the molecules and increased solubility of drug (Plaizier-Vercammen & De Neve, Citation1981). Thus, increasing the temperature decreases the nucleation rate.
E. Solution viscosity: Low solution viscosity favors drug precipitation. Increasing the viscosity of the aqueous medium decreases the rate of drug diffusion from bulk solution to the crystal nuclei or surface, inhibiting crystallization, during both nucleation and growth phases (Porter et al., Citation2007).
Mechanism to inhibit drug precipitation
The mechanism by which these precipitation inhibitors act is not fully understood. However, a number of probable mechanisms are proposed, which include the following factors.
A. Reticulate formation: Creation of widely spaced cellulosic polymer network has been proposed to generate a supersaturated state of HPMC with supersaturable SMEDDS formulation. These networks created by the HPMC chains in water consist of cellulosic bundles resulting in a tenuous network of swollen clusters with hydrophobic substituent surrounded by sheaths of structured water. HPMC polymer chain may inhibit nucleation as well as crystal growth by adsorption of HPMC molecules onto surface of nuclei or on to the surface of crystals (Gao & Morozowich, Citation2007).
B. Hydrogen bonding: Approximately 60% of the hydroxyl groups in HPMC are not substituted. HPMC or the hydrophilic polymers can thus form both intramolecular and intermolecular hydrogen bonds with drug, which is likely to retard drug precipitation. Indirubin-PVP hydrogen bonds were formed between the carbonyl group of the pyrrolidone ring of PVP K17 and the amino group in the pyrolle ring as well as the amido-hydrogen of Indirubin (Chen et al., Citation2012).
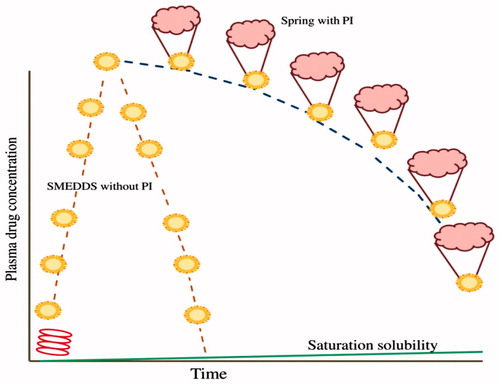
C. Spring and Parachute mechanism: Supersaturable approach is a promising approach for utilization of drug compounds with poor aqueous solubility. Supersaturable formulations are thermodynamically stable formulations which could induce a supersaturated concentration in aqueous environment of gastrointestinal tract. These supersaturable formulations work on spring and parachute mechanism (Warren et al., Citation2010; Bevernage et al., Citation2013). The most common ways to initiate supersaturation is through salts which will rapidly dissolve amorphous solids, co-solvents and self-emulsifying formulations. All these formulations are referred to as springs. The spring brings the dissolved drug concentrations above the saturate (equilibrium) solubility. The disadvantage of such formulations is that supersaturated formulations induce precipitation when administered in vivo. Therefore, supersaturable formulations should be optimized to minimize variability in performance. A formulation component which hinders nucleation or crystal growth acts a parachute to stabilize the metastable supersaturated formulations for sufficient time period for absorption to take place. Parachute slowly settles down concentration to the saturation solubility (). The generation of a supersaturated state and subsequent inhibition of precipitation have been referred to as a “spring and parachute” approach. Excipients which can affect supersaturation include cellulosic polymer (HPMC, HPC and MC) and other rheological polymer (PVP), surfactants (Tweens, Cremophor and TPGS) and cyclodextrins (Mathews & Sugano, Citation2010). Celluloses are the most commonly used precipitation inhibitors.
Figure 2 Spring with Parachute mechanism.
Supersaturable SMEDDS
Supersaturable SMEDDS are designed to minimize precipitation of drug from SMEDDS in the gastrointestinal tract. Supersaturable SMEDDS are thermodynamically stable formulations which contain a reduced amount of surfactant and a polymeric precipitation inhibitor. SMEDDS prevent precipitation of drug by generating and maintaining the supersaturated state in vivo following dilution with water. Supersaturable formulations differ from the supersaturated formulations, as the latter formulations are not thermodynamically stable and drugs in supersaturated formulations can crystallize on storage. Therefore, the physical stability of supersaturated formulations is fundamentally challenging and this limits their practical utility (Gao & Morozowich, Citation2006; Beverange et al., Citation2013).
Polymeric precipitation inhibitors employed in supersaturable SMEDDS formulation are mainly water soluble cellulosic polymers like HPMC, PVP, Methyl cellulose, HPMC phthalate, sodium CMC which can sustain the supersaturated state by preventing the precipitation of drug (Xu & Dai, Citation2013). The precipitation inhibiting capacity of the three hydrophilic polymers has been found to be in the order PVP K17 > PEG 4000 > HPMC. PVP K17 (0.5%) has been found to efficiently retard precipitation and maintain a higher indirubin concentration for approximately 2 h or longer (Chen et al., Citation2012). Among 41 materials evaluated, hydroxypropylmethylcellulose acetate succinate (HPMCAS) was the most effective polymer in maintaining drug supersaturation. The other advantage of supersaturable formulation approach is the reduced amount of surfactant in formulation, thereby achieving an improved toxicity/safety profile with supersaturated SMEDDS formulation.
Encapsulation in HPMC capsule or gelatin capsule
Majority of the marketed SMEDDS formulations are available as soft gelatin capsule. However, gelatin capsules are associated with few drawbacks. Gelatin cross-linking, crystallization of solubilized components, deterioration of mechanical strength of shell, migration of solutes between fill and shell components, transmissible spongiform encephalopathy (TSE) and consumer preference/religion are the few issues associated with gelatin capsule (Sirois, Citation2007).
Gelatin cross-linking
Unpredictable dissolution and rupture of the softgel shell may become apparent upon aging, when capsules are exposed to extreme physical conditions, such as heat (Yannas & Tobolsky, Citation1967; Welz & Ofner, Citation1992) high temperature and humidity, UV radiation, gamma-radiation (Bessho et al., Citation2007), rapid drying (Reich, Citation1995) and exposure to chemical substances such as aldehydes, ketones, imines and carbodiimides (Sheehan & Hlavka, Citation1957; Davis & Tabor, Citation1963; Digenis et al., Citation1994; Tomihata & Ikada, Citation1996; Bottom et al, Citation1997; Gold et al., Citation1998; Fan & Dash, Citation2001; Liang et al., Citation2004). These problems are attributed to cross-linking of gelatin (pellicle formation) which causes the gelatin shell to become swollen, tough, rubbery and insoluble in water. Chemically, aldehydes are known to form methylene bonds between two amino groups on adjacent gelatin chains or within the same chain. The aldehyde and other carbonyl impurities in softgels may originate from the auto-oxidation of materials containing polyoxyethylene moieties in their structures, e.g. polyethylene glycols, methoxypolyethylene glycols, polyoxyethylene fatty acid esters, polyoxyethylene sorbitan fatty acid esters, polyoxyl 40 hydrogenated castor oil (Azaz et al., Citation1973; Hamburger et al, Citation1975; McGinity et al, Citation1975; Donbrow et al, Citation1978; Johnson & Taylor, Citation1984; Bindra et al., Citation1994). The rate of cross-linking in gelatin by aldehydes is strongly influenced by humidity with maximum cross-linking occurring around 60–70% humidity (Albert et al., Citation1991; Roy et al., Citation2001).
The cross-linking of gelatin in softgel shell can be reduced by using excipients with low aldehyde content in the fill formulation. Crosslinking of gelatin can also be reduced using excipients containing abundant free amino groups (e.g. glycine, lysine) in the shell formulation which can compete with amino groups present in gelatin chain for available aldehydes originating from components used in softgel (i.e. aldehyde scavengers) (Gullapalli, Citation2010). Masking the number of amino groups available along the molecular chain of the gelatin through covalent bonds with suitable masking agents is another approach to reduce gelatin crosslinking. Succinic acid is often used to mask the amino groups present in gelatin chain through covalent bonds (succinization). However, a disadvantage of succination approach is that gelatin shell prepared using succinated gelatin is usually highly permeable to volatile solvents (e.g. ethyl alcohol) and migratable ingredients (e.g. propylene glycol). Incorporation of both amino acid (e.g. glycine) and carboxylic acid (e.g. citric acid) into powder fill of hydrochlorothiazide hard gelatin capsule was found to provide a reduction in the cross-linking of gelatin (Adesunloye & Stach, Citation1998).
Crystallization of solubilized components
High initial water content of the capsule shell formulation at the time of encapsulation (30%), causes migration of water from shell into fill (content) or vice versa, during drying of water and subsequent equilibrium processes (Seager, Citation1985; Brox, Citation1988a; Mullin, Citation2001). The rate and extent of migration is influenced by the composition of capsule shell formulation (e.g. type and concentration of plasticizer, presence of additives) and composition of filled formulation (Serajuddin et al., Citation1986). Propylene glycol, having low viscosity and high plasticizing efficacy for gelatin, requires less water during gel mass production as compared to glycerin or sorbitol, as the plasticizer (Serajuddin et al., Citation1986; Roy et al., Citation2001). Gross crystallization of a poor water soluble test compound in the softgel occurred when the compound was encapsulated as PEG-400-based solution. However, crystallization of compound was not observed in softgel, when test compound was encapsulated as Gelucire 44/14-PEG-400 (6:1) based solution (Serajuddin et al., Citation1986). Water migration from capsule shell into lipid content, even in small amounts, as compared to polyethylene glycol-based content, may have profound consequences on the solubility of compounds in lipid vehicles (Cao et al., Citation2004; Land et al., Citation2005; Rane & Anderson, Citation2008). The presence of water in a lipid vehicle may alter the solubility of a compound. The presence of even ultra low amount of water in the lipids is sufficient to induce hydrate formation and reduce the lipid solubility of compounds, such as anhydrous testosterone (Land et al., Citation2005).
Deterioration of mechanical strength of gelatin capsule shell
Polyethylene glycols of lower molecular weight (400D), used as fill vehicles, have a higher affinity for water and glycerin used in shell formulation. This causes migration of the shell components into polyethylene glycol fill. The migration of water from shell into the polyethylene glycol fill may be reduced by using higher molecular weight polyethylene glycol (substituting PEG 600 for PEG 400) which has lower hygroscopicity. Migration of a plasticizer from shell into fill in a softgel could result in reduced elasticity (flexibility) and increased brittleness of the shell shortly after production or storage, especially when exposed to cold temperatures (Brox, Citation1988b; Shah et al., Citation1992). The elasticity of the shell in such case could be markedly improved by inclusion of small amount of glycerin in polyethylene glycol fill.
Migration of solutes between capsule content and gelatin shell components
Softgels, having dynamic nature, could potentially give way to considerable migration (partitioning) of solutes between shell and encapsulated fill. Ethyl alcohol, commonly used co-solvent in SEDDS and SMEDDS formulation, has been shown to diffuse readily through conventional softgel shell. This occurs at a rate that most of the ethyl alcohol would disappear from the fill by the end of drying processes (Moreton & Armstrong, Citation1995, Citation1998). Loss of ethyl alcohol from the encapsulated fill could potentially result in precipitation of dissolved compound (Hauer et al., Citation1994). Loss of volatile component from fill formulation could be minimized by packaging softgel product in a tight packaging material, such as an aluminum–aluminum blister. A variety of non-volatile co-solvents have been investigated to substitute ethyl alcohol in the preparation of SEDDS and SMEDDS for softgel encapsulation, including propylene glycol, diethylene glycol monoethyl ether (Transcutol), tetrahydrofurfuryl alcohol polyethylene glycol (Glycofurol; Hauer et al., Citation1994, Citation2007).
These problems drive the market requirement to find a suitable substitute for gelatin. The current alternative material of choice, to animal gelatin capsules are those prepared from HPMC. The HPMC capsule shell has been explored as an alternative approach for encapsulating supersaturable SMEDDS formulation (Ku et al., Citation2010). The SEDDS or SMEDDS formulation of a drug filled in a HPMC capsule essentially showed the same drug concentration time profile as the SEDDS formulation containing suspended HPMC powder filled in gelatin capsules. In both cases, where HPMC was present either as suspended powder in SEDDS liquid or as HPMC provided by the capsule shell, the drug concentration was found to be maintained at a level about five fold higher than that of a SEDDS liquid alone in a hard gelatin capsule (Gao & Morozowich, Citation2006). This can be explained suitably with the spring with parachute mechanism ().
SMEDDS in HPMC capsule shell
Stability of powdered meloxicam SEDDS formulation on storage in a gelatin capsule and HPMC capsules was evaluated to study the effect of water migration from the content into the capsule shells. Capsules softened to a different extent as a function of fill material, with HPMC capsules showing greater resistance to water migration. Formulations with low water and propylene glycol content might have less impact on shells (Agarwal et al., Citation2012).
Handling and storage issues with liquid SMEDDS
SMEDDS are normally viscous liquids that are administered in hard or soft gelatin capsule. Lipid formulation may leach into and interact with the capsule shells. This may cause brittleness or softness of the capsule shell, leakage of the content and precipitation of the drug or excipients, especially at lower temperatures. To address these problems, several attempts have been made to transform liquid SMEDDS into solid SMEDDS dosage forms (Tang et al., Citation2008). These approaches combine the advantages of SMEDDS with those of solid dosage forms while overcoming the disadvantages of liquid SMEDDS (Nekkanti et al., Citation2010). Formulating solid SMEDDS combine the advantages of SMEDDS (i.e. enhanced solubility and bioavailability) with those of solid dosage forms (e.g. low production cost, convenience of process control, high stability and reproducibility, better patient compliance). Thus, the presentation of liquid SMEDDS in a solid dosage form provides a more stable and robust dosage form with lower manufacturing costs (Milovic & Djuris, Citation2012; Mu et al., Citation2013).
Dosage forms of Solid SMEDDS
Dry emulsions. Dry emulsions are powdered solid dosage forms which spontaneously emulsify with the addition of water. Dry emulsions could be obtained by rotary evaporation, freeze drying and spray drying. The technique of spray drying is more frequently used in preparation of dry emulsions. The o/w emulsion is formulated and then spray-dried to remove the aqueous phase (Christensen et al., Citation2001).
Capsules. Solid SMEDDS prepared by various techniques such as spray drying, adsorption to solid carriers etc. can be filled into capsule shells. This prevents physical incompatibility of liquid SMEDDS with the capsule shell (Janga et al., Citation2012; Kallakunta et al., Citation2012).
Tablets. Liquid SMEDDS could be adsorbed on to porous carriers in different proportions. The surface adsorbed liquid SMEDDS is then mixed with other suitable excipients. The mixture is then compressed using compression machine. Eutectic-based self-emulsifying tablets inhibited irreversible precipitation of the drug within the formulation (Nazzal & Khan, Citation2006).
Pellets. Pellets are convenient multiple unit dosage forms, which are made by extrusion/spheronization technique (Bansal et al., Citation2008; Wang et al., Citation2010; Hu et al., Citation2012).
Techniques employed to produce solid SMEDDS
A. Encapsulation of liquid and semisolid SMEDDS. Liquid SMEDDS can be simply filled into the capsules and sealed using a microspray or a banding process. For a semisolid SMEDDS, encapsulation is a four step process: (1) heating the semisolid excipients to at least 20 °C above its melting point; (2) adding the drug in the molten mixture while stirring; (3) filling the drug loaded molten mixture into the capsule shell; and (4) cooling the product to room temperature. Encapsulation of SMEDDS in capsules is suitable for low dose highly potent drugs and allows high drug incorporation (Tang et al., Citation2008).
B. Spray drying. In this method, the liquid SMEDDS is mixed with a solid carrier in a suitable solvent. The solvent is then atomized into a spray of fine droplets. These droplets are introduced into a drying chamber, where the solvent is evaporated, forming dry particles under a controlled temperature and airflow conditions (Yi et al., Citation2008).
C. Adsorption onto solid carriers: The liquid SMEDDS could be adsorbed onto free flowing powders that possess very large surface area and are capable of adsorbing high quantities of oil material. The adsorbents are capable of adsorbing liquid SEDDS/ SMEDDS up to 70% w/w of their own weight (Ito et al., Citation2005). The commonly used adsorbents are enlisted in . Grades of Neusilin, Neusilin US2 and Neusilin UFL 2, along with Florite R have been reported to have good oil adsorbing capacity (Kallakunta et al., Citation2012). Compressibility index (CI) of Florite R is poor, although it has good flow property. Thus, Florite R can be a suitable adsorbent for capsule filling. However, due to poor CI it cannot be preferred for compression into a tablet dosage form. On the other hand, Neusilin US2 has good flow property and better oil adsorbing capacity along with good CI, thus making it an ideal adsorbent for capsule filling as well as tablet dosage form (Hentzschel et al., Citation2012; Kang et al., Citation2012).
D. Melt granulation. Melt granulation is a process in which powder agglomeration is obtained through the addition of a binder that melts or softens at relatively low temperatures. As a one step operation, melt granulation offers several advantages as compared with conventional wet granulation, since the liquid addition and the subsequent drying phase are omitted. The variables to be controlled in this process are impeller speed, mixing time, binder particle size and the viscosity of the binder (Tang et al., Citation2008).
E. Extrusion spheronization. In this method, the liquid SMEDDS are first mixed with an extrusion aid followed by addition of water until the wet mass is suitable for extrusion. The extruded mass is then spheronized to form uniform pellets. The pellets are then dried and size separated (Wang et al., Citation2010; Hu et al., Citation2012).
Table 2 List of commonly used adsorbents used in solid SMEDDS formulation.
Various techniques employed to produce solid SMEDDS depicting maximum lipid exposure along with maximum drug loading are enlisted in (Jannin et al., Citation2008).
Table 3 Different techniques to produce solid SMEDDS formulation.
Limited lymphatic uptake of SMEDDS
Intestinal lymphatic transport has been found to be an absorptive pathway for oral administration of lipophilic drugs. Lipophilic drugs diffuse across the intestinal enterocyte and associate with secretable enterocyte lipoprotein chylomicron. Lymph is a mixture of fluid and protein that has been filtered and extracted/squeezed out of the blood (i.e. blood plasma). The ducts of the lymphatic systems interconnect the lymph organs including the bone marrow, lymph nodes, spleen, as well as the thymus gland (Alexandera et al., Citation2010; Yanez et al., Citation2011). Lymphatic capillaries play a vital role in the particulate absorption and their lymphatic uptake. The walls of capillary lymphatics are made up of a single layer of non-fenestrated endothelial cells that are extensively gapped and overlapped, forming numerous clefts and pores (Charman et al., Citation1986; Porter & Charman, Citation2001). These pores allow passage of macromolecules into the capillary lumen when interstitial pressure exceeds the intraluminal lymphatic pressure. Factors controlling particulate uptake into the lymphatics include size, composition, dose, surface charge, molecular weight and particle size of drug. For example, an optimum range for lymphatic uptake of subcutaneously injected particles is 10–80 nm while particle greater than 100 nm in diameter will remain largely at the injection site and smaller particles are absorbed by the capillary network that drains into the systemic circulation (Oussoren et al., Citation1997; Oussoren & Storm, Citation2001). Small neutral liposomes ( ≥ 40 nm) injected subcutaneously, results in more than 60–70% of the liposomes cleared from the injection site by 24 h, with only 30–40% of the injected dose remaining at the site of injection. However, liposomes larger than 500 nm have 60–80% remaining at the injection site (Oussoren & Storm, Citation2001). Thus, nanocarriers administered subcutaneously have great potential to deliver anticancer drugs to lymphatic metastases.
The carrier systems for targeted lymphatic delivery are chylomicrons which are the lowest density lipoproteins, primarily composed of triacylglycerols. Chylomicron associated drug is secreted from enterocyte into lymphatic circulation rather than portal circulation and compounds in intestinal lymph gain accesses to systemic circulation at the junction of left internal jugular and subclavian veins. Therefore, drugs that are transported lymphatically avoids first pass metabolism (Charman & Stella, Citation1986). This results in an increase in lymphatic drug concentration which can be the site of therapeutic drug action. On the other hand, hepatic portal system starts from alimentary canal and terminates into liver. Drugs transported through hepatic portal system reaches liver, undergoes first pass metabolism before reaching systemic circulation. Lymphatic uptake of SMEDDS formulation is depicted in .
Figure 3 Lymphatic uptake of SMEDDs.
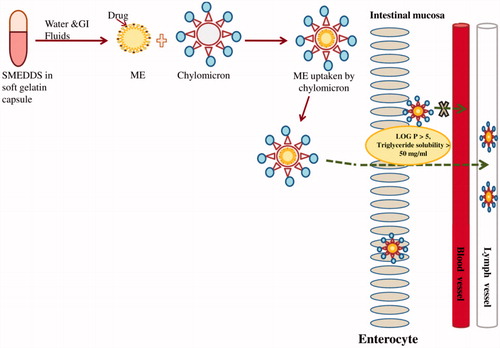
Intestinal lymphatic transport has been found to be more effective for immunomodulatory and chemotherapeutic drugs. This is because lymphatic system is the primary route for metastasis of solid tumors and transport pathway for T and B lymphocytes. The development, sequestration, and spread of the human immunodeficiency virus (HIV), hepatitis B and C virus, morbillivirus and severe acute respiratory syndrome (SARS) associated coronavirus are associated with lymphocytes present in the lymph and lymphoid tissue. This parallel and potentially alternative absorptive pathway reduces first pass metabolism and increases lymphatic drug exposure. The normal lymph flow in 24 h is 2–4 liter (Barrett et al., Citation2009). The blood flow versus lymph flow is estimated to be 500:1, thus in the absence of a targeting strategy most drugs will tend to be diverted preferentially towards the blood system (Driscoll, Citation2002).
On oral administration of different types of triglycerides [LCT (long chain triglycerides), MCT (medium chain triglycerides) and SCT (short chain triglycerides)] to the lymph cannulated fasted rats, the amount of LCT present in the lymph was found to be 3 to 9 fold higher as compared to that of MCT and SCT. This is thought to be due to the stimulatory effect of exogenously administered LCT on the secretion and lymphatic transport of endogenous LCT. The effect is hypothesized to diminish with a decrease in the chain length of the exogenously administered triglyceride, i.e. LCT > MCT > SCT (Caliph et al., Citation2000; Thomas et al., Citation2012). Relatively, even small quantity of LCT is thought to be capable of stimulating the gall bladder contraction and thereby elevating levels of intestinal bile salts, phospholipids and cholesterol (Dahan & Hoffman, Citation2007). The extent of this stimulatory response appears to increase as the amount of LCT administered increases. In contrast, administration of similar quantities of MCT has been found to have relatively limited effects on the gallbladder contraction and do not stimulate appreciable increase in the intestinal concentration of biliary derived lipids. Thus, fatty acids with medium chain length are transported primarily by the portal blood, whereas ones with longer chain length are incorporated primarily into the chylomicrons and transported in to the lymph.
The globule size of microemulsion formed from LCT SMEDDS tends to be of larger size as compared to MCT SMEDDS. Thus, a combination of both LCT and MCT would produce a desired size range with higher lymphatic uptake. For lymphatic uptake of a drug, it should have log P > 5 and long chain triglyceride solubility >50 mg/ml (Charman et al., Citation1986). However, amount of drug transported into lymphatics is variable (Trevaskis et al., Citation2008).
Significant lymphatic transport (43.7% of the dose) of the poorly lipid soluble (∼1 mg/ml) hydrochloride salt (HCl) of halofantrine (Hf) i.e. Hf-HCl was observed following oral post-prandial administration to dogs (Khoo et al., Citation1998). High level of lymphatic transport of Hf-HCl was due to conversion of Hf-HCl in the intestinal lumen during lipolysis to the more lipophilic free base which then gets associated with chylomicron. However, combination of a high log P and high triglyceride solubility does not always guarantee significant lymphatic transport. Penclomedine, an experimental cytotoxic agent with a log P of 5.48 and a triglyceride solubility of 175 mg/ml has poor transportation in the intestinal lymph (∼3% of the dose; Myers & Stella, Citation1992). Similarly, very low levels (<1% dose) of lymphatic transport using CI-976, a lipophilic lipid regulator with a log P of 5.83 and a triglyceride solubility greater than 100 mg/ml was observed (Hauss et al., Citation1994). It can be observed that the lipophilicity and triglyceride solubility correlation with lymphatic transport is not completely adequate and that a more adequate predictive model is required. However, occurrence of lymphatic transport even to extent of less than 1% of the administered dose can significantly affect the cumulative plasma exposure.
Evaluation of lymphatic animal model
Animal models are employed to study lymphatic drug transport by cannulation of the intestinal lymphatic duct for direct measurement of drug. The most commonly used animal model is rat but other large species such as dogs, pigs and sheep have also been reported. The unconscious (anesthetized) rat model has been used in various studies with various degrees of modification in the site of cannulation, lymph fistulation, feeding and rehydration, pre- and post-operative procedures and dosing (Edwards et al., Citation2001). Briefly, a triple-cannulation of the carotid artery (systemic blood collection), the mesenteric lymph duct (intestinal lymph collection), and the duodenum (administration of rehydration solution) is performed. However, it can also include a fourth cannula in the jugular vein if intravenous administration is required. The mesenteric lymph cannula allows the collection of all the lymph draining to the duodenum and consequently the total amount of drug transported lymphatically can be calculated (Boyd et al., Citation2004). The systemic blood collected from the carotid artery allows the calculation of systemic plasma exposure that can be interpreted as the absorption of drug via the portal blood. This method or model is advantageous because it allows the assessment of portal blood absorption contribution to bioavailability by comparing the plasma exposure of orally administered drug to rats that are lymph duct cannulated rats with the plasma exposure of intravenously administered drug rats that are not lymph duct cannulated rats. However, as expected with any animal model that employs cannulation, the most common complication can be cannula occlusion (Edwards et al., Citation2001; Trevaskis et al., Citation2008). This makes, the method tedious and difficult to use.
Recently, an indirect pharmacological approach has been described, utilizing intestinal chylomicron flow inhibitors; Pluronic-L81 and colchicines. In this approach, systemic drug exposure is compared in the presence and absence of co-administered Pluronic-L81 or colchicines to indirectly assess the impact of intestinal lymphatic drug transport in the oral bioavailability. This approach is advantageous because it does not require a lymph duct cannulation. However, the impact of blocking chylomicron flow in lipid processing and synthesis has to be fully characterized. For instance, Pluronic-L81 lowers plasma VLDL and low-density lipoprotein cholesterol and reduces lipid and apoprotein secretion (Dahan & Hoffman, Citation2005). Hyaluronic acid, an anionic non-sulfated glycosaminoglycan, is a biocompatible polymer that follows lymphatic drainage from the interstitial spaces. A cisplatin hyaluronic acid conjugate was synthesized for intra-lymphatic delivery of the drug (Cai et al., Citation2008). Various contrast agents including nanoparticles have been developed to improve contrast in MRI imaging of lymphatic system these include, gadolinium based agents such as gadolinium-diethylenetriamine penta-acetic acid (Gd-DTPA), iron oxide nanoparticles, liposomes and dendrimers containing Gd(III)/or iron oxides (Waters & Wickline, Citation2008; Hasebroock & Serkova, Citation2009; Nune et al., Citation2011). However, employing cannulae is still the most common approach in the small and large animal models.
Lack of effective in vitro tests
Majority of SMEDDS formulations have not been able to enter the market is due to lack of effective in vitro tests that are representative of actual in vivo performance. The performance parameters of a lipid-based system vary quite distinctly between different physiological environments as the performance of SMEDDS is significantly dependent on in situ factors (Porter & Charman, Citation2001). In some circumstances, in vitro, dissolution can be used as surrogate indicator of the likely in vivo dissolution profile and can act as a tool to predict extent of absorption, where dissolution is the rate limiting step (Dressman et al., Citation1998; Horter & Dressman, Citation2001). The simple aqueous media used to assess the dissolution profile of poor water soluble drugs is often limited by the low intrinsic aqueous solubility of the drug and is therefore difficulty in maintaining sink conditions. Problems of sink condition can be overcome, especially for dissolution tests by adding surfactants, co-solvents in the dissolution media (Park & Choi, Citation2006; Delalonde & Ruiz, Citation2008; Siepmann & Siepmann, Citation2013). To improve the accuracy of in vivo dissolution prediction using in vitro dissolution, defined dissolution media that are more accurately reflective of the solubilization process in vivo needs to be developed. The components of these various dissolution media have been modeled primarily on the likely levels of endogenous bile salts and phospholipids in the fasted and postprandial GI tract. For non-ionizable drugs, good correlation was found between the difference in dissolution profiles in simulated fasted-state intestinal media and simulated fed-state media and the difference in plasma profiles obtained after fasted and postprandial administration (Dressman & Reppas, Citation2000; Nicolaides et al., Citation2001). For compounds with appreciable ionization over the likely physiological range, the situation is complicated by the impact of both ionization and solubilization on solubility (Dressman & Reppas, Citation2000).
Assessment of lipid based formulations using in vitro lipolysis
The design of SEDDS/SMEDDS formulation has prime focus on optimizing the solubility of the drug in formulation, on in vitro emulsification efficiency and particle size of the dispersion obtained on dilution in aqueous media. In recent years, in vitro dispersion tests and in vitro lipid digestion models, reflective of the gastrointestinal environment have been developed to predict the in vivo performance of poorly water soluble drugs (Venkatesh et al., Citation2010). In traditional dissolution testing, dissolution of drug from solid state into simple and biorelevant media is measured. However, assessment of Lipid-based formulations should be based on evaluation of the rate and extent of drug precipitation with respect to time rather than solubilization. This is because the drug is already present in solubilized state in the formulation (Porter et al., Citation2007, Citation2008).
Lipid digestion model is one such model that can be used for predicting the efficiency of SEDDS/SMEDDS formulation. In this method, lipid based formulations are introduced to a vessel contains digestive buffers and bile salts. Lipid digestion is initiated by the addition of pancreatic lipase. The onset of lipid digestion results in the liberation of fatty acids which in turn causes transient decrease in pH. The drop in pH is quantified by a pH electrode that is coupled to a pH controller. Desired pH is maintained by the addition of equimolar quantity of sodium hydroxide, from autoburette, to titrate the liberated fatty acids. This enables the pH sensitive digestion process to continue and facilitates indirect quantification of the extent of digestion process. Extent of lipid digestion is determined via quantification of the rate of sodium hydroxide addition and assumption of stoichiometric reaction between fatty acid and sodium hydroxide. Throughout digestion process, samples are taken and ultra centrifuged to separate the digested products into a poorly dispersed oil phase, highly dispersed aqueous phase and a precipitated pellet phase (Porter et al., Citation2007; Dahan & Hoffman, Citation2008; Fatouros & Mullertz, Citation2008; Porter et al., Citation2008). Quantification of mass of drug in highly dispersed aqueous phase is an indication of solubilized drug in aqueous phase which do not precipitate in vivo. This could be good method to rank in vivo performance of a series of lipophilic formulations.
The distribution and solubilization pattern of dexamethasone and griseofulvin was investigated across the different phases of the in vitro lipolysis model resulting from LCT, MCT and SCT-based lipid systems, followed by in vivo evaluation of these formulations (Dahan & Hoffman, Citation2007). They concluded that MCT lipid led to higher amounts of solubilized drug in the aqueous phase (>95%), while the LCT lipid resulted in a smaller amount of drug solubilized in aqueous phase and SCT exhibited the lowest.
Dynamic gastric model (DGM)
DGM is the first dynamic in vitro model of human stomach developed by Institute of Food Research (Norwich, UK). This model is able to replicate the human digestive functions of transforming the bolus into chyme. The DGM is composed of a main body and antrum. The DGM replicates physiological condition of gastric secretion, general pulsatile contractions etc. of stomach. The gastric secretions present the same composition of the in vivo secretions, and the enzymatic digestion is accomplished by gastric lipase and pepsin. In analogy to human stomach, the ingested material is subjected to acid, enzymatic and mechanical processing. At predetermined time intervals, samples are collected for droplet size analysis (Mercuri et al., Citation2011; Vardakou et al., Citation2011).
Oxidation and polymorphism of the lipids used in formulating SEDDS/SMEDDS
Oxidation of lipids
Lipid excipients containing unsaturated fatty acids and its derivatives are prone to lipid oxidation. The rate of oxidative degradation is proportional to the degree of unsaturation in the fatty acid molecule. Polyethylene glycol and materials containing polyoxyethylene moieties in their structures are known to undergo auto-oxidation in presence of oxygen and produce reactive organic peroxides and hydrogen peroxides (Ha et al., Citation2002; Kumar & Kalonia, Citation2006; Li et al., Citation2007; Wasylaschuk et al., Citation2007). Lipid oxidation is catalyzed by impurities including peroxides, metal ions and photochemical sensitizers such as chlorophyll and riboflavin, which interact with light to produce triplet or singlet oxygen. These highly reactive oxygen species catalyze the peroxidation of many types of lipids resulting in subsequent degradation to alcohols, furans, aldehydes, ketones and acids, which give oxidized or rancid lipids (Hauser & Poupart, Citation1992). The organic peroxides are further degraded to produce short chain carboxylic acids and aldehydes. It has been postulated that polyoxyethylene moieties undergo oxidative decomposition at high temperatures in the presence of water to ethylene glycol, which may then oxidize further to formaldehyde (Chafetz et al, Citation1984; Frontini & Mielck, Citation1995). The amount of these reactive peroxides, carboxylic acids and aldehydes present in polyethylene glycol or related material are dependent upon molecular weight (higher is molecular weight of polymer lower is its concentration) and age of polymer (older is the polymer greater is its concentration) and extent of its exposure to air during its storage (Barrio et al., Citation2006; Hamburger et al., Citation1975; Li et al., Citation2007). The reactive products generated from auto-oxidation of polyoxyethylene polymers have been implicated in degradation of several compounds formulated in polyethylene glycols (McGinity et al., Citation1975; Johnson & Taylor, Citation1984; Bindra et al., Citation1994; Frontini & Mielck, Citation1995). The carboxylic acids so-formed during auto-oxidation reaction could lower apparent pH of an aqueous polyethylene glycol solution. The extent of decrease in pH is dependent on how much auto-oxidation of polyethylene glycol have taken place during its handling and storage (Johnson & Taylor, Citation1984).The low pH degrades the compounds that are susceptible to hydrolytic degradation under acidic conditions (Bindra et al., Citation1994). On the other hand, reactive aldehydes can cross-link compounds containing amino groups in their structures through methylene groups (Chaw et al., Citation1980; Huang et al., Citation1992). The minor impurities present as a result from amide hydrolysis of asparagines and glutamine residues during acid or alkaline hydrolysis of collagen could potentially induce degradation of encapsulated compounds (Patel et al., Citation1997).
The degradation of encapsulated compounds can be minimized through the use of gelatin with reduced ammonia content in a gelatin shell. This also, requires inclusion of Lipid soluble antioxidant in capsule formulation [e.g. tertiary-butyl hydroquinone (TBHQ), 250–500 ppm, or propyl gallate, 25–500 ppm; Bowtle, Citation2007]. Addition of free radical quenching agents such as α-tocopherol, β-carotene, ascorbic acid, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), propyl gallate or the addition of metal chelators such as ethylenediaminetetraacetate (EDTA) have also been proved effective for controlling lipid oxidation (Zhu et al., Citation2012). Alternatively, these formulations may be protected from oxidation by the hard gelatin capsule shell. Hard gelatin capsules afford greater protection to lipid formulation from oxidation due the inherent, low permeability of oxygen, which is augmented by the capsule sealing process (Laguerre et al., Citation2007). Further protection against oxidation can be achieved by treating the hard gelatin capsule headspace with nitrogen. This can be easily achieved for hard gelatin capsule by incorporating nitrogen feed device at the capsule closing station. The presence of plasticizers in soft gelatin capsules renders them permeable to oxygen thereby, increasing liability for product oxidation. HPMC capsules, due to the inherent physicochemical properties of the material possess relatively high permeability to oxygen, as well. Protection from oxidation in either soft gelatin capsule or HPMC capsules is therefore limited to the inclusion of an appropriate lipid-soluble antioxidant in the formulation or in shell. For example, evening primrose oil in HPMC capsules can be protected against oxidation by the inclusion of TBHQ at 250 ppm in formulation (Bowtle, Citation2007; Ahn et al., Citation2012; Schaich et al., Citation2013).
Polymorphism
Simple lipids, such as alcohols, fatty acids, methyl and ethyl esters of fatty acids, crystallize predominantly as bilayers. They exhibit polymorphism, which is primarily based on different modes of hydrocarbon chain packing. Other parameters in which these crystal structures differ are the tilt angle of the hydrocarbon chains and the hydrogen bonding within the polar group layer (Tilcock, Citation1986). Lipids with a larger head group area and a small hydrocarbon area have a cone-like geometry, self-assembled into micelles. While lipids, which are cylindrical in shape, having nearly equal head group to hydrocarbon are self-assemble into lipid bilayers. Alternatively, lipids with small head group areas adopt “inverted” lipid phases such as inverted hexagonal (HII phase) phases or cubic phases (Hafez & Culis, Citation2001). For example, dioleoylphosphatidylethanolamine (DOPE) forms a bilayer phase below 10 °C while at elevated temperatures DOPE adopts the HII (inverted hexagonal) phase. Formulation of HII phase is required and hence, promoted by increasing acyl chain unsaturation and increasing temperature. A characteristic feature of the long-chain fatty acids is the complex polymorphism.
X-ray diffraction techniques are usually used to determine the structure of lipid phases and differential scanning calorimetry (DSC) is used to study the transition of one lipid phase to another. This combination of a direct structural technique (X-ray diffraction) with a thermodynamic technique (DSC differential scanning calorimetry) has proved extremely valuable in studies of Lipid polymorphism in both model and biological membranes (Soukharev, Citation2007).
Thermo-softening lipid excipients, particularly macrogol glycerides, require specific process controls in their application, to minimize or control a tendency towards polymorphic changes of the excipient matrix. From the processing perspective, such polymorphic changes may lead to prolongation of solidification time, which can adversely impact capsule filling and handling during production (Sato, Citation2001). From a performance perspective, polymorphic changes can produce non-uniformity of the solidified formulation matrix with resultant changes in the product release-profile, particularly upon aging. In order to control this phenomenon, these excipients should be completely melted by heating them, 20 °C above their nominal melting point and thoroughly mixing them prior to encapsulation. This serves to destroy any preformed crystalline structure and promotes homogeneous dispersion of the multiple components present in these excipients (e.g. the lauroyl macrogolglycerides). The molten material will then behave in a consistent fashion with regard to solidification rate and the final, solidified product will provide a uniform and predictable drug release profile (Bowtle, Citation2007).
Conclusion
Currently, SMEDDS are in focus for developing formulations of poorly water soluble drugs. However, a considerable gap exists between the need for lipid-based drug delivery systems and the available marketed products of SMEDDS. Various attempts have been made to overcome the problems associated with SMEDDS. Most of the marketed SMEDDS formulations are in soft gelatin capsule which manifests handling issues and also increases cost of the product. Thus, formulating solid SMEDDS could minimize handling issues, decrease cost of product and would overcome stability problem of liquid product. Also, these formulations should be designed to work in harmony with the physiological environment. Such attempts will ensure complete exploration or usage of this potential drug delivery system especially for poorly soluble drug.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. S. D. is grateful to NIPER-Ahmedabad and Ministry of Chemicals and Fertilizers, Government of India, for providing fellowship as financial assistance.
References
- Adesunloye TA, Stach PE. (1998). Effect of glycine/citric acid on the dissolution stability of hard gelatin capsules. Drug Dev Ind Pharm 24:494–500
- Agarwal V, Alayoubi A, Siddiqui A, Nazzal S. (2012). Powdered self-emulsified lipid formulations of meloxicam as solid dosage forms for oral administration. Drug Dev Ind Pharm 1–9
- Ahn JH, Kim YP, Kim HS. (2012). Effect of natural antioxidants on the lipid oxidation of microencapsulated seed oil. Food Control 23:528–54
- Albert K, Bayer E, Worsching A, Vogele H. (1991). Investigation of the hardening reaction of gelatin with 13C labeled formaldehyde by solution and solid state 13C NMR spectroscopy. Z Naturforsch 46:385–9
- Alexandera JS, Gantaa VC, Jordanb PA, Witte MH. (2010). Gastrointestinal lymphatics in health and disease. Pathophysiology 17:315–35
- Anton N, Vandamme TF. (2010). Nano-emulsions and micro-emulsions: clarification of the critical differences. Pharm Res 28:978–85
- Amri A, Clanche SL, Thérond P, et al. (2013). Resveratrol self-emulsifying system increases the uptake by endothelial cells and improves protection against oxidative stress-mediated death. Eur J Pharm Biopharm doi: 10.1016/j.ejpb.2013.10.015. [Epub ahead of print]
- Ash M. (2007). Handbook of pharmaceutical additives. New York: Synpase Information Resources, 30–2, 289, 481–2, 714–6
- Avdeef A. (2003). Absorption and drug development solubility, permeability and charge state. New Jersey: Wiley-Interscience, 91–15
- Azaz E, Donbrow M, Hamburger R. (1973). Incompatibility of non-ionic surfactants with oxidisable drugs. Pharm J 211:15
- Bansal T, Mustafa G, Khan ZI, et al. (2008). Solid self-nanoemulsifying delivery systems as a platform technology for formulation of poorly soluble drugs. Crit Rev Ther Drug Carrier Syst 25:63–116
- Barrett K, Brooks H, Boitano S, Barman S. (2009). Blood as a circulatory fluid & the dynamics of blood & lymph flow. Ganong's review of medical physiology. New York: McGraw Hill, 550–2
- Barrio MD, Hu J, Zhou P, Cauchon N. (2006). Simultaneous determination of formic acid and formaldehyde in pharmaceutical excipients using headspace GC/MS. J Pharm Biomed Anal 41:738–43
- Bessho M, Kojima T, Okuda S, Hara M. (2007). Radiation-induced cross-linking of gelatin by using γ-rays: insoluble gelatin hydrogel formation. Bull Chem Soc Japan 80:979–85
- Bevernage J, Brouwers J, Brewster ME, Augustijns P. (2013). Evaluation of gastrointestinal drug supersaturation and precipitation: strategies and issues. Int J Pharm 453:25–35
- Bindra DS, Williams TD, Stella VJ. (1994). Degradation of O6-benzylguanine in aqueous polyethylene glycol 400 (PEG 400) solutions: concerns with formaldehyde in PEG 400. Pharm Res 11:1060–4
- Bottom CB, Clark M, Carstensen JT. (1997). Dissolution testing of soft shell capsules-acetaminophen and nifedipine. J Pharm Sci 86:1057–61
- Boyd M, Risovic V, Jull P, et al. (2004). A stepwise surgical procedure to investigate the lymphatic transport of lipid based oral drug formulations: cannulation of the mesenteric and thoracic lymph ducts within the rat. J Pharmacol Toxicol Method 49:115–20
- Boyd BJ, Nguyen TH, Mullertz A. (2011). Lipids in oral controlled release drug delivery. In: Wilson CG, Crowley PJ, eds. Controlled release in oral drug delivery. New York: Springer, 299–321
- Bowtle W. (2007). Materials, process, and manufacturing considerations for lipid-based hard- capsule formats. In: Hauss DJ, ed. Oral lipid-based formulations enhancing the bioavailability of poorly water-soluble drugs. vol. 170. New York: Informa Healthcare, 79–106
- Brox W. (1988a). Gelatin capsule. US Patent 4,780,316
- Brox W. (1988b). Soft gelatin capsules and methods for their production. US Patent 4,744,988
- Cai S, Xie YM, Bagby TR, et al. (2008). Intralymphatic chemotherapy using a hyaluronan-cisplatin conjugate. J Surg Res 147:247–52
- Caliph SM, Charman WN, Porter CJH. (2000). Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph cannulated and non-cannulated rats. J Pharm Sci 89:1073–84
- Cao Y, Marra M, Anderson BD. (2004). Predictive relationships for the effects of triglyceride ester concentration and water uptake on solubility and partitioning of small molecules into lipid vehicles. J Pharm Sci 93:2768–79
- Chafetz L, Hong W, Tsilifonis DC, et al. (1984). Decrease in the rate of capsule dissolution due to formaldehyde from Polysorbate 80 autooxidation. J Pharm Sci 73:1186–7
- Charman WN, Noguchi T, Stella VJ. (1986). An experimental system designed to study the in situ intestinal lymphatic transport of lipophilic drugs in anesthetized rats. Int J Pharm 33:155–64
- Charman WN, Stella VJ. (1986). Estimating the maximal potentials for intestinal lymphatic transport of lipophilic drug molecules. Int J Pharm 34:175–8
- Chaw YFM, Crane LE, Lange P, Shapiro R. (1980). Isolation and identification of cross-links from formaldehyde-treated nucleic acids. Biochem 19:5525–31
- Chen ZQ, Liu Y, Zhao JH, et al. (2012). Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomed 7:1115–25
- Christensen KL, Pedersen GP, Kristensen, HG. (2001). Preparation of redispersible dry emulsions by spray drying. Int J Pharm 212:187–94
- Clement J. (2009). Intestinal absorption of triacylglycerols. Reprod Nutr Dev 20: 1285–307
- Dahan A, Hoffman A. (2005). Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci 24:381–8
- Dahan A, Hoffman A. (2007). The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex-vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm 67:96–105
- Dahan A, Hoffman A. (2008). Rationalizing the selection of oral lipid based drug delivery system by an in vitro lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release 129:1–10
- Dai WG. (2010). In vitro methods to assess drug precipitation. Int J Pharm 393:1–16
- Davis P, Tabor BE. (1963). Kinetic study of the cross linking of gelatin by formaldehyde and glyoxal. J Polymer Sci 1:799–815
- Delalonde M, Ruiz T. (2008). Dissolution of pharmaceutical tablets: the influence of penetration and drainage of interstitial fluids. Chem Eng Process 47:370–6
- Digenis GA, Gold TB, Shah VP. (1994). Cross-linking of gelatin capsules and its relevance to their in vitro-in vivo performance. J Pharm Sci 83:915–21
- Donbrow M, Azaz E, Pillersdorf A. (1978). Autoxidation of polysorbates. J Pharm Sci 67:1676–81
- Dressman JB, Amidon GL, Reppas C, Shah VP. (1998). Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res 15:11–22
- Dressman JB, Reppas C. (2000). In vitro-in vivo correlations for lipophilic, poorly water-soluble drugs. Eur J Pharm Sci 11:S73–80
- Driscoll CMO. (2002). Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci 15:405–15
- Eccleston GM. (2007). Emulsions and microemulsions. In: Swarbrick J, ed. Encyclopedia of pharmaceutical technology, vol. 3. New York: Informa Healthcare, 1548–65
- Edwards GA, Porter CJH, Caliph SM, et al. (2001). Animal models for the study of intestinal lymphatic drug transport. Adv Drug Deliv Rev 50:45–60
- Fan H, Dash AK. (2001). Effect of cross-linking on the in vitro release kinetics of doxorubicin from gelatin implants. Int J Pharm 213:103–16
- Fatouros DG, Mullertz A. (2008). In vitro lipid digestion models in design of drug delivery systems for enhancing oral bioavailability. Expert Opin Drug Metab Toxicol 4:65–76
- Frontini R, Mielck JB. (1995). Formation of formaldehyde in polyethylene glycol and in poloxamer under stress conditions. Int J Pharm 114:121–3
- Gao P. (2011). Design and development of self-emulsifying lipid formulations for improving oral bioavailability of poorly water- soluble and lipophilic drugs. In: Williams RO, Watts AB, Miller DA, eds. Formulating poorly water soluble drugs. New York: Springer, 243–66
- Gao P, Morozowich W. (2006). Development of supersaturable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin Drug Deliv 3:97–110
- Gao P, Rush BD, Pfund WP. (2003). Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci 92:2386–98
- Gao P, Morozowich W. (2007). Design and development of supersaturable self-emulsifying drug delivery systems for enhancing the gastrointestinal absorption of poorly soluble drugs. In: Hauss DJ, ed. Oral lipid-based formulations enhancing the bioavailability of poorly water-soluble drugs, vol. 170. New York: Informa Healthcare, 303–24
- Gao P, Morozowich W. (2009). Improving the oral absorption of poorly soluble drug using SEDDS and S-SEDDS formulations. In: Qiu Y, Chen Y, Zhang GGZ, eds. Developing solid oral dosage forms theory and practice. New York: Academic Press, 443–9
- Gao P, Shi Y. (2012). Characterization of supersaturable formulations for improved absorption of poorly soluble drug. AAPS Pharm Sci Tech 14:703–13
- Gold TB, Buice RG, Lodder RA, Digenis GA. (1998). Detection of formaldehyde-induced crosslinking in soft elastic gelatin capsules using near-infrared spectrophotometry. Pharm Dev Technol 3:209–14
- Gullapalli PR. (2010). Soft gelatin capsules (Softgels). J Pharm Sci 99:4107–48
- Ha E, Wang W, Wang YJ. (2002). Peroxide formation in polysorbate 80 and protein stability. J Pharm Sci 91:2252–64
- Hafez IM, Cullis PR. (2001). Roles of lipid polymorphism in intracellular delivery. Adv Drug Deliv Rev 47:139–48
- Hamburger R, Azaz E, Donbrow M. (1975). Autoxidation of polyoxyethylenic non-ionic surfactants and of polyethylene glycols. Pharm Acta Helv 50:10–7
- Hasebroock KM, Serkova NJ. (2009) Toxicity of MRI and CT contrast agents. Expert Opin Drug Met 5:403–16
- Hauer B, Meinzer A, Posanski U, Richter F. (1994). Pharmaceutical compositions comprising cyclosporins. US Patent 5,342,625
- Hauer B, Meinzer A, Posanski U, Richter F. (2007). Pharmaceutical compositions comprising cyclosporins. US Patent 7,235,248
- Hauser H, Poupart G. (1992). The structure of biological membranes. In: Yeagle PL, ed. The structure of biological membranes. Boca Raton: CRC Press, 3–21
- Hauss DJ. (2007). Oral lipid-based formulations. Adv Drug Deliv Rev 59:667–76
- Hauss DJ, Mehta SC, Radebaugh GW. (1994). Targeted lymphatic transport and modified systemic distribution of CI-976, a lipophilic lipid regulator drug, via a formulation approach. Int J Pharm 108:85–93
- Hentzschel CM, Alnaief M, Smirnova I, et al. (2012). Tabletting properties of silica aerogel and other silicates. Drug Dev Ind Pharm 38:462–7
- Hoener BA, Benet LZ. (2002). Factors influencing drug absorption and drug availability. Drugs Pharm Sci 121:93–117
- Horter D, Dressman JB. (2001). Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev 46:75–87
- Huang Y, Tian R, Hu W, et al. (2012). A novel plug-controlled colon-specific pulsatile capsule with tablet of curcumin-loaded SMEDDS. Carbohydr Polym 92:2218–23
- Hu X, Lin C, Chen D, et al. (2012). Sirolimus solid self-microemulsifying pellets: formulation development, characterization and bioavailability evaluation. Int J Pharm 438:123–33
- Huang H, Solomon MS, Hopkins PB. (1992). Formaldehyde preferentially interstrand cross-links duplex DNA through deoxyadenosine residues at the sequence 50-d (AT). J Am Chem Soc 114:9240–1
- Humberstone AJ, Charman WN. (1997). Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev 25:103–28
- Ito Y, Kusawake T, Ishida M, et al. (2005). Oral solid gentamicin preparation using emulsifier and adsorbent. J Control Release 105:23–31
- Janga KY, Jukanti R, Sunkavalli S, et al. (2012). In-situ absorption and relative bioavailability studies of Zaleplon loaded self-nanoemulsifying powders. J Microencapsul 30:161–72
- Jannin V, Musakhanian J, Marchaud D. (2008). Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev 60:734–46
- Johnson DM, Taylor WF. (1984). Degradation of fenprostalene in Polyethylene glycol 400 solution. J Pharm Sci 73:1414–7
- Kallakunta VR, Bandari S, Jukanti R, Veerareddy PR. (2012). Oral self emulsifying powder of lercanidipine hydrochloride: formulation and evaluation. Powder Technol 221:375–82
- Kang JH, Oh DH, Oh YK, (2012). Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self nanoemulsifying drug delivery system (solid SNEDDS). Eur J Pharm Biopharm 80:289–97
- Kashchiev D, Van Rosmalen GM. (2003). Nucleation in solutions revisited. Crystal Res Technol 38:555–74
- Khan AW, Kotta S, Ansari SH, et al. (2012). Potentials and challenges in self-nanoemulsifying drug delivery systems. Expert Opin Drug Deliv 9:1305–17
- Khoo SM, Humberstone AJ, Porter CJH, et al. (1998). Formulation design and bioavailability assessment of lipidic self emulsifying formulations of halofantrine. Int J Pharm 167:155–64
- Kohli K, Chopra S, Dhar D, et al. (2010). Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today 15:958–65
- Kumar V, Kalonia DS. (2006). Removal of peroxides in polyethylene glycols by vacuum drying: implications in the stability of biotech and pharmaceutical formulations. AAPS PharmSciTech 7:E47–53
- Ku MS, Li W, Dulin W, et al. (2010). Performance qualification of a new hypromellose capsule: Part I. Comparative evaluation of physical, mechanical and processability quality attributes of Vcaps Plus®, QualiV® and gelatin capsules. Int J Pharm 386:30–41
- Laguerre M, Lecomte J, Villeneuve P. (2007). Evaluation of the ability of antioxidants to counteract lipid oxidation: existing methods, new trends and challenges. Prog Lipid Res 46:244–82
- Land LM, Li P, Bummer PM. (2005). The influence of water content of triglyceride oils on the solubility of steroids. Pharm Res 22:784–8
- Lawrence MJ, Warisnoicharoen W. (2006). Recent advances in microemulsions as drug delivery vehicles. In: Torchilin VP, ed. Nanoparticulates as drug carriers. London: Imperial College Press, 125–60
- Li Z, Kozlowski BM, Chang EP. (2007). Analysis of aldehydes in excipients used in liquid/semi-solid formulations by gas chromatography-negative chemical ionization mass spectrometry. J Chromatogr A 1160:299–305
- Liang HC, Chang WH, Liang HF. (2004). Crosslinking structures of gelatin hydrogels crosslinked with genipin or a water-soluble carbodiimide. J Appl Polymer Sci 91:4017–26
- Mahmoud EA, Bendas ER, Mohamed MI. (2009). Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech 10:183–92
- Mathews C, Sugano K. (2010). Supersaturable formulations. Drug Deliv Sys 25–4:371–4
- McGinity JW, Hill JA, La Via AL. (1975). Influence of peroxide impurities in polyethylene glycols on drug stability. J Pharm Sci 64:356–7
- Mercuri A, Passalacqua A, Wickham MJW. (2011). The effect of composition and gastric conditions on the self-emulsification process of ibuprofen-loaded self-emulsifying drug delivery systems: a microscopic and dynamic gastric model study. Pharm Res 28:1540–51
- Milovic M, Djuris J. (2012). Characterization and evaluation of solid self microemulsifying drug delivery systems with porous carriers as systems for improved carbamazepine release. Int J Pharm 34:1–8
- Mohsin K. (2012). Design of lipid based formulations for oral administration for administration of poorly water-soluble drug fenofibrate: effects of digestion. AAPS PharmSciTech 13:637–46
- Mohsin K, Long MA, Pouton CW. (2008). Design of lipid-based formulations for oral administration of poorly water-soluble drugs: precipitation of drug after dispersion of formulations in aqueous solution. J Pharm Sci 98:3582–95
- Moreton RC, Armstrong NA. (1995). Design and use of an apparatus for measuring diffusion through glycerogelatin films. Int J Pharm 122:79–89
- Moreton RC, Armstrong NA. (1998). The effect of film composition on the diffusion of ethanol through soft gelatin films. Int J Pharm 161:123–31
- Müllertz A, Ogbonnaa A, Ren S, Rades T. (2010). New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmcol 62:1622–36
- Mu H, Holm R, Mullertz A. (2013). Lipid-based formulations of oral administration of poorly water-soluble drugs. Int J Pharm 453:215–24
- Mullin JW. (2001). Nucleation. In: Crystallization. 4 ed. London: Butterworth-Heinemann, 181–215
- Myers RA, Stella VJ. (1992). Factors affecting the lymphatic transport of penclomedine (NSC-338720), a lypophilic cytotoxic drug: comparison to DDT and hexachlorobenzene. Int J Pharm 80:511–62
- Nanjwade B, Patel DJ, Udhani R, Manvi F. (2011). Functions of lipids for enhancement of oral bioavailability of poorly water-soluble drugs. Sci Pharm 879:705–27
- Nazzal S, Khan MA. (2006). Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm 315:110–21
- Nekkanti VK, Karatg PK, Prabhu R, Pilai R. (2010). Solid self-microemulsifying formulation of candesartan cilexetil. AAPS PharmSciTech 2:9–17
- Nicolaides E, Symillides M, Dressman JB, Reppas C. (2001). Biorelevant dissolution testing to predict the plasma profile of lipophilic drugs after oral administration. Pharm Res 18:380–8
- Nune SK, Gunda P, Majeti BK, et al. (2011). Advances in lymphatic imaging and drug delivery. Adv Drug Deliv Rev 63:876–85
- Oussoren C, Storm G. (2011). Liposomes to target the lymphatics by subcutaneous administration. Adv Drug Deliv Rev 50:143–56
- Oussoren C, Zuidema J, Crommelin DJA, Storm G. (1997). Lymphatic uptake and biodistribution of liposomes after subcutaneous injection. II. Influence of liposomal size, lipid composition and lipid dose. Biochim Biophys Acta 1328:261–72
- Park SH, Choi KH. (2006). The effects of surfactants on the dissolution profiles of poorly water-soluble acidic drugs. Int J Pharm 321:35–41
- Parmar N, Singla N, Amina S, Kohli K. (2011). Study of co-surfactant effect on nano emulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf B 86:327–38
- Patel K, Kearney AS, Palepu NR. (1997). Investigation of new degradation products arising from the encapsulation of an oilbased suspension formulation of topotecan. Int J Pharm 151:7–13
- Patil P, Patil V, Paradkar A. (2007). Formulation of a self-emulsifying system for oral delivery of simvastatin: in vitro and in vivo evaluation. Acta Pharm 57:111–22
- Plaizier-Vercammen JA, De Nève RE. (1981). Interaction of povidone with aromatic compounds II: evaluation of ionic strength, buffer concentration, temperature, and pH by factorial analysis. J Pharm Sci 70:1252–6
- Porter CJH, Charman WN. (2001). Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev 50:61–80
- Porter CJH, Pouton CW, Culine JF, Charman WN. (2008). Enhancing intestinal drug solubilisation using lipid based drug delivery systems. Adv Drug Deliv Rev 60:673–91
- Porter CJH, Trevaskis NL, Charman WN. (2007). Lipids and lipid based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–8
- Pouton CW. (2000). Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci 11:S93–8
- Pouton CW. (2006). Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci 29:278–87
- Pund S, Borade G, Rasve G. (2013). Improvement of anti-inflammatory and anti-angiogenic activity ofberberine by novel rapid dissolving nanoemulsifying technique. Phytomedicine doi: 10.1016/j.phymed.2013.09.013. [Epub ahead of print]
- Pund S, Shete Y, Jagdale S. (2014). Multivariate analysis of physicochemical characteristics of lipid based nanoemulsifying cilostazol – quality by design. Colloids Surf B Biointerfaces 115:29–36
- Rahman A, Hussain A, Hussain S, et al. (2012). Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/ SMEDDS). Drug Dev Ind Pharm 39:1–19
- Rane SS, Anderson BD. (2008). What determines drug solubility in lipid vehicles: is it predictable? Adv Drug Deliv Rev 60:638–56
- Rao J, McClements DJ. (2012). Food-grade microemulsions and nanoemulsions: role of oil phase composition on formation and stability. Food Hydrocolloids 29:326–34
- Reich VG. (1995). Effect of drying conditions on structure and properties of gelatin capsules: studies with gelatin films. Pharm Ind 57:63–7
- Rodriguez-Hornedo N, Murphy D. (1999). Significance of controlling crystallization mechanisms and kinetics in pharmaceutical systems. J Pharm Sci 88:651–60
- Roy A, Winnike R, Long S, et al. (2001). Development of a soft gelatin capsule formulation of a poorly soluble compound X using a selfemulsifying delivery system. In: Saint-Priest BT, ed. Oral drug absorption: a renaissance. Cedex, France : Gattefosse Corporation, 141–9
- Sander C, Holm P. (2009). Porous magnesium aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation. AAPS PharmSciTech 10:1388–95
- Sato K. (2001). Crystallization behaviour of fats and lipids – a review. Chem Eng Sci 56:2255–65
- Schaich KM, Shahidi F, Zhong Y, Eskin NAM. (2013). Lipid oxidation. In: Eskin NAM, Shahidi F, eds. Biochemistry of foods, vol. 3. New York: Elsevier, 419–78
- Seager H. (1985). Soft gelatin capsules: a solution to many tableting problems. Pharm Technol 9:84–104
- Serajuddin ATM, Sheen PC, Augustine MA. (1986). Water migration from soft gelatin capsule shell to fill material and its effect on drug solubility. J Pharm Sci 75:62–4
- Shah NH, Stiel D, Infeld MH, et al. (1992). Elasticity of soft gelatin capsules containing polyethylene glycol 400-quantitation and resolution. Pharm Technol 3:126–31
- Shahba AAW, Mohsin K, Alanazi FK. (2012). The studies of phase equilibria and efficiency assessment for self-emulsifying lipid-based formulation. AAPS PharmSciTech 13:522–33
- Solans C, Izquierdo P, Nolla J, et al. (2005). Nano-emulsions. Curr Opin Colloid Interface Sci 10:102–10
- Soukharev AR. (2007). Stability of lipid excipients in solid lipid nanoparticles. Adv Drug Deliv Rev 59:411–8
- Sheehan JC, Hlavka JJ. (1957). The cross-linking of gelatin using a water-soluble carbodiimide. J Am Chem Soc 79:4528–9
- Siepmann J, Siepmann F. (2013). Mathematical modeling of drug dissolution. Int J Pharm 453:12–24
- Singh AK, Chaurasiya A, Singh M, et al. (2008). Exemestane loaded self- microemulsifying drug delivery system (SMEDDS) development and optimization. AAPS PharmSciTech 9:628–34
- Singh B, Bandopadhyay S, Kapul R, et al. (2009a). Self-emulsifying drug delivery systems (SEDDS): formulation development, characterization, and applications. Expert Opin Drug Deliv 26:427–1
- Singh SK, Verma PRP, Razdan B. (2009b). Development and characterization of a carvedilol-loaded self-microemulsifying delivery system. Clin Res Regul Aff 26:50–64
- Sirois P. (2007). Feasibility assessment and considerations for scaling initial prototype lipid-based formulations to phase I/II clinical trial batches. In: Hauss DJ, ed. Oral lipid-based formulations enhancing the bioavailability of poorly water-soluble drugs, vol. 170. New York: Informa Healthcare, 63–77
- Taha E, Ghorab D, Zaghloul AA. (2007). Bioavailability assessment of vitamin a self-nanoemulsified drug delivery systems in rats: a comparative study. Med Princ Pract 16:355–9
- Tang B, Cheng G, Gu JC, Xu CH. (2008). Development of solid self emulsifying drug delivery system; preparation techniques and dosage forms. Drug Discov Today 13:606–12
- Tang JL, Sun J, Guihe Z. (2007). Self-emulsifying drug delivery systems: strategy for improving oral delivery of poorly soluble drug. Curr Drug Ther 2:85–93
- Thomas N, Mullertz A, Graf A, Rades T. (2012). Influence of lipid composition and drug load on the in vitro performance of self-nanoemulsifying drug delivery systems. J Phar Sci 101:1721–31
- Tilcock PSC. (1986). Lipid polymorphism. Chem Phys Lipid 40:109–25
- Tomihata K, Ikada Y. (1996). Cross-linking of gelatin with carbodiimides. Tissue Eng 2:307–13
- Trevaskis NL, Charman WN, Porter CJH. (2008). Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev 60:702–16
- Vardakou M, Mercuri A, Naylor TA, et al. (2011). Predicting the human in vivo performance of different oral capsule shell types using a novel in vitro dynamic gastric model. Int J Pharm 419:192–9
- Venkatesh G, Majid MIA, Mansor SM, et al. (2010). In vitro and in vivo evaluation of self-microemulsifying drug delivery system of buparvaquone. Drug Dev Ind Pharm 36:735–43
- Wadhwa J, Nair A, Kumria R. (2012). Emulsion forming drug delivery system for lipophilic drugs. Acta Pol Pharm Drug Res 69:179–91
- Wang Z, Sun J, Wang Y, et al. (2010). Solid self-emulsifying nitrendipine pellets: preparation and in vitro/in vivo evaluation. Int J Pharm 38:1–6
- Warren DB, Benameur H, Porter CJH, Pouton CV. (2010). Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target 18:704–31
- Wasylaschuk WR, Harmon PA, Wagner G, et al. (2007). Evaluation of hydroperoxides in common pharmaceutical excipients. J Pharm Sci 96:106–16
- Waters EA, Wickline SA. (2008). Contrast agents for MRI. Basic Res Cardiol 103:114–21
- Wei Y, Ye X, Shang X, et al. (2012). Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf A Physicochem Eng Aspects 396:22–8
- Welz MM, Ofner CM. (1992). Examination of self-crosslinked gelatin as a hydrogel for controlled release. J Pharm Sci 81:85–90
- Woo JS, Song YK, Hong JY, et al. (2008). Reduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur J Pharm Sci 33:59–65
- Xi J, Chang CK, Meng ZY, et al. (2009). Formulation development and bioavailability evaluation of a self-nanoemulsified drug delivery system of oleanolic acid. AAPS PharmSciTech 10:172–82
- Xu S, Dai, WG. (2013). Drug precipitation inhibitors in supersaturable formulations. Int J Pharm 453:36–43
- Yanez JA, Wang S, Knemeyer IW, Wirth MA. (2011). Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev 63:923–42
- Yang S, Gursoy N, Lambert G, Benita S. (2004). Enhanced oral absorption of paclitaxel in a novel self- microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitors. Pharm Res 21:261–70
- Yannas IV, Tobolsky AV. (1967). Cross-linking of gelatine by dehydration. Nature 215:9–10
- Yao G, Li Y. (2010). Preparation, characterization and evaluation of self-microemulsifying drug delivery systems (SMEDDSs) of Ligusticum chuanxiong oil. Biomed. Prev Nutr 1(1):36–42
- Yi X, Wan J, Xu H, Yang X. (2008). A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur J Pharm Biopharm 70:439–44
- You J, Cui FD, Li QP. (2005). A novel formulation design about water-insoluble oily drug: preparation of zedoary turmeric oil microspheres with self-emulsifying ability and evaluation in rabbits. Int J Pharm 288:315–23
- Zhang P, Liu Y, Xu J. (2008). Preparation and evaluation of self-emulsifying drug delivery system of oridonin. Int J Pharm 355:269–76
- Zhu X, Schaich KM, Chen X, et al. (2012). Target release rate of antioxidants to extend induction period of lipid oxidation. Food Res Int 47:1–5
- Zidan AS, Sammour OA, Hammad MA, et al. (2007). Quality by design: understanding the product variability of a self-nanoemulsified drug delivery system of cyclosporine A. J Pharm Sci 96:2409–23