
Abstract
The objective of this paper is to introduce some strategic guidance to a rational formulation strategy of new molecules as oral dosage forms, based on a sound scientific understanding of factors determining the oral bioavailability. The critical implication of permeability and solubility is discussed along with the efficient dose of the drug. The concept of dose-solubility number is introduced as a tool for chemists to assess the develop-ability of different molecules very early during discovery stage. Based on this understanding, a rational formulation strategy for preclinical and early clinical phases is provided. The technical considerations and limitations of different formulation technologies are discussed and illustrated via concrete examples. This approach has the advantage of streamlining the formulation process in order to avoid delaying the development of new drugs due to formulation related issues.
Introduction
The advent of modern discovery tools such as high throughput screening (HTS) and rational drug design approaches (approaches not based on empirical screening) have facilitated the selection of molecules with increased potency and selectivity against a range of challenging biological targets. However, the widespread use of these tools has favored the selection of lead molecules with higher molecular weight and/or increased lipophilicity. Such changes in physicochemical properties have a negative impact on the develop-ability attributes of lead molecules, with adverse impacts on aqueous solubility and permeability. Essentially, the trend toward increased lipophilicity is generally worsening the aqueous solubility while the trend of increasing the molecular weight is worsening both aqueous solubility and permeability (Lipinski, Citation2000).
This tendency is well reflected in the Biopharmaceutical Classification System (BCS) where molecules are classified according to their aqueous solubility and permeability (Amidon et al., Citation1995). This system was first presented as a regulatory tool for generic bioequivalence waiver. However, several criticisms had been raised in publications about the high-severity of this classification, especially for Class II compounds. In fact, it adopted a relatively conservative approach, particularly in the assessment of when solubility and/or dissolution rate are critical in limiting oral absorption. This is because in the context of protecting patients, it is more important to ensure changes to products that result in altered in vivo performance are correctly classified rather than the misclassification of those that do not. The limitation of its use as a development tool was highlighted by several authors (Butler et al., Citation2010; Rinaki et al. Citation2003).
Nevertheless, using BCS as a comparison tool for the evaluation of permeability and solubility properties, we can observe that there is a significant increase in the number of Class II (poorly soluble molecules) and Class IV (poorly soluble/permeable) molecules in clinical development compared to the distribution seen for marketed drugs (Benet et al., Citation2006).
As solubility and permeability are major constraints to bioavailability, the role of the formulation scientist within the drug development process becomes more and more crucial to correct the intrinsic poor drug-like properties of clinical candidates and improve their bioavailability.
The previously dominant strategy of using a simple drug filled capsule/bottle approach to accelerate timelines for Phase I/First-in-Human (FIH) clinical studies is no longer possible for a large number of new molecules. Indeed, many molecules require so-called “enabling formulations” to achieve a reasonable bioavailability. For some very challenging molecules, the enabling formulation effort should be initiated as early as preclinical studies, in order to achieve an acceptable exposure for pharmacology and/or toxicology studies.
Facing this new situation, pharmaceutical firms adopted diverse strategies to address these formulation challenges. Formulation development is generally started earlier and the “Develop-ability” of molecules is better integrated in the choice of the clinical candidate. The main goal remains to increase the chance of clinical success without increasing the time to market of the new drugs.
Several publications had been released in the past decade describing how the different pharmaceutical companies handled this new challenging situation.
Different approaches, with multiple decision trees, have been presented. These approaches take into consideration the physicochemical characteristics of the New Chemical Entity (NCE), the stage of the project (preclinical or clinical), the route of administration and several other scientific and practical aspects.
Ku (Ku, 2008) suggested a simple formulation strategy for early drug development based on BCS. Butler (Butler et al., Citation2010) presented a Develop-ability Classification System (DCS) linking regions of biopharmaceutical space to formulation strategy. He assessed compounds in regard to their difficulty in formulating to maximum oral absorption. The Class II is devised to IIa (dissolution limited absorption compounds) and to IIb (solubility limited absorption compounds). Class IIa compounds could be formulated by standard technologies with a specific attention to particle size distribution. Class IIb compounds require more challenging formulation technologies such as solid dispersion and lipid formulations.
The objective is to introduce some strategic guidance to a rational formulation strategy of new molecules as oral dosage forms, based on scientific principles and factors determining the oral bioavailability. In addition, the industrial constraints are considered as increasing the productivity of pharmaceutical research is one of the major current challenges of the industry.
Understanding factors determining the bioavailability of oral formulations
According to the Biopharmaceutical Classification System (BCS), the solubility and the permeability are the predominant factors determining the ability of drugs to be absorbed in the Gastrointestinal (GI) tract and thus impacting the oral bioavailability.
Permeability
The tendency towards less permeable molecules could be explained by the drug discovery trend of producing leads with higher molecular weight and/or increased H-bond count (Lipinski, Citation2000). Thus, permeability is the function of inherent determinants of medicinal chemistry process.
Several in vitro models are available to evaluate the permeability of new drug candidates in discovery phase (Buckley et al., 2012). Caco-2 system is very commonly used to assess the permeability of new drugs candidates then comparing the result with different FDA permeability references drugs. For compounds transported via the passive transcellular route, Caco-2 permeability is considered to be a good predictor for human permeability. For compounds transported via paracellular or transporter-mediated process or very insoluble, the Caco-2 permeability tends to underestimate human permeability (Ku, 2006).
The permeability problem could be overcome by using different permeability enhancers but their toxicity is often a limiting factor (Kathryn et al., Citation2008).
If the effective permeability is very low (less than 5% of Metoprolol, FDA high permeability reference) the barrier for GI absorption is too high for oral delivery. Thus, an alternative route of administration may be considered (Sherry, Citation2006).This is especially true for high-dose molecules.
As poor permeability is less likely to be addressed by formulation technologies, this paper does not handle the case where the bioavailability is mainly permeability limited.
Solubility
Numerous technologies were developed in the recent years to overcome the increasing solubility problem of new molecules. Understanding the underlying origin of poor solubility is a key factor to choose the most appropriate one.
The revised General Solubility Equation (GSE) (Ran et al., 2001) not only help predicting the solubility but also can give very useful information to determine factors controlling the solubility:
where log Sw is the log of the aqueous solubility, log P is the log of the octanol-water partition coefficient and MP is the melting point of the drug.
A rearrangement of this equation to better show the contribution of the two variables, Partition coefficient and Melting point, will give the following form of the equation:
This equation clearly illustrates the contribution of each variable. Thus, we can use it to determine the limiting factor for drug solubility as following:
If log P > MP/100 then the influence of log P is predominant and the solubility is rather log P limited. In other words, the poor solubility could be attributed to the high-lipophilicity of the molecule. Thus, formulation technologies that can improve the wettability may be very helpful in overcoming the solubility problem. This may be achieved, for example, by the simple addition of a surfactant, or more challenging, the Self Emulsifying Drug Delivery System (SEDDS) which involves the use of lipidic solvent and co-solvent.
If log P < MP/100 then the solubility is rather melting point limited. In other words, the poor-solubility could be attributed to the high crystal lattice forces of the molecule. Thus, high-energy formulation technologies such as solid dispersion and nano-crystals may reduce this crystal lattice force and improve the solubility. The general expectation that lowering melting point increases solubility is based on this simplified solubility equation (Wouters et al., 2011):
An important point should be taken into consideration when evaluating the impact of solubility on oral bioavailability. In fact, the relationship between solubility and bioavailability is not systematic. This means that we can see some molecules with poor aqueous solubility and still have high bioavailability (Rinaki et al., Citation2003; Lindenberga et al., 2004). This could be explained by the fact that the gastrointestinal medium is a dynamic one where the physiological factors such as enzymes and the compensatory influence of high permeability can be enough to correct the poor solubility.
This leads to the important question that the author often received from his medicinal chemistry colleagues, when a recently synthesized poorly soluble molecule is being discussed: “Could we improve the bioavailability of this molecule by increasing the aqueous solubility?” The real underlying question should be: “Is the bioavailability of this molecule solubility limited?”
To answer this question in an easy manner, the author established a simple decision tree which handles the different scenarios of poor bioavailability ().
Figure 1. Evaluate the influence of solubility in the bioavailability (BA) of a compound.
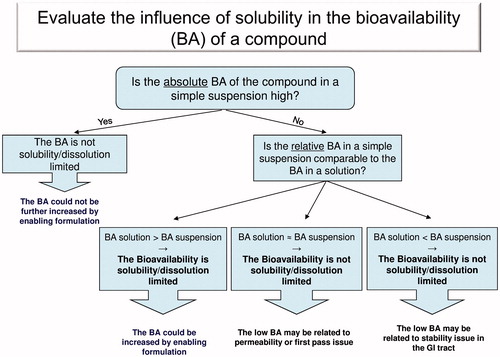
This decision tree makes use of the usually available pharmacokinetic (PK) studies during lead optimization process. The message is that before initiating a long and expensive formulation study to improve the solubility, the relationship between oral bioavailability and solubility for a specific compound should be clearly established.
Further prediction of the human relative bioavailability (Frel), based on the Frel rat data, shows that roughly Frel rat >50% translates into Frel human of >90% (Muenster et al., 2011).
Understanding the relationship among solubility, permeability and efficient dose to optimize drug absorption
To answer the question of what level of permeability and solubility is needed to achieve the required exposure; scientists should consider both factors concomitantly at the specific efficacious dose.
Lipinski (Lipinski, Citation2000) explained that a compound with an efficient animal dose of 1 mg/kg and high permeability requires a minimum acceptable solubility of 10 µg/ml while a compound with an efficient animal dose of 10 mg/kg requires a minimum solubility of 100 µg/ml.
In order to correlate the aqueous solubility (Sw – mg/ml) and the efficient dose (ED – mg/kg) at a specific permeability level (high, medium or low), the use of dose-solubility number (DS – ml/kg) is introduced as following:
This DS number represents the quantity of solvent required to solubilize the efficient dose. It could be very convenient tool to classify the lead candidates and to set an acceptance guiding criterion, taking into consideration together the three parameters of solubility, permeability and efficient dose.
For high permeability compounds, a DS number of <100 ml/kg could be used as a guideline for discovery chemist to assess the develop-ability of different molecules. This number should be reduced to <20 ml/kg for medium permeability compounds and down to <5 ml/kg for poor permeability compounds. These suggested DS numbers are calculated based on the profiling values of nominated clinical candidates in a big pharmaceutical organization (Lipinski, Citation2000).
The classification of compounds’ permeability could be done based on easily accessible Caco-2 assay (Muenster et al., 2011; Muenster, 2010) as summarized in .
Table 1. Permeability classification using Caco-2 assay.
If, after all chemistry effort, the best lead candidate has the desired profile but does not achieve this targeted solubility in aqueous medium (at pH 6.5 or 7.0), the solubility in standard vehicle for toxicology study could be considered instead (Ku et al., 2010).
Rational formulation strategy in support to preclinical studies
Although formulating a molecule for animal or human involves the use of similar technologies, some specific challenges for animal formulations appear early in drug discovery. This is related to the very small quantity of the new molecules available for formulation trials, the limited characterization of molecules at this early stage, which may lead to a significant variability from batch to batch, and the need of high drug substance load for the toxicology studies.
An additional challenge is that even the most commonly used excipients for human administration may interfere with the pharmacology test or induce toxicity in animal.
Physicochemical characterization:
Pre-formulation studies are highly important to characterize new molecules before starting any formulation effort. It helps in understanding the critical properties of the NCE and its deficiencies that need to be overcome by the formulation process.
There is a general agreement about the importance of this step but the list of tests and especially when to do them is still debatable. Efforts to perform rigorous physicochemical characterization are often compromised by the large number of molecules to be profiled in the lead optimization process and the fact that quantities of drug substance remain limited at this very early stage of development.
A rational approach to the required tests at this early stage may include: melting point, microscopy, solubility (pH 1, pH 7.4), log P, pKa, permeability and chemical stability.
The pharmaceutical literature is providing a large number of methodologies to perform these tests (Gaud et al., Citation2008). In addition, several of these characteristics could be estimated using commercially available softwares (Li et al., 2007).
Salt/polymorph state screening
It is well-known that a compound’s phase (i.e. salt or neutral, crystalline or amorphous) can have profound effects on solubility and subsequent oral absorption (Palucki et al., 2010).
As different polymorphic forms could co-exist for the same NCE, it is very important to identify a stable polymorphic form and make sure that the same form is used during the different stages of drug development, so that adequate exposure can be maintained. This is especially important for molecules whose absorption is only limited by their dissolution (Guidance for industry, ANDAs, 2007).
Thus, it is recommended to perform polymorphic screening prior to animal PK studies for non-BCS 1 compound irrespective of free or salt form. Adoption of a salt form should be based on the enhanced PK performance and/or manufacturability against the free form (Ku, 2008).
Strategy of formulation development for solubility limited absorption molecules in preclinical phase:
Several formulation approaches to handle the problem of poor solubility compounds has been published, including different decision trees.
Maas (Maas et al., 2007) designed the formulations according to the study objective (PK, pharmacology or toxicology). Different formulation approaches are proposed based on the level of solubility.
Li (Li et al., 2007) suggested starting by simple solution or suspension formulation before moving to more complex technologies such as micronization and the use of co-solvent/surfactant. If these approaches do not allow enough solubility enhancements, one needs to move to “novel formulation” such as nano-suspension or solid dispersion.
Rabinow (Rabinow, Citation2004) presented a decision tree for the selection of formulation approach which emphasize on the advantages of nano-suspension technology.
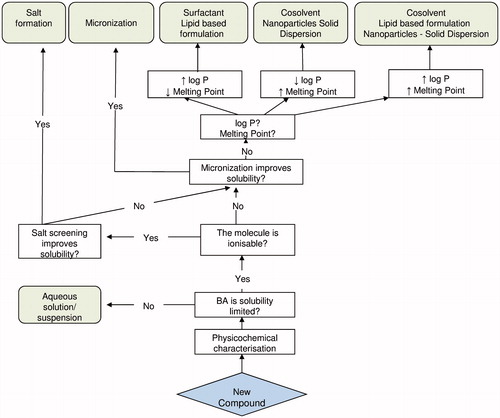
The formulation strategy presented henceforth is based on a sound understanding of the origin of the poor solubility. In addition, it replies to a basic objective of keeping the formulation as simple as possible, providing that the required exposure for the preclinical study is achieved.
After characterizing the NCE, as described above, testing the bioavailability of a simple aqueous solution/suspension preparation is the logic start. This is to verify if the bioavailability is solubility limited and to have an “intrinsic” bioavailability value of the molecule.
If improving the solubility is necessary, evaluating the possibility of salt formation is the first recommended approach. Then, the use of micronized compound is highly recommended too, whenever a suspension is prepared. This is to improve the dissolution rate and helps obtaining a homogeneous suspension when the complete solubility is not obtained.
If these basic approaches do not lead to sufficient exposure, some more complex approaches should be considered. But first, the contribution of lipophilicity and/or lattice force to the poor solubility should be identified, as discussed previously.
For molecules with high log P and low melting point, improving the wettability would enhance the absorption. This could be achieved by using surfactants, oily vehicles or lipid based surfactants. Several publications presented useful lists of recommended excipients for animal dosing (Maas et al., 2007; Neervannan, 2006). presents some selected efficient ones based on authors’ experience.
Table 2. Recommended excipients for oral administration in preclinical studies (Dose: 10 ml/kg).
For challenging molecules, advanced lipidic formulation such as Self Emulsifying Drug Delivery System (SEDDS) and Self Micro Emulsifying Drug Delivery System (SMEDDS) could be evaluated.
For molecules with low log P and high melting point, solubilization in co-solvent should be evaluated first. As the solute risks precipitating in logarithmic proportion upon dilution in the GI fluid, the addition of surfactant help keeping the compound as a fine suspension. If the solubility improvement is not enough, nanoparticles or solid dispersion formulation would be necessary to overcome the lattice force.
For molecules with both high melting point and high log P, where co-solvent does not permit enough exposure, enabling technologies such as solid dispersion, lipid based formulation and nanoparticles should be considered.
Although a clear categorization of high log P and high melting point is not universally established, values of log P ≥ 2–3 and values of melting point ≥160 °C could be considered as high, within the scope of this formulation strategy.
The formulation strategy is summarized in the decision tree in .
Figure 2. Decision tree for preclinical formulation.
Rational formulation strategy for FIH up to clinical proof of concept phase
Generally, two different organizations for FIH formulation development could be often observed in the practice of pharmaceutical firms.
Some companies have a designated scientific group to perform the physicochemical characterization of preclinical molecules. In addition to this assignment, they are supporting the formulation of difficult molecules in the preclinical phase. When a molecule is declared as clinical candidate, the responsibility is transferred to the formulation group where the formulation scientists are in charge of providing the formulated drug in support of all clinical phases until industrialization.
The second approach is the result of moving the pharmaceutical development effort early in drug discovery stage. In this model, an early formulation team is in charge of pre-formulation as well as formulation work from lead optimization until clinical proof of concept (PoC) in Phase IIa.
If the molecules survive the bottleneck of PoC phase, a second formulation team is in charge of carrying out the formulation and the clinical trial material supply until the commercial phase.
The latter approach may better handle the current challenges of formulation development. It permits to streamline the formulation process by using the gained experience from early formulation and bridging among pre-formulation, preclinical formulation and FIH formulation.
Physicochemical characterization
Before deciding the best formulation technology to bring the NCE to clinical phase, the physicochemical characterization started in preclinical phase should be completed. This is to allow a better understanding of properties that will guide the formulation choice. The additional characterization tests are summarized in .
Table 3. Recommended physicochemical test as per the development phase.
Strategy of formulation development
As attrition rate through clinical phases is very high (89%) (Kola et al, 2004) the logic question would be at which stage the maximum formulation effort should be placed.
Hariharan (Hariharan et al., 2003) suggested a practical approach. He proposes using the simple API in capsule/bottle formulation if the objective is to go rapidly for human to determine the PK/tolerance profile of the molecule. Alternatively, he recommends the use of enabling formulation if there is risk of low exposure or if there is a need to accelerate Phases II/III
Ku (Ku, 2006) suggested a formulation strategy based on the BCS classification of molecules. A decision tree is proposed for each of four classes.
The approach presented henceforth tries to turn the knowledge acquired in the preclinical phase to good accounts to move rapidly and efficiently to FIH formulation. The idea here is not to choose automatically the simplest approach such as drug substance in capsule/bottle, neither to try all possible formulation technologies in this early stage.
We have seen that based on the physicochemical characterization of the NCE, several formulation technologies had been proposed for preclinical dosing. Thus, a rational approach would first evaluate if one of these technologies is appropriate for FIH formulation. For example, if a suspension including surfactant was used in preclinical phase to improve the wettability of the compound, a conventional tablet dosage form with surfactant has good chance to give the required exposure in human. The same reasoning would be applied if solid dispersion was employed as enabling formulation in preclinical studies.
This formulation strategy for FIH and beyond, along with the correlation with preclinical formulation, is summarized in .
Table 4. Formulation strategy for FIH based on preclinical formulation.
It is assumed that the same polymorphic and salt form is used for preclinical and clinical formulations. In addition, if micronization was applied in preclinical formulation, this should be maintained when keeping the same formulation moving to clinical phase.
The advantage of this strategy is to concentrate the maximum effort on the technology that has the highest chance for producing the required PK profile. At the same time, the costly bridging bioequivalence studies would be avoided by keeping the same formulation through the different clinical phases up to commercialization.
Technical considerations in choosing the proper formulation
It is certainly rare that only one formulation technology is permitting achieving the targeted PK profile for a given molecule. Thus, several technical and practical aspects should be considered when choosing an enabling technology. This includes physicochemical properties of the drug substance, targeted drug loading in the formulation, patent freedom to operate and the industrial scale production with its cost of goods (COGs).
Typically for nanoparticles technology, the neutral form is the preferred starting form. Possible liabilities of the salt form are (i) risk of disproportionation between the salt and the free base during nanomilling, (ii) risk of aggregation due to charge-based interactions in the small intestine, such as those with bile salts, and (iii) rapid solubilization and turnover of nanoparticles of a salt form into larger particles of the neutral form due to the pH changes in the gastrointestinal (GI) tract. In the same manner, active pharmaceutical ingredients (API) with ionizable groups and pKa between 2 and 7 (e.g. physiological pH range) run the risk of charge-based interactions even if not presented as a salt (Kesisoglou et al., 2007).
For solid dispersion, the ease of crystallization of a drug substance from its amorphous state is an important limiting factor for the use of this technology. It depends on the driving force for crystallization given by the free energy difference between the amorphous and the crystalline state. Thus, very high melting points (Tm) molecules will have a high tendency to crystallize, being difficult to stabilize the amorphous solid dispersion. Values of Tm> 300 °C or Tm/Tg (°K/°K) >1.4 could be used as an indicator of this physical stability problem formulating an API in solid dispersion.
On other hand, glass transition temperature (Tg) of the drug/solid dispersion should be high enough to decrease molecular mobility, thus, to avoid crystallization over time.
Lipid formulations are the optimal drug delivery technology if the poorly soluble drug is as well liposoluble, but lipid-based formulation is often limited by the dose that can be solubilized in the different lipidic solvents. The commonly used size 0 capsule can contain maximum of 0.68 ml. Considering a human dose in the range of 200 mg, this means that the solubility of a molecule in this dose range should not be less than 294 mg/ml, which is not easily achievable. Even for molecules with high solubility in lipidic solvents, the tendency of precipitating upon dilution in the gastrointestinal fluid should be evaluated. The objective is to avoid significant reduction and variability of exposure when the solubilized molecule is precipitating as less soluble crystalline form.
In addition to these technical aspects, the freedom to operate of the chosen drug delivery system is an important financial component as using a patented drug delivery system involves royalty’s payment to inventor.
Prediction of formulation behavior in Human
When the applied technology gives the expected results in vitro, the most important question remains how this formulation will behave in human. Of course, it is not affordable neither ethically acceptable to test each formulation in human. Thus, predicting the human performance and establishing in vivo in vitro correlation (IVIVC) is very important to mitigate the risk of failure in FIH study. In addition, IVIVC can be used to obtain bio-waiver for changes in the production of a drug product during clinical development or post marketing approval (Guidance for Industry Extended Release Oral Dosage Forms, Citation1997).
Animal models, such as rat, dog, and monkey are very commonly used to evaluate the new formulations before going to human. Each one has its advantage and disadvantage in term of capability predicting the formulation’s behavior in humans. Alternatively, several physiologically-based PK modeling (PBPK) softwares, such as GastroPlus™ and PK-SIM™, could be used for this purpose. The quality of prediction depends on the precision of the input data and especially on the model used for predicting the PK profile (Kuenz, 2008). Thus, a rational approach starts by validating the PBPK model using the animal data, before using it to predict the behavior of the formulation in the FIH study.
Examples of successful formulations and the limitations of this strategy
Some examples from literature are confronted with this suggested strategy to see how it is fitting with the formulation practice and to try to understand the discrepancy and the limitations.
ADX73943 is an example of the high melting point (232 °C) and low log P (1.1) active ingredients (Ayad et al., 2012). Its poor solubility in water and almost in all solvents oriented the formulation effort towards an amorphous solid dispersion formulation. Consequently, 20-fold bioavailability increase was achieved compared with the crystal form of the compound. Another example of high melting point and low log P molecules successfully formulated as solid dispersion is prednisolone (Palanisamy et al., 2011) (melting point is 235 °C − log P is 1.6).
Halofantrine is an example of low melting point (80 °C) and high log P (8.0). The lipid-based formulations of the base afforded a six- to eight-fold improvement in absolute oral bioavailability relative to tablet formulation (Shui-Mei et al., Citation1998). Another example within this category which was successfully formulated as lipid-based formulation is Simvastatin (Patil et al., 2007) (melting point is 135 °C – log P is 4.7).
An example of both high log P (4.6) and high melting point (160 °C) is BMS-708163 (Hussein, 2011). Compared with a simple micronized suspension, amorphous solid dispersing improved the bioavailability by 8-fold and soluble lipid formulation increased it by 10-folds.
The above five examples represents formulation approaches which match very well the strategy explained in this paper. Nevertheless, as each general rule, exceptions are drawing its limits of application. An example of these exceptions is fenofibrate. One of its formulations is commercialized as solid dispersion using the patented technology Meltdose®. The solid dispersion approach was chosen instead of a lipid formulation although the low melting point (80.5 °C) and the high log P (5.3).
Other interesting case is the poorly soluble HIV protease inhibitor drug Ritonavir, which has high logP (3.9) and low melting point (122 °C). This drug was first introduced to the market as lipid-based formulation before a second dosage form was introduced using solid dispersion technology. The PK performance of the two forms shows similar bioavailability (Klein et al., Citation2008). This indicate that in some cases even if the high lattice force is not the major contributor for low solubility, overcoming this lattice force by introducing an amorphous form may be enough to improve the bioavailability of poorly soluble molecule.
Conclusion
A rational formulation strategy can reduce the development time cycle and increase the chance of success moving a new drug through the different preclinical and clinical phases. This paper presents a scientific based approach to select the best formulation technology as early as preclinical or FIH phase. Consequently, the same formulation approach, with some adjustment, could be used in the proof of concept phase and eventually up to commercial scale. This is possible if the characterization of NCE is properly used to define the hurdle to be overcome during the formulation development process. As a result, a limited number of formulation technologies could be evaluated to select the most adapted one to the molecule.
This approach has the advantage of streamlining the formulation process in order to avoid delaying the development of new drugs due to formulation related issues. In addition, it takes into consideration the practical constraints of the pharmaceutical industry related to the need of reducing the time to market while keeping the cost of development under control.
Notice of Correction:
In the original version of this article, published online ahead of print on 27 March 2014, two reference citations were written incorrectly in the section ‘Understanding factors determining the bioavailability of oral formulations; Permeability’. These have been corrected in this version.
Acknowledgements
The author wishes to express sincere thanks to Mark McAllister (Pfizer), Burkhard Dickenhorst, May Darwich (Berlin University) for reviewing this Manuscript. The author also wishes to thank Roland Bodmeier, Andriy Dashevskiy, Muhammad Irfan (Berlin University), Claude Blaser (Gattefossé), Macro Gil (Hovione), Munir Hussain (BMS), Sherry Ku (TWi Anchen Pharmaceutical) for their useful discussion during the formulation of the manuscript.
Declaration of interest
The author reports no conflicts of interest. The author alone is responsible for the content and writing of this article.
References
- Amidon GL, Lennernas H, Shah VP, Crison JR. (1995). A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 12:413–20
- Barrett Rabinow E. (2004). Nanosuspensions in drug delivery. Nat Rev Drug Discov 3:785–96
- Butler JM, Dressman JB. (2010). The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci 99:4940–54
- Filippos K, Santipharp P, Yunhui W. (2007). Nanosizing – Oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev 59:631–44
- Gaud SR, Yeole PG, Yadav AV, Gokhale SB. (2008). Pharmaceutics. 10th Edition. : Nirali Prakashan
- Guidance for Industry Extended Release Oral Dosage Forms. (1997). Development, evaluation, and application of in vitro/in vivo correlations. : Food and Drug Administration
- Guidance for Industry, ANDAs. (2007). Pharmaceutical solid polymorphism. : Food and Drug Administration
- Ismail K, John L. (2004). Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–16
- Jochen M, Walter K, Gerrit H. (2007). An integrated early formulation strategy – From hit evaluation to preclinical candidate profiling. Eur J Pharm Biopharm 66:1–10
- Johan W, Luc Q. (2011). Pharmaceutical Salts and Co-crystals. : Royal Society of Chemistry Publishing
- Kathryn W, Natalie K, Samir M. (2008). Safe and effective permeation enhancers for oral drug delivery. Pharm Res 25:1782–8
- Klein C, Y-Chiu L, Awni W, et al. (2008). A pivotal biostudy comparing ritonavir 100 mg film-coated tablet to a ritonavir 100 mg soft gelatin capsule in healthy adult subjects. Oral Abstract Session: AIDS 2008 – XVII International AIDS Conference: Abstract no. THAB0405, Mexico City, Mexico
- Leslie Benet Z, Chi-Yuan Wu, Joseph MC. (2006). Predicting drug absorption and the effects of food on oral bioavailability, Bulletin technique Gattefosse Number 99, Académie des Alpilles
- Lipinski CA. (2000). Drug-like properties and causes of poor solubility and poor permeability. J PharmacolToxicol Methods 44:235–49
- Madhusudan H, Loksidh DG, Gregory EA, et al. (2003). Reducing the time to develop and manufacture formulations for first oral dose in humans. Pharm Technol 27:68
- Marc L, Sabine K, Jennifer Dressman B. (2004). Classification of orally administered drugs on the world health organization model list of essential medicines according to the biopharmaceutics classification system. Eur J Pharm Biopharm 58:265–78
- Martin K. (2008). Drug absorption modeling as a tool to define the strategy in clinical formulation development. AAPS J 10:473–9
- Michael P, John Higgins D, Elizabeth K, Allen Templeton C. (2010). Strategies at the interface of drug discovery and development: early optimization of the solid state phase and preclinical toxicology formulation for potential drug candidates. J Med Chem 53:5897–905
- Mohamad Ayad H, Beatrice B, Jacques Q, et al. (2012). Amorphous solid dispersion successfully improved oral exposure of ADX71943 in support of toxicology studies. Drug Dev Ind Pharm 39:1–6
- Mohanraj P, Jasmina K. (2011). Solid dispersion of prednisolone: solid state characterization and improvement of dissolution profile. Drug Dev Ind Pharm 37:373–86
- Munir Hussain A.(2011). Oral drug delivery: solubility & dissolution rate, oral absorption, and strategies to enable a successful and speedy transition from FIH to drug product, oral presentation AAPS annual meeting, Washington DC
- Ping L, Luwei Z. (2007). Developing early formulation: practice and perspective, Int. J. Pharm 341:1–9
- Pradeep P, Vandana P, Anant P. (2007). Formulation of a self-emulsifying system for oral delivery of simvastatin: in vitro and in vivo evaluation. Acta Pharm 57:111–22
- Ran Y, Jain N, Yalkowsky SH. (2011). Prediction of aqueous solubility of organic compounds by the general solubility equation (GSE). Chem J Inf Comput Sci 41:1208–17
- Rinaki E, Valsami G, Macheras P. (2003). quantitative biopharmaceutics classification system: the central role of dose/solubility ratio. Pharma Res 20:1917–25
- Sashadri N. (2006). Preclinical formulations for discovery and toxicology: physicochemical challenges. Expert Opin Drug Metab Toxicol 2:715–31
- Sherry MK. (2008). Use of the biopharmaceutical classification system in early drug development. AAPS J 10:208–12
- Sherry MK. (2006). An oral formulation decision tree based on biopharmaceutical classification system for first-in-human clinical trial. Bulletin technique Gattefosse Number 98, Académie des Alpilles
- Sherry MK, Wendy D. (2010). A biopharmaceutical classification-based Right-First-Time formulation approach to reduce human pharmacokinetic variability and project cycle time from First-In-Human to clinical Proof-Of-Concept. Pharm Dev Technol 17:285–302
- Shui-Mei K, Andrew Humberstone J, Christopher J, et al. (1998). Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm 167:155–64
- Stephen Buckley T, Sarah Fischer M, Gert F, Martin B. (2012). In vitro models to evaluate the permeability of poorly soluble drug entities: challenges and perspectives. Eur J Pharm Biopharm 45:235–50
- Uwe M, Christian P, Thomas B, et al. (2011). Volume to dissolve applied dose (VDAD) and apparent dissolution rate (ADR): tools to predict in vivo bioavailability from orally applied drug suspensions. Eur J Pharm Biopharm 78:522–30
- Uwe M. (2010). Volume to dissolve applied dose (VDAD) and apparent dissolution rate (ADR) – tools to predict in vivo bioavailability from orally applied drug suspensions. Phys Chem Forum 9, Barcelona, Spain