
Abstract
Uniform mesoporous carbon spheres (UMCS) were used as a carrier to improve the bioavailability of the model drug, celecoxib (CEL). Furthermore, we investigated the mechanism responsible for the improved bioavailability of CEL. The association, adhesion and uptake of UMCS by intestinal epithelial cells were studied by transmission electron microscopy (TEM), fluorescence-activated cell sorting (FACS) and laser confocal scanning microscopy (LCSM). UMCS was found to promote cellular uptake of CEL. Drug transport in Caco-2 cell monolayers proved that UMCS can significantly reduce the rate of drug efflux and improve CEL permeability. The dissolution rate of CEL from drug-loaded samples was markedly improved compared with pure crystalline CEL; moreover, oral bioavailability of CEL loaded into UMCS was also markedly improved compared with that of commercially available capsules. UMCS indicates the advantages and potential of this method to achieve improved oral absorption by increasing the dissolution rate, cellular uptake and permeability of the drug.
Introduction
An increasing number of new chemical drugs are being synthesized due to the development of high throughput screening technology and combinatorial chemistry. However, some of these drugs (about 40%) already in the market exhibit low solubility, dissolution-limited adsorption and poor bioavailability. Hence, this poses a great challenge for pharmaceutical researchers who need to improve their poor gastrointestinal adsorption and bioavailability. Common ways of improving the formulation of poorly water-soluble drugs include the use of micronization, nanosuspension, co-solvent and liposome (Dressman & Reppas, Citation2007; Hauss, Citation2007; Karavas et al., Citation2007; Vogt et al., Citation2008).
In recent years, porous nano-materials (pore diameter 2–50 nm), such as porous starch foam, porous silica and porous clay have been used as drug delivery vehicles due to controllable morphology, high drug adsorption and a controllable pore size, which could give drugs an amorphous form and allow controlled drug release kinetics.
In contrast, porous carbon exhibits many special advantages such as a large surface area, large pore volume and higher absorption (Oh et al., Citation2010; Zhang et al., Citation2010; Deng et al., Citation2011; Zhu et al., Citation2011). In addition, mesoporous carbon materials have found many applications in biomedicine mainly due to their attractive properties, such as good biocompatibility, low toxicity and good chemical stability (Wisniewski et al., Citation2011). Therefore, a number of researches have described the use of porous carbon as a carrier (Kim et al., Citation2008; Wang et al., 2011; Ji et al., Citation2012) and we have previously investigated the effect of different pore structures and morphologies on drug loading and controlled release (Zhao et al., Citation2012a,Citationb). However, there is little information about in vivo studies conducted using mesoporous carbon spheres (particle diameter about 900 nm) to control the release of poorly water-soluble drugs (Liu et al., Citation2009; Zhu et al., Citation2010; Wang et al., 2011). In addition, to our knowledge, there are no published reports about any investigations of their effect on reducing drug side effects, improving intestinal epithelial mucosa drug absorption and permeability.
In the present study we have prepared uniform mesoporous carbon spheres (UMCS) which have a continuous 3-D pore system. Celecoxib (CEL), belonging to the Biopharmaceutical Classification System (BCS) II class of drugs, was chosen as a model drug for our investigations. The present research was aimed at investigating the effect of UMCS as a drug delivery vehicle on the oral bioavailability and permeability of CEL. In addition, the in-vitro cytotoxicity was also investigated. The cellular uptake by Caco-2 cell monolayers of CEL from UMCS were investigated by fluorescence-activated cell sorting (FACS), transmission electron microscopy (TEM) and laser confocal scanning microscopy (LCSM). The pharmacokinetic behavior of CEL-loaded UMCS was compared with the commercial product, CELEBREX, in beagle dogs and the CEL permeability was studied using Caco-2 cell monolayers.
Materials and methods
Materials
Caco-2 cells were purchased from Jiahe Biotechnology Co., Ltd. (Shanghai, China). Coumarin 6 was purchased from Sigma-Aldrich (St. Louis, MO). MTT was purchased from Amreso (Solon, OH). Fetal bovine serum was purchased from Tian Hang Biotechnology Co., Ltd., Beijing, China. Hexadecylamine was obtained from Best-reagent company (Chengdu, China). Tetraethylorthosilicate (TEOS), sulphuric acid, isopropanol, sucrose, ammonia, ethanol and hydrofluoric acid were obtained from Yu Wang Chemical Reagent Company (Shandong, China). Pepsin and pancreatin were obtained from Sigma-Aldrich. Deionized water was prepared by ion exchange and used in all procedures.
Synthesis of UMCS and Coumarin-UMCS
UMCS was synthesized according to the procedure reported by Zhao et al. (2012a,b). Briefly, 3 g hexadecylamine was added to a mixture of 175 mL isopropanol and 225 mL deionized water under continuous stirring. Then, 15 mL tetraethoxysilane (TEOS) and 4.0 mL ammonia were added and then purified by centrifugation. The precipitate was washed several times with ethanol and deionized water. Finally, mesoporous silica spheres (MSS) were obtained by calcination at 550 °C for 5 h. Then, 1 g of MSS was added to a mixture of 0.5 g sucrose, 4 g H2O and 0.1 mL H2SO4. The mixtures were placed in a drying oven, maintained at 92 °C for 6 h, and then the temperature was increased to 160 °C for another 6 h. This was followed by the addition of 0.3 g sucrose, 0.09 mL H2SO4, and 4 g H2O, and the mixtures were again subjected to the same process. Then, the samples were calcined at 900 °C for 6 h under N2, and carbon–silica compounds (C–Si) were obtained. Finally, these C–Si samples were placed in15% hydrofluoric acid for 24 h to remove silica, and UMCS samples were obtained. Coumarin-UMCS was prepared by mixing 20 mg of UMCS in 3 mL of a chloroform solution with coumarin 6 (2 mg/mL) for 2 h and then the mixture was centrifuged. The supernatant was discarded and the sediment was washed several times with ethanol and deionized water. Finally, the precipitate was dried to give coumarin-UMCS.
Drug-loading performance
CEL was loaded into the pores of UMCS by a solvent deposition method. In detail, UMCS was added to an ethanol solution of CEL (50 mg/mL). After gentle stirring for 12 h at room temperature, ethanol was allowed to be completely removed by evaporation at 50 °C for 24 h. The loaded samples were referred as CEL-UMCS.
Characterization methods
The morphology and the nanostructures of UMCS were studied using an SEM apparatus (ZEISS, SUPRA 35, Jena, Germany) and a TEM instrument (FEI, Tecnai G2 F20, Eindhoven, The Netherlands). The pore volumes and size distributions were obtained using a surface area analyzer (SA3100, Beckman Coulter, Pasadena, CA). The Brunauer–Em-mett–Teller (BET) method and the Barett–Joyner–Halenda (BJH) were used to investigate the surface properties, pore size and other nanostructural features. DSC (DSC-60SHIMADZU) was used to obtain information about the physical state of the drugs in the carrier and the CEL-UMCS after stored for 12 months at the heating rate of 10 °C/min. The crystalline characteristics of the samples were determined by an X-ray diffractometer (PW3040/60 PANALYII CALB.V, Almelo, The Netherlands) using Cu-Kα radiation as the X-ray source.
Degree of drug loading
UV Spectrophotometry (Unico, UV-2000, Lincoln, NE) was used to determine the amount of loaded CEL in the UMCS. The drug loading capacity was analyzed by extracting drug loaded samples with ethanol. The extracting solution was then filtrated and examined by UV at 254 nm. The degree of drug loading was calculated according to the following equation:
(1)
In vitro release profile testing
The USP І basket method was used to study the dissolution profiles and the dissolution testing was carried out using a dissolution apparatus (Tianjin University Radio Factory, D 800 LS, Tianjin, China). The dissolution medium consisted of enzyme-free simulated intestinal fluid (pH 6.8) which contained 0.1% sodium-dodecyl-sulfate (SDS) and enzyme-free simulated gastric fluid (pH1.2) obtained by adding 0.2% SDS to achieve sink conditions, and the experiments were conducted at a rate of 100 rpm for 1 h at 37 °C. Samples were taken at intervals and analyzed by UV.
Cellular uptake and cytotoxicity study in Caco-2 cells
In-vitro cytotoxicity study
Caco-2 cells were chosen for cytotoxicity assays of UMCS. Caco-2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, high glucose, 10% fetal bovine serum, l-glutamine, pyruvate, non-essential amino acids) in culture flasks and incubated at 37 °C in an atmosphere of 5% CO2, and the medium was renewed every two days. In each test, the cell suspension diluted with DMEM was seeded onto 96-well plates at 100 μL per well and incubated for 24 h. Then, carrier suspensions with different concentrations, pure CEL and CEL-UMCS suspensions were added to 96-well plates at 20 μL per well and incubated for 12, 24 and 48 h. Following this, 5 mg/mL MTT (Thiazolyl Blue Tetrazolium Bromide) solution was added to 96-well plates at 10 μL per well and incubated for 24 h. The solution was then removed and replaced with 150 μL DMSO. Finally, the absorbance was determined at 492 nm (n = 6) and the cell survival was calculated according to Equation (Equation2(2) ). An optical microscope (Olympus, IX 51, Tokyo, Japan) was used to examine the morphology of the Caco-2 cells.
(2)
LSCM Study
To investigate the cellular uptake of Coumarin-UMCS, 6-well plates with cover glasses were used and Caco-2 cells were plated onto 6-well plates at 2 mL per well with a density of 5.0 × 105 cells/mL and incubated for several days until reaching a confluence of more than 80%. The culture medium was renewed every two days. Following this, the dispersed Coumarin-UMCS suspensions were added to the cells at a concentration of 10 µg/mL and the coumarin solution with the same concentration was added to the cells as a control. After exposure for 2 h, the culture medium was removed and the plates were washed with PBS for several times, then 1 mL of PBS solution containing 3.7% formaldehyde was added to every well and allowed to stand for 25 min. The formaldehyde solution was then removed followed by washing with PBS several times. The cells were then incubated with 1% bovine serum albumin plus 0.1% Triton X-100 in PBS at room temperature for 25 min, then stained with rhodamine-phalloidin in PBS for 60 min. The plates were washed with Hanks balanced salt solution (HBSS) for several times, then stained with 1 µg/mL Hoechst 33258 in PBS for 25 min, finally the fixed cells were determined under LSCM instrument (Leica, TCS NT Sp5, Wetzlar, Germany).
Flow cytometry analysis
Caco-2 cells were plated onto 6-well plates at 2 mL per well with a density of 5.0 × 105 cells/mL and incubated for 24 h. Then, culture medium was removed and 2 mL coumarin solution and dispersed Coumarin-UMCS suspensions were added to every well at a concentration of 100 µg/mL. After exposure for 2 h, the culture medium was removed and the plates were washed with PBS several times. Then, culture medium was added to the 6-well plates at 2 mL per well and incubated for 12 h. This was followed by digestion with pancreatic enzymes for 10 min, centrifugation at 1000 rpm for 6 min and dispersion in PBS. Finally, the samples were examined using a flow cytometer (Becton Dickinson, FACS Calibur, Franklin, MA).
TEM observation
The cellular uptake of UMCS into Caco-2 cells was also observed with TEM. Caco-2 cells were plated onto the 6-well plates at a density of 5.0 × 104 cells/mL and incubated for several days, and the culture medium was then removed followed by the addition of 100 μL UMCS suspension (80 µg/mL in serum-free DMEM) to each well, then incubated for 5 h and following removal of the culture medium, the cells were washed three times with PBS. Cells were collected and centrifuged at 1200 rpm for 6 min, and the supernatant was removed. Then, the cells were fixed with 0.1 mol L−1 PBS solution (containing 2% glutaraldehyde) for 12 h at 4 °C. The cells were then rinsed with 0.1 mol L−1 PBS (containing 2.5% agarose gel) several times, and then post-fixed with 4% osmium tetroxide solution for 1 h. After this, the cells were stained with uranyl acetate for 1 h and dehydrated with ethanol, then embedded in epoxy resin which was polymerized for 48 h at 60 °C. The samples were then cut into ultrathin sections (50–60 nm) with an ultra-microtome, and stained with 5% uranyl acetate and lead citrate solution. Finally, the samples were examined using a TEM instrument (Hitachi, JEOL-200CX-H9000, Tokyo, Japan).
Caco-2 permeability experiments
To investigate the Caco-2 permeability of CEL, Caco-2 cells were plated into Transwell polycarbonate inserts and incubated for 22–27 days, and the culture medium was renewed every two days for the first week, then the culture medium was renewed every other day. The transepithelial electrical resistance (TEER) values should be more than 300 Ω/cm2. Before experiment, the culture medium was removed followed by washing with Hank’s balanced salt solution (HBSS), then incubated with HBSS for 40 min. To measure the basolateral to apical (BL → AP) drug permeation, 1500 μL drug solution was added to the basolateral side and 500 μL HBSS was added to the apical side. For the apical to basolateral (AP → BL) drug permeability, 500 μL drug solution was added to the apical side and 1500 μL HBSS was added to the basolateral side. The drug solutions used were 5 µg/mL CEL and 5 µg/mL CEL with 12 µg/mL UMCS. The plates were shaken (50 rpm) at 95% humidity, 5% CO2, and a temperature of 37 °C. Then, 100 μL samples were taken from the acceptor compartment at predetermined time intervals, and the volume of the acceptor compartment was maintained by immediately adding an equal volume of HBSS. All samples were analyzed in triplicate by HPLC and the apical-to-basolateral and basolateral to apical permeability (apparent permeability coefficient, Papp) were calculated according to the following equation:
(3)
where dQ/dt (mol/s) is the rate of appearance of CEL in the acceptor compartment, A is the surface area of the monolayers (1.13 cm2), and C0 is the initial drug concentration.
Absorption enhancement ratios (AR) were calculated from Papp values using the following equation:
(4)
Efflux ratios (ER) were calculated according to the following equation:
(5)
In vivo pharmacokinetics
The in vivo experiments were performed using male pure-bred beagle dogs (12–15 kg). The animal experiments were carried out according to the guidelines for the use of Laboratory Animals approved by Shenyang Pharmaceutical University. The protocol was approved by the Chinese Animal Ethics Committee. Animals were fed according to the institutional guidelines. Dogs (n = 6) were fasted for 12 h prior to the experiments. A CEL capsule (commercial product CELEBREX which was obtained from Pfizer Pharmaceuticals LLC, NY) was given orally as a control and CEL-UMCS was also given orally in capsules at a dose of 5 mg/kg. Blood samples (approximately 4 mL) were taken from the jugular vein at approximately 0.5, 1, 1.5, 2, 2.5, 3, 3.5 4, 6, 8, 12, 24 and 48 h after oral doses. Then, the samples were centrifuged at 5000 rpm for 10 min, and the supernatant was stored at −20 °C until analysis. For analysis, the supernatant (500 µL) was vortex-mixed with 50 µL internal standards (Carbamazepine, 10 µg/mL), then 2 mL acetidin was added and vortex-mixed for 8 min. The mixture was centrifuged at 12 000 rpm for 6 min. Then, the supernatant were transferred to 1.5 mL Eppendorf tubes for drying under vacuum. The residues were redissolved in 50 µL mobile phase involving of methanol:water (77:23) and vortexed for 2 min. Then, the mixture was centrifuged at 5000 rpm for 5 min. The supernatant (20 µL) was subjected to HPLC. The calibration curve for CEL was linear (R2 > 0.996) over the range 10–1000 µg/L.
Results and discussion
Preparation and characterization of ordered mesoporous carbon carriers
In this work, the hard-template method reported by (Lund et al., Citation2010) was employed to synthesize ordered mesoporous carbon using mesoporous silica as a template. We can synthesize UMCS using mesoporous silica spheres (MSS) as a template. As shown in Figure S1, MSS, carbon-silica complex (C-Si) and UMCS consist of spherical nanoparticles with relatively uniform particle sizes of about 900 nm in diameter. To further investigate the nanostructure of the samples, TEM was used. A mesoporous structure on the surface and within the particles of UMCS was clearly visible (), confirming its 3-D pore system. And it was clear that the pore size and pore volume of UMCS was larger than that of MSS (). For the drug-loaded sample CEL-UMCS (), the pore channels are not as clear as in the case of unloaded carriers, demonstrating that most of the pore channels were filled with the model drug CEL.
Figure 1. TEM images of (A) MSS, (B) UMCS and (C) CEL-UMCS.

The nitrogen adsorption/desorption isotherms are shown in Figure S2, and the surface area, pore volume, pore size and degree of drug loading are presented in . The nitrogen adsorption/desorption properties of UMCS display typical type IV isotherms which indicates a mesoporous structure. The drug-loading capacity of UMCS (up to 0.349 g g−1) was high which may due to its high surface area, large pore volume and stronger adsorbability. This illustrates the potential and advantages of mesoporous carbon for application in drug delivery systems. In addition, the pore volume of CEL-UMCS is much lower than that of UMCS, which indicates that most of the porous channels have been filled by model drug CEL ().
Figure 2. (A) DSC profiles of CEL, UMCS, CEL-UMCS and CEL-UMCS after stored for 12 months. 2(B) XRD pattern of CEL, UMCS, CEL-UMCS and CEL-UMCS after stored for 12 months.
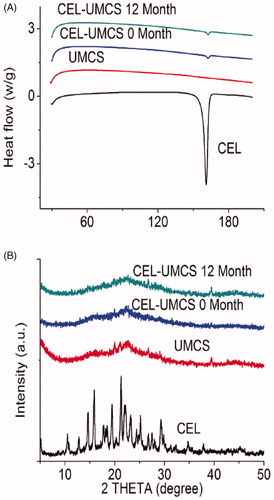
Table 1. Nanoparticle characterization and degree of drug loading of samples.
DSC measurement and XRD analysis
The solid state of UMCS, crude drugs and the drugs loaded in UMCS were assessed by DSC measurement and XRD analysis. As shown in , the endothermic peak of crude CEL was at 162 °C. After drug loaded in UMCS and then stored for 12 months, CEL-UMCS exhibit weaker and wider endothermic peaks. This indicates a transfer in the crystallinity of CLE after entrapment into UMCS. The same results were obtained by XRD analysis. As shown in , the diffractogram of crude CEL showed many distinctive peaks between 10 °C and 35 °C, indicating the highly crystalline characteristics of CEL. The absence of crystalline peaks in the samples of CLE–UMCS confirmed that CLE absorbed into the pores of carriers was in an amorphous form. This agreed with the results of DSC characterizations. Based on the above analysis, we can see that the majority of the drugs were dispersed in the carriers in a noncrystalline state.
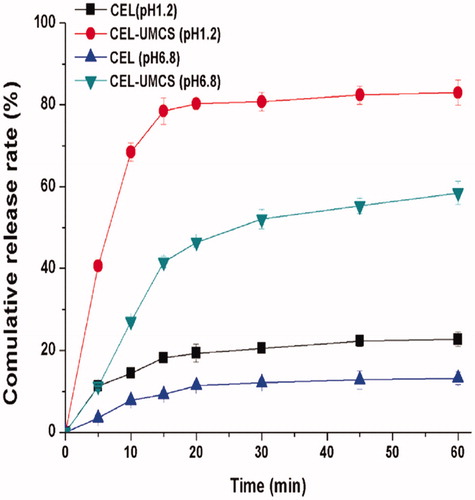
Figure 3. Release profiles of CEL-UMCS and pure crystalline CEL in enzyme-free simulated intestinal fluid (pH 6.8) and enzyme-free simulated gastric fluid (pH 1.2).
In vitro release studies
The in vitro release tests of crude CEL and CEL loaded in UMCS were performed in enzyme-free simulated gastric fluid (pH 1.2) and enzyme-free simulated intestinal fluid (pH 6.8). As shown in , the cumulative amounts of dissolved CEL in pH 1.2 and pH 6.8 medium at 60 min was 26.8% and 14.3%, respectively. However, for CEL-UMCS, the cumulative amounts of CEL in the same conditions improved to 83.4% and 58.8%. CEL is a weakly acidic drug, therefore it has a higher cumulative release rate in the medium of pH 6.8 than pH 1.2. The clearly enhanced dissolution rate of CEL loaded in UMCS may be attributed to the noncrystalline state of CEL loaded in UMCS.
Cellular uptake and cytotoxicity in Caco-2 cells
In-vitro cytotoxicity study
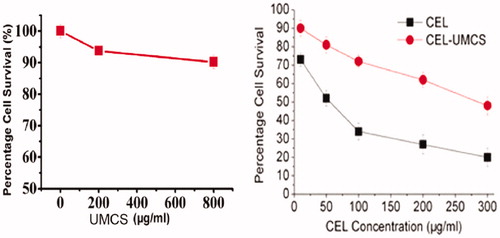
The cytotoxicity of UMCS, pure CEL and CEL-UMCS was determined using Caco-2 cells by incubating them with different concentrations of the samples for 48 h. As shown in , after a 48 h of exposure, UMCS exhibits no apparent cytotoxic effects on Caco-2 cells at the tested concentrations (10–800 μg/mL) indicating that UMCS is fairly biocompatible in vitro which was in accordance with reports by other authors (Kim et al., Citation2008). When the concentration of the carriers increased, the cell survival was slightly reduced, and this weak toxic effect observed at high concentrations might be due to carrier agglomeration (Wick et al., Citation2007) in aqueous culture medium, as a result of strong Vander Waals interactions (Zhang et al., Citation2007). The in-vitro cytotoxicity of pure CEL and CEL-UMCS after incubation with Caco-2 cells for 48 h was shown in . The cell survival was reduced markedly from 73% to 20% by increasing the CEL concentration from 10 to 300 μg/mL. However, compared with pure CEL suspension, the cytotoxicity of CEL-UMCS was much lower with the same molar drug concentration. This may be attributed to the encapsulation and dispersion of CEL by UMCS, suggesting its effect on reducing the toxicity of the loaded drug.
Figure 4. Cytotoxicity study of UMCS, pure CEL and CEL-UMCS on Caco-2 cells. (A) Cells were treated with different concentrations of UMCS for 48 h. (B) Cells were treated with different concentrations of pure CEL and CEL-UMCS suspensions with the same drug molar concentrations for 48 h.
Cellular uptake and flow cytometry analysis
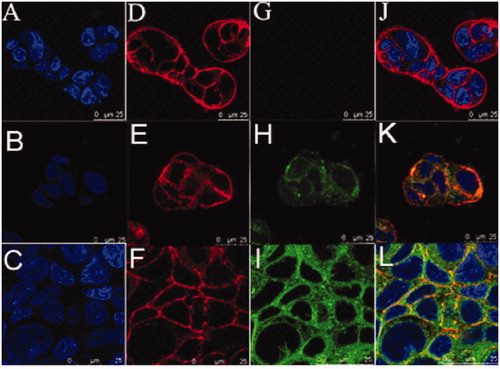
The cellular binding and transport of coumarin in Coumarin-UMCS were investigated with LSCM. The uptake of coumarin by Caco-2 cells after 2 h incubation is shown in . After a 2 h exposure, coumarin can be seen in the cytoplasm () indicating that coumarin was internalized into Caco-2 cells, and the density of coumarin around the nucleus increased markedly after Caco-2 cells incubation with coumarin-UMCS for 2 h, which demonstrates that UMCS could promote the cellular uptake of drug loaded. This was also confirmed by flow cytometric analysis.
Figure 5. Time-course confocal images of Caco-2 cells untreated (A, D, G, J) and Caco-2 cells incubation with coumarin at 2 h (B, E, H, K) and Caco-2 cells incubation with coumarin-UMCS at 2 h (C, F, I, L). Images of the cell nucleus (A, B, C), images of the cell membrane (D, E, F), images of coumarin fluorescence in cells (G, H, I), image of coumarin fluorescence superimposed on the cell nucleus and cell membrane (J, K, L).
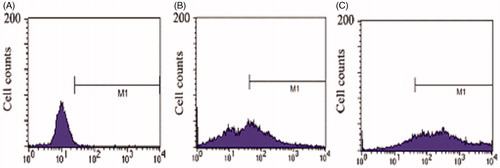
Compared with negative control (), the cellular uptake percentage of Caco-2 cells after incubation with coumarin for 2 h was 43.04% (), indicating that coumarin could be endocytosed by the Caco-2 cells. However, after Caco-2 cells were incubation with Coumarin-UMCS for 2 h, the cellular uptake percentage had a significant increase to 80.57% (). The possible explanation for this enhanced courmain uptake by Caco-2 cells could be explained as followed. First, it has been previously reported by other authors that the smaller the size, the better bioadhesion to enterocytes. For 10 μm particles, only fair deposition were observed while 1μm particles showed higher binding (Lamprecht et al., Citation2001). In addition, the hydrophobic surface of UMCS will also help to increase intestinal epithelium adhesion through hydrophobic interaction (Lai et al., Citation2009). Therefore, UMCS with a diameter of 900 nm and the hydrophobic surface will increase CEL-UMCS binding and association with the underlying epithelium, thereby increasing the courmain concentration at the site of uptake. Second, due to the successful incorporation of coumarin into the pores of UMCS, the porous carrier could probably create a “depot effect” and when the coumarin was released from UMCS in the intestinal tract, a higher concentration gradient was obtained in the gastrointestinal fluids to facilitate the uptake of coumarin. Therefore, it is proposed that the increased coumarin uptake by intestinal epithelium is correlated with the small particle size, hydrophobic surface and porous structure of UMCS.
Figure 6. Flow cytometric histograms of (A) untreated Caco-2 cells (B) Caco-2 cells incubated with coumarin for 2 h (C) Caco-2 cells incubated with coumarin-UMCS for 2 h.
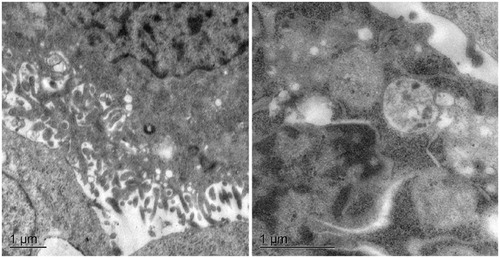
The cellular binding and transport of UMCS were investigated with TEM. As shown in , after incubation with UMCS for 5 h, none of UMCS was observed in the cytosolic or vesicular compartments of the Caco-2 cells, and the result showed that UMCS cannot be endocytosed into Caco-2 cells. So, UMCS cannot accumulate and most of the UMCS is excreted through the digestive tract after oral administration, and this confirms the safety of UMCS as a drug delivery vehicle. Therefore, it seems that particles with a diameter in the 900 nm range could achieve equilibrium between reducing the particle size to promote cell adhesion and choosing an appropriate diameter to guarantee a safety.
Figure 7. TEM images of the cellular uptake of UMCS.
Drug transport in Caco-2 cell monolayers
As is well known, human Caco-2 cell monolayers are usually used to mimic the intestinal epithelium in drug penetration studies. In this research, the results of TEER measurements showed that the cell monolayers are intact and no cellular damage occurred. The transport of CEL across Caco-2 cell monolayers is shown in . The transport of the CEL was linear over the time period studied, and it increased with time. The efflux ratio (ER) and apparent permeability coefficient (Papp) values for the model drug (CEL), alone or with UMCS are shown in . For CEL alone, Papp(BL → AP) is much (about 4.76 times) higher compared with Papp(AP → BL), indicating the high efflux and transport of the model drug. For CEL loaded in UMCS, efflux was reduced sharply, leading to a reduced efflux ratio (ER) about 1.44 and a significant (2.21-fold) increase in CEL absorption enhancement ratio (AR). UMCS carrying a high dose of drug payloads that can overwhelm the drug efflux kinetics may contribute to the sharply reduced ER.
Figure 8. The cumulative transport versus time curves of 13.12 μmol/L CEL in Caco-2 cell monolayer. (A) AP → BL direction, (B) BL → AP direction. Each data point represents the mean ± SD (n = 3).
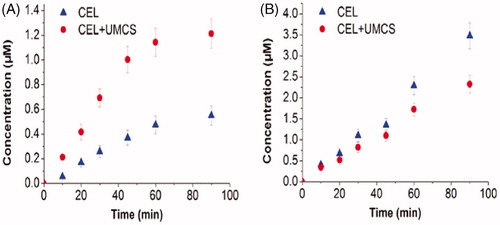
Table 2. Mean Papp of Celecoxib (CEL) in the Direction of BL → AP and AP → BL.
In vivo pharmacokinetics
In order to evaluate the oral bioavailability of CEL, the in vivo pharmacokinetics of CEL Capsules and CEL-UMCS was studied in beagle dogs. The concentration versus time profiles of CEL are shown in and the pharmacokinetic parameters are presented in . It can be clearly seen that the absorption rate of CEL-UMCS was much faster compared with CEL capsules and maximum plasma concentrations were reached to 2.0 h. In addition, the Cmax of CEL-UMCS was much higher than that of CEL capsules, and this may result from the faster dissolution rate of CEL from CEL-UMCS as shown in the in vitro release studies. Moreover, the AUC0–24h of CEL-UMCS was about 1.7-fold higher than that of CEL Capsules, demonstrating an increase in the oral absorption of CEL when administered using mesoporous carbon as carriers.
Figure 9. Plasma concentration–time profiles of CEL capsules and the drug loaded sample (CEL-UMCS Capsules).
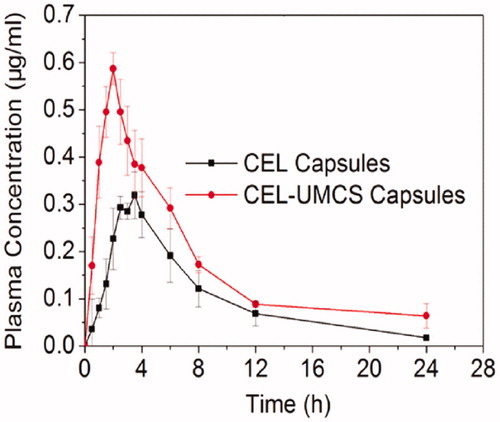
Table 3. Pharmacokinetic parameters of celecoxib after oral administration of CEL Capsules and the drug loaded sample (CEL-UMCS Capsules), mean ± SD, n = 6.
CEL-UMCS was found to have a better oral bioavailability compared with CEL capsules, the possible explanation may be as followed. First, CEL loaded in UMCS is in a noncrystalline state therefore has a greater solubility and faster dissolution rate in the gastrointestinal tract, which was also in agreement with the in vitro release. Second, the moderate particle size, hydrophobic surface and “depot effect” of UMCS could improve the drug uptake by improving the concentration gradient at intestinal epithelium cells. Third, the increased permeability of CEL loaded in UMCS, by overwhelming the drug efflux kinetics, may be one of the reasons for the improved bioavailability.
Conclusions
In summary, we have successfully prepared UMCS and investigated their potential as a carrier for poorly water-soluble drugs. The oral bioavailability of CEL-UMCS capsules increased markedly compared with the commercial product, CELEBREX, in beagle dogs. The oral bioavailability was heavily dependent on the architecture of carrier, which is related to the drug dissolution rate, cellular uptake of drug loaded in UMCS and the permeability of the drug. So, this work proves the potential of UMCS as a drug delivery system and explains the mechanism for the increased oral bioavailability of the model drug, CEL.
Supplemental Material.pdf
Download PDF (29.5 KB)Acknowledgements
We would like to thank Dr. David Jack who is a native English-speaker with sufficient scientific knowledge for correcting language.
Declaration of interest
This work was supported by the National Basic Research Program of China (973 Program) (No. 2009CB930300), National Natural Science Foundation of China (No. 81273449), Major National Platform for Innovative Pharmaceuticals (No.2009ZX09301-012).
References
- Deng ZW, Zhen ZP, Hu XX, et al. (2011). Hollow chitosan-silica nanospheres as pH-sensitive targeted delivery carriers in breast cancer therapy. Biomaterials 32:4976–86
- Dressman J, Reppas C. (2007). Drug solubility: how to measure it, how to improve it. Adv Drug Deliv Rev 59:531–2
- Hauss DJ. (2007). Oral lipid-based formulations. Adv Drug Deliv Rev 59:667–76
- Ji Z, Lin G, Lu Q, et al. (2012). Targeted therapy of SMMC-7721 liver cancer in-vitro and in-vivo with carbon nanotubes based drug delivery system. J Colloid Interface Sci 1:143–9
- Karavas E, Georgarakis E, Sigalas MP, et al. (2007). Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug–polymer interactions. Eur J Pharm Biopharm 66:334–47
- Kim S, Shibata E, Sergiienko R, et al. (2008). Purification and separation of carbon nanocapsules as a magnetic carrier for drug delivery systems. Carbon 12:1523–9
- Lai SK, Wang YY, Hanes J. (2009). Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev 61:158–71
- Lamprecht A, Schäfer U, Lehr CM. (2001). Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res 18:788–93
- Liu Z, Tabakman S, Welsher K, et al. (2009). Carbon nanotubes in biology and medicine: in vitro and in vivo detection, imaging and drug delivery. Nano Res 2:85–120
- Lund K, Muroyama N, Terasaki O. (2010). Accidental extinction in powder XRD intensity of porous crystals: mesoporous carbon crystal CMK-5 and layered zeolite-nanosheets. Micropor Mesopor Mater 128:71–7
- Oh WK, Yoon H, Jang J. (2010). Size control of magnetic carbon nanoparticles for drug delivery. Biomaterials 31:1342–8
- Vogt M, Kunath K, Dressman JB. (2008). Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: comparison with commercial preparations. Eur J Pharm Biopharm 68:283–8
- Wang T, Zou M, Jiang H, et al. (2011a). Synthesis of a novel kind of carbon nanoparticle with large mesopores and macropores and its application as an oral vaccine adjuvant. Eur J Pharm Sci 44:653–9
- Wang XF, Liu P, Tian Y. (2011b). Ordered mesoporous carbons for ibuprofen drug loading and release behavior. Micropor Mesopor Mater 142:334–40
- Wick P, Manser P, Limbach LK, et al. (2007). The degree and kind of agglomeration affect carbon nanotube cytotoxicity. Toxicol Lett 168:121–31
- Wisniewski M, Pacholczyk A, Terzyk AP, et al. (2011). New phosphorus-containing spherical carbon adsorbents as promising materials in drug adsorption and release. J Colloid Interface Sci 354:891–4
- Zhang L, Zeng L, Barron AR, et al. (2007). Biological interactions of functionalized single-wall carbon nanotubes in human epidermal keratinocytes. Int J Toxicol 26:103–13
- Zhang YZ, Zhi ZZ, Jiang TY, et al. (2010). Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J Control Release 145:257–63
- Zhao P, Jiang H, Jiang T, et al. (2012a). Inclusion of celecoxib into fibrous ordered mesoporous carbon for enhanced oral bioavailability and reduced gastric irritancy. Eur J Pharm Sci 45:639–47
- Zhao P, Wang L, Sun C, et al. (2012b). Uniform mesoporous carbon as a carrier for poorly water soluble drug and its cytotoxicity study. Eur J Pharm Biopharm 80:535–43
- Zhu M, Wang HX, Liu JY, et al. (2011). A mesoporous silica nanoparticulate/β-TCP/BG composite drug delivery system for osteoarticular tuberculosis therapy. Biomaterials 32:1986–95
- Zhu SM, Chen CX, Chen ZX, et al. (2010). Thermo-responsive polymer-functionalized mesoporous carbon for controlled drug release. Mater Chem Phys 126:357–63