
Abstract
iRGD is a tumor tissue penetrating peptide due to its targeted binding of integrin and neuropilin-1 receptors. Whether iRGD carries the liposomes in a similar way as it penetrates the cancer drugs or conjugated drugs into tumor tissues and cells has not been fully defined. Here, iRGD-modified and doxorubicin-loaded sterically stabilized liposomes (iRGD-SSL-DOX) and passive liposomes (SSL-DOX) were prepared. A series of experiments were performed to evaluate the tissue penetration, cell penetration, tumor blood vessel damage and anti-tumor effect. The results of flow cytometry and confocal microscopy studies showed that iRGD-SSL-DOX with 5% DSPE-PEG2000-iRGD achieved higher cellular uptake level than that of SSL-DOX on B16 melanoma cells. iRGD-SSL-DOX also exhibited stronger cell growth inhibition in cytotoxicity experiments. The tumor penetrating effect of iRGD was further confirmed by imaging and cellular uptake studies in vivo, in which higher distribution of iRGD-modified liposomes in tumor tissue and tumor cells was observed. Moreover, iRGD-SSL-DOX displayed improved tumor growth inhibition and anti-angiogenesis with less systemic toxicity in an armpit B16 melanoma model. In conclusion, iRGD reserved its tumor-penetrating properties well when modified on the surface of liposomes at optimal density and iRGD-SSL-DOX would be a promising drug delivery system for active targeting tumor therapy.
Introduction
Liposomes have been widely studied in drug delivery systems (DDS) because of their advantages in biocompatibility, stability, all kinds of drugs loading and favorable pharmacokinetic properties (Allen, Citation1998). Particularly, PEGylated liposomes, also known as sterically stabilized liposomes (SSL) (Lasic, Citation1996; Silvander, Citation2002), have significantly prolonged circulation time of drugs in vivo due to less phagocytosis by reticuloendothelial system (RES), and therefore improved the efficacy of cancer therapy. To date, several liposomal formulations have been approved for marketing (Allen, Citation2004), of which the most famous one should be sterically stabilized liposomes loading doxorubicin (SSL-DOX), also known as Doxil. Though SSL achieved more drug accumulation in tumor region by enhanced permeability and retention (EPR) effect (Allen, Citation2004), passive targeting effect could not guarantee the increasing cellular uptake of drugs (Lu et al., Citation2007; Xiong et al., Citation2005a). In addition, some serious side effects still existed in the application of SSL formulations (Slingerland et al., Citation2012).
Recently, more and more attention has been paid to active targeting liposomes, which were modified with ligands on their surface. This kind of active targeting could increase intracellular drug amounts through the interaction between surface ligands and specific or up-regulated receptors on tumor cells. Various targeting ligands have been investigated, including oligopeptides such as RGD or cyclic RGD (Xiong et al., Citation2005a,Citationb; Amin et al., 2013), NGR (Wang et al., Citation2009; Dunne et al., Citation2011), OCT (Huo et al., Citation2012), folate (Gabizon et al., Citation2004), transferrin (Yue et al., Citation2012) and cell penetrating TAT (Sethuraman & Bae, Citation2007). However, these targeting ligands performed poorly in drug penetration into tumor parenchyma (Sugahara et al., Citation2009).
iRGD (CRGDRGPDC) was a defined tumor homing peptide, which could achieve favorable drug distribution and penetration in tumor tissue (Sugahara et al., Citation2010). iRGD homes to tumors through a step-by-step process: the RGD motif binds to integrins on tumor endothelium first and then iRGD is proteolyticly cleaved to a binding motif for neuropilin-1. Integrins receptors were highly involved in tumor metastasis, angiogenesis and signal transduction between tumor cells and the extracellular matrix (Hood & Cheresh, Citation2002; Jin & Varner, Citation2004) and neuropilin receptors were also related to tumor metastasis and angiogenesis (Bielenberg & Klagsbrun, Citation2007). Particularly, neuropilin-1 (Nrp-1) receptor could regulate endothelial permeability through VEGF binding property (Becker et al., Citation2005). Conjugation to iRGD or co-administration with iRGD significantly improved the sensitivity of tumor-imaging agents and enhanced the activity of an antitumor drug (Teesalu et al., Citation2013). Based on the penetration function of iRGD, efforts have also been made to explore enhanced anti-tumor effect by linking iRGD with polymers (Zhu et al., Citation2011), liposomes (Ye et al., Citation2011; Liu et al., Citation2013; Yu et al., Citation2013), nanoparticles (Sugahara et al., Citation2009). Though the improved anti-tumor effects of liposomal doxorubicin has been achieved by iRGD modification in one previous study (Yu et al., Citation2013), it is worth of making further research and validation. What’s more, the studies of the referred research are still not enough, especially on iRGD density optimization, cellular penetration in vivo, the toxicity reduction and in vitro–in vivo correlation.
In this study, iRGD was conjugated to DSPE-PEG2000-NHS and its targeting effect was evaluated by iRGD-modified and DOX-loaded sterically stabilized liposomes (iRGD-SSL-DOX). In order to investigate more comprehensive resulting information from iRGD modification, a series of experiments were performed to evaluate the tissue penetration, cellular uptake, tumor blood vessel damage and integrated anti-tumor effect. B16 melanoma cells were used a tumor cell model in vitro and in vivo which express both integrin and neuropilin receptors (Mizejewski, Citation1999; Bielenberg & Klagsbrun, Citation2007).
Materials and methods
N-hydroxysuccinimidyl-PEG2000-DSPE (DSPE-PEG2000-NHS) and DSPE-PEG2000 were purchased from NOF Corporation (Tokyo, Japan). Cholesterol (Chol) and Sephadex G-50 were from Pharmacia Biotech (Piscataway, NJ), and EPC was from Lipoid GmbH (Ludwigshafen, Germany). iRGD (Mw 978.06) was synthesized by GL Biochem Co., Ltd. (Shanghai, China). Doxorubicin hydrochloride (DOX) was kindly provided as a gift by Haizheng Pharmaceutical Co., Ltd. (Zhejiang, China). Sulforhodamine B (SRB), trichloroacetic acid (TCA) and Tris base were from Sigma-Aldrich (St. Louis, MO). Fluorescent probe DiR was purchased from Biotium Inc. (Hayward, CA), and Hoechst 33258 was from Molecular Probes Inc. (Eugene, OR). Collagenase III was purchased from Worthington Biochemical Corporation (Lakewood, NJ). Nrp-1 rabbit polyclonal IgG, integrin α5β1 rat monoclonal IgG and CD31 rat monoclonal IgG were from Abcam (Cambridge, UK). FITC-conjugated goat anti-rabbit IgG was from Protein Tech Group, Inc. (Chicago, IL), and FITC-labeled goat anti-rat IgG was from KPL, Inc. (Gaithersburg, MD). All other chemicals and reagents were of analytical or HPLC grade.
Mouse melanoma cell line B16 was obtained from Institute of Basic Medical Science, Chinese Academy of Medical Science (Beijing, China). Cells were cultured in RPMI-1640 medium (Macgene Biotech Co., Ltd, Beijing, China) supplemented with 10% fetal bovine serum (FBS) and antibiotics (penicillin 100 U/ml and streptomycin 100 mg/ml) at 37 °C with 5% CO2.
Male C57BL/6 mice (6 weeks old, 18–20 g) were from Vital River Laboratory Animal Center (Beijing, China), and kept under SPF condition with free access to standard food and water. All experiments were performed by the principles of care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of Peking University.
Synthesis and identification of the targeting compound DSPE-PEG2000-iRGD
DSPE-PEG2000-iRGD was synthesized through esterification of the primary amino group in iRGD with NHS group in DSPE-PEG2000-NHS. In brief, DSPE-PEG2000-NHS was mixed with iRGD at 2:1 molar ratio in DMF, and then pH was adjusted to 10.0 with triethylamine (TEA). The reaction was maintained for 48 h at room temperature with moderate stirring and monitored by RP-HPLC (Shimadzu, LC-10AT, Japan). The mobile phase consisted of water, acetonitrile and trifluoroacetic acid (92:8:0.1, v/v/v), and UV detection wavelength was 220 nm. Finally, the reaction mixture was dialyzed (MW cutoff = 3500 Da) against deionized water for 48 h to remove free iRGD and DMF solvent. The final product was lyophilized and stored at −20 °C until use. The product was identified by 1H-NMR and MALDI-TOF MS, and the content of DSPE-PEG2000-iRGD was measured by element analysis method.
Preparation of liposomal formulations
Lipids of EPC/Chol/DSPE-PEG2000 (62.0:31.8:6.2, molar ratio) and EPC/Chol/DSPE-PEG2000/DSPE-PEG2000-iRGD (61.9:31.8:6.0:0.3, molar ratio) were used for SSL-DOX and iRGD-SSL-DOX, respectively. Liposomes were prepared by thin lipid film hydration followed by sonication method as previously reported (Xiong et al., Citation2005a). Briefly, lipids were dissolved in chloroform and evaporated at 37 °C under reduced pressure. The dried lipid film was hydrated with 123 mM ammonium sulfate, followed by sonication at 37 °C for 20 min. The ammonium sulfate gradient was established by eluting through a Sephadex G-50 column equilibrated with PBS (pH 7.4). DOX was loaded by ammonium sulfate gradient method. In brief, DOX was added to the blank liposomes (lipids: drug = 20:1, w/w) and incubated at 37 °C for 15 min with gentle shaking, then unencapsulated DOX was separated by Sephadex G-50 column again. For liposomes containing DiR, lipids of above formulations and DiR (lipids: DiR = 1000:1, w/w) were dissolved in chloroform and evaporated to form thin lipid film. The dried film was hydrated with PBS, followed by sonication at 37 °C for 40 min. Free DiR was removed by eluting through Sephadex G-50 column.
Characterization of liposomes
Size distribution and zeta potential of liposomes were measured by dynamic light scattering (DLS) using Malvern Zetasizer Nano ZS (Malvern, UK). The morphology of liposomes was identified by transmission electron microscope (TEM, JEM-200CX, JEOL, Japan). The samples were placed on a 200-mesh, carbon-coated, copper grid and negative stained with uranyl acetate solution (1%, w/v). The observations were operated at an acceleration voltage of 120 kV. The entrapment efficiency (EE) of liposomes was calculated with the formula: EE = (We/Wtotal) × 100%, where We was the amount of DOX in liposome suspensions after eluting through Sephadex G-50 column and Wtotal was the amount of DOX in liposome suspensions with the same volume before eluting through Sephadex G-50. The in vitro release profile of DOX liposomes was measured as previously reported (Xiong et al., Citation2005a). Briefly, 0.5 ml liposome was mixed with 0.5 ml FBS and placed in a dialysis bag (Mw cutoff 12 000–14 000 Da). Bags were incubated in 50 ml PBS (pH 7.4) at 37 °C with gentle shaking (100 rpm). Aliquots of 0.5 ml medium were removed at predetermined time points (0.5, 1, 2, 4, 8, 12, 24, 36, 48 h) and replaced with fresh medium. The concentration of DOX or DiR in liposomes was measured by UV-Vis spectrophotometer (UV-1100, Mapada instruments, China) at 485 nm or fluorescence spectrophotometer at 748/780 nm, respectively. More specifically, the liposomal formulations were broken with Triton X-100 (final concentration 1% v/v) and diluted with PBS to volume. The absorbance of prepared samples was measured with 1% Triton X-100 solution served as the blank control. In in vitro release study, the released DOX was quantified by RP-HPLC with mobile phase containing methanol, water and acetic acid (55:40:5, v/v/v), and with detection wavelength at 233 nm.
Integrin and Nrp-1 receptor expression on B16 cells
B16 cells were selected to evaluate the targeting effect of iRGD modified liposomes. To identify the expression of both integrin and Nrp-1 receptors, cells cultured on coverslips were fixed by paraformaldehyde, followed by blocking with 10% goat serum and 1% bovine serum albumin (BSA). Cells were incubated with primary antibody at 4 °C overnight to label integrin receptors or at 37 °C for 2 h to label Nrp-1 receptors. Negative controls (incubated with 1% BSA) were also included. Cells were then incubated with FITC-labeled secondary antibodies at 37 °C for 1 h. Receptor expression was investigated by confocal laser scanning microscope (CLSM, Leica SP5, Germany).
Optimization of iRGD density on liposome surface
To optimize the iRGD ligand content achieving the best targeting effect, formulations with different iRGD modification density (DOX concentration of 10 μg/ml) were added to cells cultured on coverslips. After incubation at 37 °C for 1 h, cells were washed with cold PBS, followed by fixation with paraformaldehyde and nuclei staining with Hoechst 33258. Different uptake profiles were determined by CLSM.
In vitro uptake and competition experiments
To confirm the penetrating effect of iRGD in vitro, B16 cells cultured on 6-well plates were incubated with SSL-DOX, iRGD-SSL-DOX at 37 °C for 3 h, or with 5 mg/ml free iRGD for 1 h followed by iRGD-SSL-DOX at 37 °C for 3 h. The concentration of DOX was adjusted to 10 μg/ml. Cells were then washed with cold PBS and trypsinized. After washing with PBS again, the cellular DOX fluorescence was immediately analyzed by flow cytometry using FACScan (BD). The uptake and competition experiment was also detected by CLSM with procedures described in above-mentioned method, except the incubation time was 3 h.
In vitro cytotoxicity assay
Sulforhodamine B method was applied to investigate the cytotoxicity of free DOX, SSL-DOX and iRGD-SSL-DOX against B16 cells. Briefly, cells were cultured on 96-well plates at 5000 cells/well for 24 h, and then cultured with different formulations with a series of DOX concentration for another 48 h. Cells were fixed with 10% TCA at 4 °C for 1 h, followed by washing and drying. Fixed cells were stained with 0.4% SRB at room temperature for 30 min, and the excess dye was washed out by 1% acetic acid. The protein-bound dye was dissolved by 10 mM Tris base, and absorbance was measured at 540 nm using a 96-well plate reader (Thermo Scientific Multiskan, FC). The drug concentration inhibiting cell growth by 50% (IC50) was calculated by SPSS software.
In vivo distribution study by living imaging investigations
In vivo tumor penetrating and tissue distribution of iRGD modified liposome was investigated by DiR loaded liposomes. The armpit tumor model was established by subcutaneously inoculating 1 × 106 B16 cells/mouse in the right flank of C57BL/6 mice. Hair on the abdomen was removed on the 12th day after inoculation, and experiment was carried out on the next day. Mice were injected with 0.2 ml free DiR, DiR-loaded liposome (SSL-DiR) or iRGD modified DiR liposome (iRGD-SSL-DiR) with DiR concentration of 4 μg/ml through tail veins. Mice were anaesthetized by isoflurane at 2.5, 4.5, 8 and 12 h after injection, and the distribution of different formulations was visualized by in vivo imaging system (Carestream, Fx Pro). Mice were then sacrificed and tumors and major organs were excised. The ex vivo fluorescent images of tumors and organs were also obtained.
In vivo tumor inhibition and toxicity study
Mice were implanted with B16 melanoma as described above. Mice were randomly assigned to four groups (n = 6) treated with: PBS, free DOX, SSL-DOX or iRGD-SSL-DOX. The dosage of DOX was 1 mg/kg, and four times of treatment was carried out every other day, beginning on the 12th day after tumor inoculation. Tumor width and length were measured every 2 days and body weight was measured every day. Tumor volume was calculated as: [0.5 × (length) × (width)2]. At the end point, 20 μl blood sample was collected from retro-orbital sinus of each mouse. Complete blood counts were used to evaluate the blood toxicity of DOX formulations. Mice were sacrificed after blood collection and tumors were excised, weighted and pictured.
In vivo cellular penetration study
In vivo cellular penetration of SSL-DOX and iRGD-SSL-DOX were analyzed by flow cytometry. Tumors freshly harvested from the treated mice of experiment of “In vivo tumor inhibition and toxicity study” were cut into pieces, followed by incubation with collagenase III at 37 °C for 1 h with gentle stirring to dissociate enzymes in tumor tissue. Function of collagenase was ended by adding RPMI-1640 medium containing 10% FBS. The mixture was filtered through 200 mesh screen to obtain single cell suspension. Cells were washed and resuspended with PBS, filtered through 400-mesh screen, and then DOX fluorescence was measured by FACScan.
Tumor vasculature staining
Changes of tumor vasculature were investigated by CD31 immunohistochemistry. In brief, frozen sections of tumors harvested from the Section “In vivo cellular penetration study” were fixed with ice cold acetone, blocked by goat serum and BSA, followed by incubation with CD31 primary antibody at 4 °C overnight. Secondary antibody labeling was performed at 37 °C for 1 h, followed by nuclei staining with Hoechst 33258. Tumor vasculature was visualized using CLSM and quantified by Leica Qwin image analysis software.
Statistical analysis
All experiments were repeated at least three times. All data were shown as means ± SD. Two-tailed Student’s t test or one-way analyses of variance (ANOVA) were performed in statistical evaluation. A p value <0.05 was considered to be statistically significant, and a p value <0.01 was considered to be highly significant.
Results and discussions
Conjugation of iRGD to DSPE-PEG2000-NHS
iRGD was linked to DSPE-PEG2000 to prepare the iRGD-modified liposomes. Conditions of the reaction were relatively simple. RP-HPLC was used to monitor iRGD conjugation. As shown in , the retention time of iRGD was ∼20.1 min and the disappearance of the peak of iRGD demonstrated the complete linking with DSPE-PEG2000 at 48-h time point. In case that there might be decompositions of peptide in the reaction, which would damage its recognition sites, blank reaction in which only free iRGD underwent the same reaction process was also performed. Mw of free iRGD was measured by mass spectrum before and after the reaction. The raw material and blank reaction product exhibited the same Mw (Figure S1), indicating the conditions used above could not bring structural changes for iRGD. The structure of reaction product was identified by MALDI-TOF MS. Mass spectrum of the product presented an average Mw 4000 (), which was approximately the Mw sum of DSPE-PEG2000-NHS and iRGD.
Figure 1. Identification of iRGD conjugation and characteristics of DOX liposomes. (A) Synthetic reaction monitored by HPLC at the beginning, 12 and 48 h later. (B) Identification of the reaction product by MALDI-TOF MS. (C) Morphology of iRGD-SSL-DOX measured by TEM. (D) In vitro release rate of SSL-DOX and iRGD-SSL-DOX. 0.5 ml liposome and 0.5 ml FBS were placed in a dialysis bag (Mw cutoff 12 000–14 000 Da) and incubated in 50 ml PBS at 37 °C with gentle shaking (100 rpm). The released DOX was measured by HPLC at predetermined time points (n = 3).
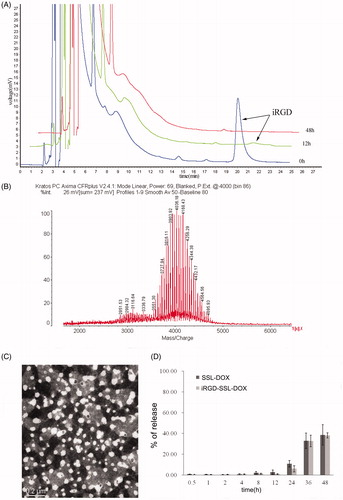
Preparation and characterization of liposomes
Next, DSPE-PEG2000-iRGD was used to prepare liposomes characterized with both prolonged circulation in vivo and tumor targeting. Average size, zeta potential and entrapment efficiency of SSL-DOX and iRGD-SSL-DOX are summarized in . The diameter of liposomes was ∼100 nm with PDI <0.2 and slightly negative surface charge. Loading efficiency was >95%. The morphology of liposomes was identified by TEM image of iRGD-SSL-DOX (), which is spherical shape. Release percentage of DOX was found <10% within the first 12-h incubation, and increased to ∼40% in 48 h (). In all, SSL-DOX and iRGD-SSL-DOX presented similar size distribution, zeta potential value, entrapment efficiency and in vitro release profile. Consequently, the modification with iRGD had little influence on the physicochemical properties of liposomes.
Table 1. Characteristics of SSL-DOX and iRGD-SSL-DOX (mean ± SD, n = 3).
Optimization of iRGD density and in vitro cellular uptake studies
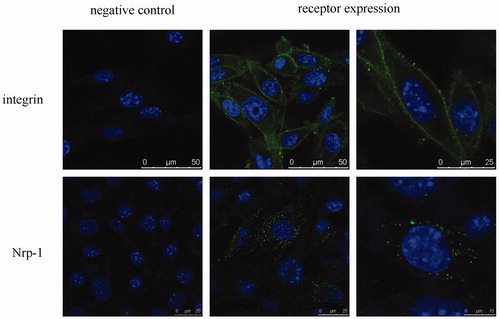
Expression of the two kinds of receptors involved in the targeting and penetrating function of iRGD has been investigated (Sugahara et al., Citation2009). Our results showed B16 cells expressed high and moderate level of integrin and Nrp-1 receptors, respectively (). The different expression levels could be compared by the number of positive cells and were in accordance with previous reports (Kramer et al., Citation1991; Graeven et al., Citation2000; Pellet-Many et al., Citation2008; Yang et al., Citation2010).
Figure 2. Receptor expression of integrin α5β1 and Nrp-1 on B16 cells. Receptors and nuclei were stained with fluorescein and Hoechst 33258, respectively. Circled or spotted staining surrounding the nucleus demonstrated the expression of these membrane receptors.
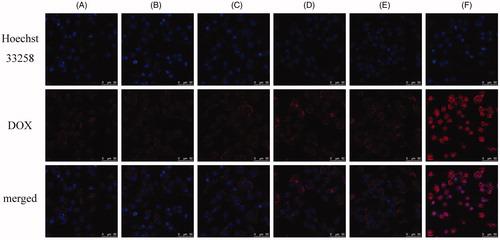
Previous researches showed the modifying density could affect the targeting efficiency of peptide ligand (Gu et al., Citation2008; Waite & Roth, Citation2011). The best density of iRGD for liposomal formulation was investigated by cellular uptake experiment. As shown in , no obvious enhancing uptake was found with 1–2% DSPE-PEG2000-iRGD modification compared with that of SSL-DOX. Cellular uptake of iRGD-SSL-DOX with 5% DSPE-PEG2000-iRGD modification achieved better effect, while further penetrating effect was not found with higher percentage. This was possibly caused by the limited receptor density on cell surface and the steric hindrance between ligands and receptors (Cairo et al., Citation2002).
Figure 3. Comparison of the cellular uptake of liposomes modified with different iRGD content by B16 cells at 37 °C for 1 h: 0% DSPE-PEG2000-iRGD (A), 1% DSPE-PEG2000-iRGD (B), 2% DSPE-PEG2000-iRGD (C), 5% DSPE-PEG2000-iRGD (D), 10% DSPE-PEG2000-iRGD (E), free DOX (F, positive control). The content of DSPE-PEG2000-iRGD was the molar percentage in total DSPE-PEG2000.
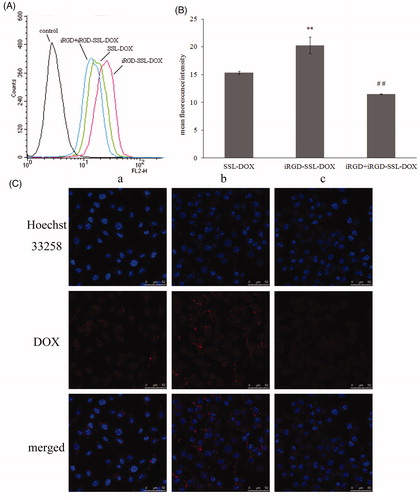
In vitro targeting effect was then investigated with the optimal iRGD-SSL-DOX formulation. As shown in flow cytometry experiment, the cellular uptake of iRGD-SSL-DOX was significantly more than that of SSL-DOX (). There was nearly 1.32-fold enhancement in the quantification result (). The cellular uptake of iRGD-SSL-DOX in competition experimental group was significantly (p < 0.01) lower than that in the non-competition group, which demonstrated the iRGD contributed to the targeting effect of modified liposome. Similar results were also observed using CLSM (). Furthermore, combined with the little released DOX from liposomes within 4 h in vitro release test (), it was believed that the enhanced cellular DOX uptake of iRGD-SSL-DOX was resulted from the targeting function of iRGD rather than other factors.
Figure 4. Uptake of SSL-DOX, iRGD-SSL-DOX or iRGD + iRGD-SSL-DOX by B16 cells after incubation at 37 °C for 3 h. (A) Representative result of three experiments by flow cytometry. (B) The quantitative result of flow cytometry. Results are means ± SD (n = 3). **p < 0.01 versus SSL-DOX and ##p < 0.01 versus iRGD-SSL-DOX. (C) Uptake studies measured by confocal microscopy: (a) SSL-DOX, (b) iRGD-SSL-DOX and (c) iRGD + iRGD-SSL-DOX.
Cytotoxicity assay
As shown in , after 48-h incubation with B16 cells, the sequence of IC50 values was: free DOX < iRGD-SSL-DOX < SSL-DOX. Although free DOX exhibited the smallest IC50 value, possibly owing to the rapid uptake and direct effect on cells (Yang et al., Citation2012), iRGD-SSL-DOX achieved significantly (p < 0.05) better cell killing effect than SSL-DOX. This improved cytotoxicity was likely to be resulted from more adhesion and easier cellular uptake mentioned above.
Table 2. IC50 values of DOX, SSL-DOX and iRGD-SSL-DOX on B16 cells in vitro after 48-h incubation (mean ± SD, n = 6).
In vivo distribution study by living imaging investigations
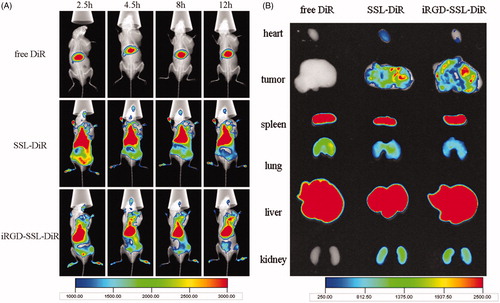
iRGD targeting effect was confirmed at in vivo level using near infrared (NIR) imaging. Compared with traditional distribution studies, in vivo imaging, being simple, sensitive and illustrative, makes continuous measurement with the same animal possible. DiR was chosen to be the fluorescent probe in this study because of its NIR excitation and emission wavelength, which could effectively reduce the interference of animal auto-fluorescence (Sharma et al., Citation2006). SSL-DiR and iRGD-SSL-DiR exhibited similar size distribution (134.9 ± 9.66 nm and 131.6 ± 11.6 nm, respectively) and zeta potential (−2.46 ± 0.85 mV and −3.19 ± 0.30 mV, respectively). As shown in , though liposomal formulations significantly improved circulation profiles and had EPR effect owing to PEG modification (Minko et al., Citation2006), there was high accumulation in liver and spleen as shown in the ex vivo image (), possibly because of the arrestment of DiR-loaded formulations by RES system (Schädlich et al., Citation2011; Xiang et al., Citation2011). However, superior distribution of liposomes, especially iRGD-SSL-DiR at 12-h post-injection, in tumors could still be observed. In ex vivo image (), more distribution of DiR florescence in tumor in iRGD-SSL-DiR group was observed than that in SSL-DiR group, especially much more DiR florescence in the deep of tumor, which confirmed the penetrating function of iRGD.
Figure 5. In vivo (A) and ex vivo (B) fluorescent images of B16 tumor-bearing mice treated with free DiR, SSL-DiR or iRGD-SSL-DiR. The distribution of fluorescent formulations at different time points after administration is shown. Tumor regions were located in the armpit of right forelimbs, as indicated by arrows.
In vivo cell penetration study
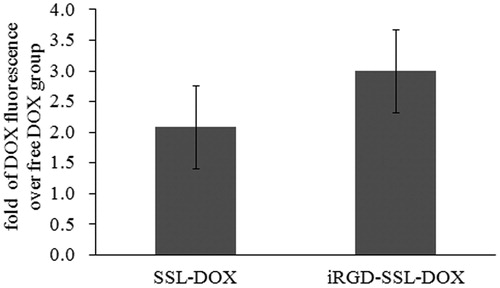
To further verify the tumor cell penetrating effect of iRGD, cellular uptake experiment was performed at in vivo level. The penetration of DOX from both SSL-DOX (nearly 2-fold) and iRGD-SSL-DOX (nearly 3-fold) were more than free DOX, and iRGD modification led to 1.44-fold increase of cellular uptake compared to SSL-DOX (). This result from tumor cellular level, together with living imaging study at tumor tissue level, strongly demonstrated the tumor penetration of liposoma DOX by iRGD could be achieved in vivo, which is not full elaborated in the previous study (Yu et al., Citation2013).
Figure 6. In vivo DOX cellular uptake in tumor tissues from tumor-bearing C57BL/6 mice treated with SSL-DOX or iRGD-SSL-DOX. Results are means ± SD (n = 3).
Tumor growth inhibition study
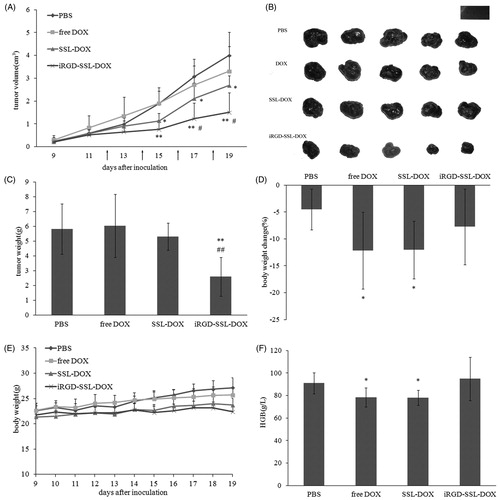
B16 melanoma armpit tumor model was used to evaluate the in vivo anti-tumor efficacy. After four times of administration with a relatively low dosage (1 mg/kg DOX), iRGD-SSL-DOX exhibited the best therapeutic effect (). Tumor volume of iRGD-SSL-DOX group became significantly smaller than PBS control group (p < 0.01) and SSL-DOX group (p < 0.05). This efficacy was also confirmed by tumor weight measurement (). The improved chemotherapeutic efficacy of iRGD-SSL-DOX was as same as the literature reports (Yu et al., Citation2013), while the B16F10 melanoma models rather than B16 melanoma models was used in the anti-tumor experiment. For toxicity evaluation, body weight measurement and blood test were performed. As shown in , there was no evident body weight loss for each group during the treatment, while only iRGD-SSL-DOX group exhibited a similar level of body weight change without tumor (the percentage change of body weight compared to weight before treatment) with control group, indicating better toleration. Previous results claimed myelotoxicity (Morelli et al., Citation1997) and damage to erythrocytes and hemoglobin (Marczak et al., Citation2006) caused by DOX treatment. Here, different from free DOX and SSL-DOX, iRGD-SSL-DOX could not significantly change hemoglobin (HGB) levels during the treatment (). In all, iRGD-SSL-DOX achieved the best therapeutic effect on B16 tumor with the least side effects.
Figure 7. In vivo anti-tumor efficacy and toxicity. (A) Tumor volume of tumor-bearing C57BL/6 mice treated with: PBS, free DOX, SSL-DOX or iRGD-SSL-DOX. Arrow points indicated the time for administration. Results are means ± SD (n = 6). *p < 0.05 versus PBS control, **p < 0.01 versus PBS control, #p < 0.05 versus SSL-DOX group. (B) Picture of the excised tumors. (C) Tumor weight of C57BL/6 mice with different treatments. Results are means ± SD (n = 6). **p < 0.01 versus PBS control, ##p < 0.01 versus SSL-DOX group. (D) Body weight changes during different treatments. Results are means ± SD (n = 6). (E) Changes of body weight with tumors removed compared to initial weight before treatment. Results are means ± SD (n = 6). *p < 0.05 versus PBS control. (F) Comparison of HGB level of different treatments. Results are means ± SD (n = 6). *p < 0.05 versus PBS control.
Tumor vasculature assay
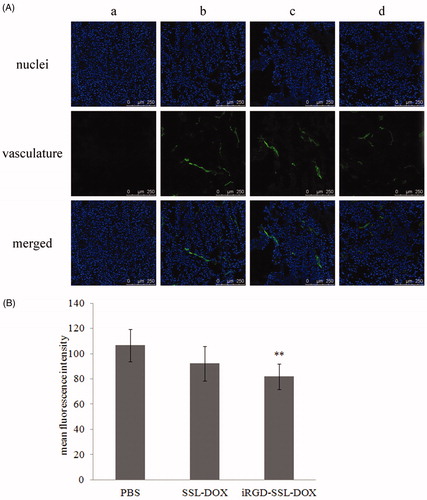
Previous studies showed tumor endothelial cells also expressed both integrin (Nikolopoulos et al., Citation2004) and Nrp-1 (Stephenson et al., Citation2002) receptors. Other studies found Nrp-1 was involved in tumor angiogenesis and progression through VEGF binding property (Bagri et al., Citation2009), and blocking Nrp-1 function could reduce angiogenesis and vascular remodeling (Geretti et al., Citation2008; Bagri et al., Citation2009). More importantly, iRGD could also cause increased binding with tumor endothelial cells (Zhang et al., Citation2011). Since Nrp-1 receptors were involved in the targeting of iRGD, status of tumor vasculature was investigated following the anti-tumor growth study. It was found that iRGD-SSL-DOX could significantly (p < 0.01) inhibit tumor angiogenesis, while SSL-DOX could not (). Consequently, besides the direct tumor cell killing effect, anti-angiogenesis also contributed to the improved therapeutic efficacy of iRGD-SSL-DOX. Free DOX was also found to be anti-angiogenic, which was possibly due to the direct endothelial cell killing effect when transported to tumor vasculature through blood circulation.
Figure 8. Tumor vasculature changes after different treatments. (A) Representative CLSM images of tumor vasculature from mice treated with: PBS (b), SSL-DOX (c) or iRGD-SSL-DOX (d). Vasculature and nuclei were labeled by FITC and Hoechst 33258, respectively. Negative control (a) for vasculature staining is also shown. The stripped staining demonstrated the tumor vasculature. (B) Quantitative result of CLSM images. 6 visual fields of each group were used for statistical analysis. **p < 0.01 versus PBS control group.
Conclusions
In this study, the tumor targeting and penetrating functions of iRGD were evaluated both in vitro and in vivo when it was modified on the surface of liposomal DOX. Though iRGD modification had little effect on physicochemical properties of liposomes, iRGD-SSL-DOX with the optimal 5% DSPE-PEG2000-iRGD surface density materialized more cellular uptake and higher cytotoxicity than SSL-DOX in vitro on B16 melanoma cells, which were found to express both integrin and Nrp-1 receptors involved in the targeting mechanism. The penetrating of liposomes brought by iRGD was further confirmed in vivo with living imaging using an NIR fluorescent probe, together with DOX cellular uptake measured by flow cytometry. In addition, tumor vasculature study indicated the anti-angiogenesis was another benefit of iRGD-SSL-DOX in tumor therapy. Combining benefits of vascular damage, tumor penetration and further in vivo cell penetration of iRGD, iRGD-SSL-DOX achieved the best anti-tumor efficacy as well as the least toxicity on B16 tumor model. Conclusively, iRGD could carry the liposomes into tumor vascular, tumor deep tissues and tumor cells in vivo, which contribute to the improved chemotherapeutic effect together.
Supplementary Figure S1
Download PDF (330 KB)Declaration of interest
The authors of this article declare no conflict of interests. Element analyses of the reaction product of iRGD conjugation; mass spectra of iRGD raw material and the product of blank reaction. This study was supported by National Science Foundation (81130059) and the Innovation Team of the Ministry of Education of China (BMU20110263).
References
- Allen TM. (1998). Liposomal drug formulations. Rationale for development and what we can expect for the future. Drugs 56:747–56
- Allen TM. (2004). Cullis PR. Drug delivery systems: entering the mainstream. Science 303:1818–22
- Amin M, Badiee A, Jaafari MR. (2013). Improvement of pharmacokinetic and antitumor activity of PEGylated liposomal doxorubicin by targeting with N-methylated cyclic RGD peptide in mice bearing C-26 colon carcinomas. Int J Pharm 458:324–33
- Bagri A, Tessier-Lavigne M, Watts RJ. (2009). Neuropilins in tumor biology. Clin Cancer Res 15:1860–4
- Becker PM, Waltenberger J, Yachechko R, et al. (2005). Neuropilin-1 regulates vascular endothelial growth factor-mediated endothelial permeability. Circ Res 96:1257–65
- Bielenberg DR, Klagsbrun M. (2007). Targeting endothelial and tumor cells with semaphorins. Cancer Metastasis Rev 26:421–31
- Cairo CW, Gestwicki JE, Kanai M, et al. (2002). Control of multivalent interactions by binding epitope density. J Am Chem Soc 124:1615–9
- Dunne M, Zheng J, Rosenblat J, et al. (2011). APN/CD13-targeting as a strategy to alter the tumor accumulation of liposomes. J Control Release 154:298–5
- Gabizon A, Shmeeda H, Horowitz AT, et al. (2004). Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev 56:1177–92
- Geretti E, Shimizu A, Klagsbrun M. (2008). Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. Angiogenesis 11:31–9
- Graeven U, Rodeck U, Karpinski S, et al. (2000). Expression patterns of placenta growth factor in human melanocytic cell lines. J Invest Dermatol 115:118–23
- Gu F, Zhang L, Teply BA, et al. (2008). Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci USA 105:2586–91
- Hood JD, Cheresh DA. (2002). Role of integrins in cell invasion and migration. Nat Rev Cancer 2:91–100
- Huo M, Zou A, Yao C, et al. (2012). Somatostatin receptor-mediated tumor-targeting drug delivery using octreotide-PEG-deoxycholic acid conjugate-modified N-deoxycholic acid-O, N-hydroxyethylation chitosan micelles. Biomaterials 33:6393–407
- Jin H, Varner J. (2004). Integrins: roles in cancer development and as treatment targets. Br J Cancer 90:561–5
- Kramer RH, Vu M, Cheng YF, et al. (1991). Integrin expression in malignant melanoma. Cancer Metastasis Rev 10:49–59
- Lasic DD. (1996). Doxorubicin in sterically stabilized liposomes. Nature 380:561–2
- Liu Y, Ji M, Wong MK, et al. (2013). Enhanced therapeutic efficacy of iRGD-conjugated crosslinked multilayer liposomes for drug delivery. Biomed Res Int 2013:378–80
- Lu Y, Wu J, Wu J, et al. (2007). Role of formulation composition in folate receptor-targeted liposomal doxorubicin delivery to acute myelogenous leukemia cells. Mol Pharm 4:707–12
- Marczak A, Kowalczyk A, Wrzesień-Kus A, et al. (2006). Interaction of doxorubicin and idarubicin with red blood cells from acute myeloid leukaemia patients. Cell Biol Int 30:127–32
- Minko T, Pakunlu RI, Wang Y, et al. (2006). New generation of liposomal drugs for cancer. Anticancer Agents Med Chem 6:537–52
- Mizejewski GJ. (1999). Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med 222:124–38
- Morelli D, Ménard S, Cazzaniga S, et al. (1997). Intratibial injection of an anti-doxorubicin monoclonal antibody prevents drug induced myelotoxicity in mice. Br J Cancer 75:659–9
- Nikolopoulos SN, Blaikie P, Yoshioka T, et al. (2004). Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell 6:471–83
- Pellet-Many C, Frankel P, Jia H, et al. (2008). Neuropilins: structure, function and role in disease. Biochem J 411:211–26
- Schädlich A, Rose C, Kuntsche J, et al. (2011). How stealthy are PEG-PLA nanoparticles? An NIR in vivo study combined with detailed size measurements. Pharm Res 28:1995–2007
- Sethuraman VA, Bae YH. (2007). TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J Control Release 118:216–24
- Sharma P, Brown S, Walter G, et al. (2006). Nanoparticles for bioimaging. Adv Colloid Interface Sci 123–126:471–85
- Silvander M. (2002). Steric stabilization of liposomes – a review. Progr Colloid Polym Sci 120:35–40
- Slingerland M, Guchelaar HJ, Gelderblom H. (2012). Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today 17:160–6
- Stephenson JM, Banerjee S, Saxena NK, et al. (2002). Neuropilin-1 is differentially expressed in myoepithelial cells and vascular smooth muscle cells in preneoplastic and neoplastic human breast: a possible marker for the progression of breast cancer. Int J Cancer 101:409–14
- Sugahara KN, Teesalu T, Karmali PP, et al. (2009). Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 16:510–20
- Sugahara KN, Teesalu T, Karmali PP, et al. (2010). Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328:1031–5
- Teesalu T, Sugahara KN, Ruoslahti E. (2013). Tumor-penetrating peptides. Front Oncol 27:216–23
- Wang X, Wang Y, Chen X, et al. (2009). NGR-modified micelles enhance their interaction with CD13-overexpressing tumor and endothelial cells. J Control Release 139:56–62
- Waite CL, Roth CM. (2011). Binding and transport of PAMAM-RGD in a tumor spheroid model: the effect of RGD targeting ligand density. Biotechnol Bioeng 108:2999–3008
- Xiang Y, Liang L, Wang X, et al. (2011). Chloride channel-mediated brain glioma targeting of chlorotoxin-modified doxorubicine-loaded liposomes. J Control Release 152:402–10
- Xiong XB, Huang Y, Lu WL, et al. (2005a). Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release 107:262–75
- Xiong XB, Huang Y, Lu WL, et al. (2005b). Intracellular delivery of doxorubicin with RGD-modified sterically stabilized liposomes for an improved antitumor efficacy: in vitro and in vivo. J Pharm Sci 94:1782–93
- Yang T, Wang Y, Li Z, et al. (2012). Targeted delivery of a combination therapy consisting of combretastatin A4 and low-dose doxorubicin against tumor neovasculature. Nanomedicine. 8:81–92
- Yang Z, Lei Z, Li B, et al. (2010). Rapamycin inhibits lung metastasis of B16 melanoma cells through down-regulating alpha-v integrin expression and up-regulating apoptosis signaling. Cancer Sci 101:494–500
- Ye Y, Zhu L, Ma Y, et al. (2011). Synthesis and evaluation of new iRGD peptide analogs for tumor optical imaging. Bioorg Med Chem Lett 21:1146–50
- Yu KF, Zhang WQ, Luo LM, et al. (2013). The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine 8:2473–85
- Yue J, Liu S, Wang R, et al. (2012). Transferrin-conjugated micelles: enhanced accumulation and antitumor effect for transferrin-receptor-overexpressing cancer models. Mol Pharm 9:1919–31
- Zhang HT, Li HC, Li ZW, et al. (2011). A tumor-penetrating peptide modification enhances the antitumor activity of endostatin in vivo. Anticancer Drugs 22:409–15
- Zhu Z, Xie C, Liu Q, et al. (2011). The effect of hydrophilic chain length and iRGD on drug delivery from poly(ɛ-caprolactone)-poly(N-vinylpyrrolidone) nanoparticles. Biomaterials 32:9525–35