
Abstract
Context: Angiotensin II receptor blockers (ARBs), angiotensin-converting enzyme inhibitors (ACEIs) are some of the most commonly prescribed medications for hypertension.
Objective: Most of all conventional dosage forms of ARBs and ACEIs undergo extensive first-pass metabolism, which significantly reduces bioavailability. Majority of ARBs and ACEIs are inherently short acting due to a rapid elimination half-life. In addition, oral dosage forms of ARBs and ACEIs have many high incidences of adverse effects due to variable absorption profiles, higher frequency of administration and poor patient compliance.
Methods: Many attempts have been made globally at the laboratory level to investigate the skin permeation and to develop transdermal therapeutic systems of various ARBs, ACEIs and other anti-hypertensives, to circumvent the drawbacks associated with their conventional dosage form.
Results: This manuscript presents an outline of the transdermal research specifically in the area of ARBs, ACEIs and other anti-hypertensives reported in various pharmaceutical journals.
Conclusion: The transdermal delivery has gained a significant importance for systemic treatment as it is able to avoid first-pass metabolism and major fluctuations of plasma levels typical of repeated oral administration. As we can experience from this review article that transdermal delivery of different ARBs and ACEIs improves bioavailability as well as patient compliance by many folds. In fact, the rationale development of some newer ARBs, ACEIs and other anti-hypertensives transdermal systems will provide new ways of treatment, circumventing current limitations for conventional dosage forms.
Introduction
Transdermal drug delivery is defined as self-contained, discrete dosage forms which, when applied to the intact skin, deliver the drug, through the skin at controlled rate to the systemic circulation. Transdermal therapeutic system (TTS) established itself as an integral part of novel drug delivery systems. The concept of delivering drugs via transdermal route for systemic treatment of diseased states is gaining increasingly great importance due to several well-known advantages that include: (i) avoidance of pre-systemic metabolism, (ii) allowing lower daily doses, (iii) reducing the dosing frequency, (iv) blood or plasma levels of the drug can be kept within the therapeutic window for extended periods of time, (v) prolonging drug action, (vi) lesser inter- and intra-patient variability, as a consequence patient compliance and acceptability improved (Aqil et al., Citation2007; Ahad et al., Citation2009). The TTS has experienced a tremendous growth over the past decade, because of the increasing number of drugs that can be delivered to the systemic circulation in clinically effective concentrations via the skin (Shin & Choi, Citation2003; Mundargi et al., Citation2007). So far, transdermal delivery has been approved for the following drugs: scopolamine (motion sickness), nitroglycerin and isosorbide dinitrate (angina pectoris), clonidine (hypertension), estradiol (hormone replacement therapy), fentanyl (analgesia), nicotine (smoking cessation), testosterone (hypogonadism), norelgestromin + ethinyl estradiol (contraception), oxybutynin (incontinence), selegiline (depression), methylphenidate (attention deficit hyperactivity disorder), buprenorphine (analgesia), rivastigmine (dementia), rotigotine (anti-Parkinsonian) and granisetron (antiemetic) (Prausnitz et al., Citation2004; Prausnitz & Langer, Citation2008).
Hypertension is one of the main factors for increasing the risk of developing kidney disease, hardening of the arteries, eye damage and stroke (Cubillos-Garzon et al., Citation2004). Hypertension affects approximately 78 million individuals in the United States and approximately 1 billion worldwide (Basile, Citation2003; Berkow & Barnard, Citation2005; Go et al., Citation2014). As the population ages, the prevalence of hypertension will increase even further unless broad and effective preventive measures are implemented. The report from the Framingham heart study suggest that individuals who are normotensive at the age of 55 years have a 90% lifetime risk for developing hypertension (Vasan et al., Citation2001, Citation2002).
Patients with hypertension needs long-term treatment and sometimes lifelong therapy is advised, as TTS offer a better quality of life, they are more accepted than the oral dosage forms (Lake & Pinnock, Citation2000; Saroha et al., Citation2011); therefore, drug deliveries via transdermal route are ideally suited for ailments like hypertension that need chronic treatment (Ahad et al., Citation2011a; Harries & Armstrong, Citation2012; Bairwa et al., Citation2014). In addition, good number of anti-hypertensive drugs undergo extensive first-pass metabolism which too can be avoided by transdermal therapy (Jain et al., Citation2008). Hence, anti-hypertensive agents of both therapeutic and prophylactic usage have been subjected to transdermal investigation.
Several studies have been investigated to develop TTS of anti-hypertensive drug(s). Ahad et al. (Citation2014a) and Aqil et al. (Citation2006b) reviewed and detailed the studies focused on the transdermal delivery of various β-blockers. While Gungor & Ozsoy (Citation2012) and Selvam et al. (Citation2010) have done a brief overview on the different anti-hypertensive drugs delivered via skin in the literature. Recently, Ahad et al. (Citation2013a), reviewed and detailed the transdermal research specifically on calcium channel blockers for the management of hypertension. Present review shows an outline of the transdermal research in the area of some class of newer anti-hypertensives, viz. angiotensin receptor blockers (ARBs), e.g. losartan (LS), valsartan (VS), olmesartan (OS), telmisartan (TS), candesartan (CS); angiotensin-converting enzyme inhibitors (ACEIs), e.g. captopril (CP), enalapril (EP), lisinopril (LP), ramipril (RP); thiazide-like diuretic, e.g. indapamide (IP); and potassium channel openers, like pinacidil (PD) reported in various pharmaceutical journals. The advances and innovations in the transdermal delivery of these classes of drugs for hypertension are discussed in the text and a summary is presented in .
Table 1. Research advances in percutaneous delivery of ARBs, ACEIs and others via skin.
Angiotensin receptor blockers
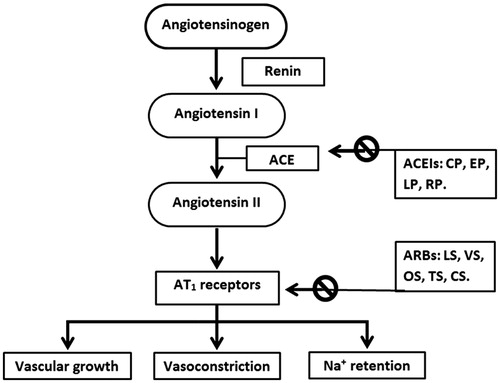
Angiotensin II receptor antagonists, also known as ARBs, AT1-receptor antagonists or sartans, are a group of pharmaceuticals that modulate the renin–angiotensin–aldosterone system (). ARBs may be used alone or combined with other medicine often a diuretic; to treat high blood pressure (BP). ARBs are primarily used for the treatment of hypertension where the patient is intolerant of ACEIs therapy (Barreras & Gurk-Turner, Citation2003). ACEIs can cause an annoying cough. ARBs do not inhibit the breakdown of bradykinin or other kinins, and are thus only rarely associated with the persistent dry cough and/or angioedema that limit ACEIs therapy (Terra, Citation2003). ARBs use has been shown to decrease the rate of progression of kidney disease in patients with diabetes, independent of their ability to lower BP. ARBs also have additional benefits in patients with congestive heart failure and possible coronary artery disease (Tronvik et al., Citation2003). Because of these benefits; several national and international guidelines recommend the use of ARBs as a first-line antihypertensive agent when patients have a diagnosis of diabetes. ARBs should be considered first-line antihypertensive agents for all patients with diabetes without contraindications for use, i.e. angioedema, bilateral renal artery stenosis. ARBs should also be considered for patients already on thiazide diuretics who require second-line therapy. Their main uses are in the treatment of hypertension, diabetic nephropathy and congestive heart failure (Lewis et al., Citation2001; Gales et al., Citation2010).
Figure 1. Site of action of ARBs and ACEIs. The ARBs can act on the cardiac, renal, vascular or central adrenergic receptors. The formation of angiotensin II involves a consecutive breakdown of angiotensinogen. The angiotensinogen is converted into angiotensin I by the action of renin, which is then hydrolyzed into angiotensin II by the activity of angiotensin-converting enzymes. The ACEIs inhibit the conversion of angiotensin I into angiotensin II, decreasing its level in the blood. The ARBs act on the AT1 receptors, responsible for all known effects of angiotensin II, including vasoconstriction, aldosterone release and effects on the myocardium and vasculature. In the end, there will a fall in the sympathetic activation. ACE: Angiotensin converting enzyme; CP: Captopril; CS: Candesartan; EP: Enalapril; LP: Lisinopril; LS: Losartan; OS: Olmesartan; RP: Ramipril; TS: Telmisartan; VS: Valsartan.
Losartan
Losartan was the first ARB and it is widely used for the management of hypertension (Velasquez, Citation1996). LS is the drug of choice for sustained release formulation since it has a short half-life of about 1.5 to 2 h, which requires frequent dosing necessary to maintain the therapeutic blood level for long-term treatment. LS shows considerable first-pass metabolism in the liver and thereby has poor bioavailability (25–35%) when administered orally (Carr & Prisant, Citation1996). LS has a molecular weight of 422.91 and logarithm partition coefficient (log P) of 4.5, again indicates its suitability for administration by the transdermal route (Goa & Wagstaff, Citation1996). In 2012, Baviskar and Parik, developed a transdermal patch of LS using different blends of polymers such as ethyl cellulose (EC):polyvinyl pyrrolidone (PVP) (hydrophobic and hydrophilic polymers), eudragit (E)-RL100:E-RS100 and polyvinyl alcohol; PVP using different ratios by the solvent casting technique. The formulation containing E-RL100:E-RS100 (hydrophobic polymers) showed satisfactory drug release amount and pattern as compared to the combination of EC:PVP and polyvinyl alcohol:PVP. Hence, the study showed that transdermal patch of LS with hydrophobic polymers exhibit good release properties as compared to that of hydrophilic polymers and combination of both hydrophobic and hydrophilic polymers (Baviskar et al., Citation2012). In another study, a monolithic TTS was developed for sustained anti-hypertensive effect of LS using the polymers E-E100 and PVP-VA64. It was observed that the prepared transdermal formulation enhance the relative bioavailability of LS by 2.2 times with reference to an oral delivery. The increased bioavailability might be due to elimination of hepatic first-pass metabolism. Thus, the transdermal formulation with polymeric composition of E-E100 and PVP-VA64 (5:3) was found to provide prolonged steady-state concentrations of LS with minimal fluctuations and improved bioavailability (Shams et al., Citation2010b). The anti-hypertensive activity of the developed formulation was also investigated (Shams et al., Citation2010a). The BP lowering activity was carried out using tail cuff method in Wistar albino rats. Hypertension was induced by methyl prednisolone acetate subcutaneously for 2 weeks. Authors claimed a significant decrease in BP (25.42% reduction in mean systolic BP of rats), which is in proximity of the normal value and it was maintained for 24 h. It was concluded that the developed transdermal matrix patch holds promise for the management of hypertension upto 24 h.
In another report, the matrix-type TTS containing LS was prepared with different polymers like EC, hydroxy propyl methyl cellulose (HPMC) and E-RL100 in varied ratios. All formulations carried dimethyl sulfoxide as penetration enhancer and propylene glycol as plasticizer in chloroform and ethanol as solvent system. The formulation, F1 with combination of polymers (4:1) emerging to be ideal formulations for LS. The optimized formulation containing HPMC:E-RL100 (4:1), with enhancer dimethyl sulfoxide showed 87.13% drug release after 24 h. It was observed that the prepared TTS of LS had shown good promising results for all the evaluated parameters and increase the efficacy of LS for the therapy of hypertension (Rokade et al., Citation2012).
The feasibility of proniosomes as TTS for LS was also reported, different preparations of proniosomes were fabricated using different non-ionic surfactants, such as Span 20, Span 40, Span 60, Span 80, Tween 20, Tween 40 and Tween 80. Different formulae were prepared and coded as proniosomal gel-1 (PNG-1) to PNG-7. The best in vitro skin permeation profile was obtained with PNG-2 in 24 h. This optimized PNG-2 was fabricated as TTS using HPMC gel as a suitable base. Proniosomal TTS prepared with HPMC (PNP-H) was found to be the optimized, as it presented improved release of drug and better permeation in a steady-state manner up to 24 h across rat skin. Pharmacokinetic study of PNP-H showed a significant increase in bioavailability (1.93 times) compared with oral formulation of LS (Thakur et al., Citation2009).
Valsartan
Valsartan is a highly selective angiotensin II type 1 receptor blocker that has been widely used for the treatment of hypertension. When administrated orally in humans, VS is rapidly absorbed. It has a favorable log P (4.5) and mean half-life (7.5 h) (McInnes, Citation1999; Beg et al., Citation2012). Its Cmax occurs at 2 to 4 h, and it is then excreted into bile. The oral bioavailability of VS is 10–35%, which is mainly due to poor absorption in the gastrointestinal tract. Additionally, food intake is known to reduce the Cmax and AUC of VS by 50 and 40%, respectively (Israili, Citation2000). The transdermal administration of VS is a possible solution to overcome these problems. In 2008, Rizwan et al., investigated the feasibility of VS for transdermal delivery and examined the effect of various terpenes, namely forskolin, 1,8-cineole, d-limonene, l-menthol and linalool on skin permeation of VS. Authors exhibited that, no apparent skin irritation (erythema, edema) was observed on treatment of skin with aforementioned terpenes. Authors concluded that 1,8-cineole was found to be the most effective enhancer for diffusion of VS through rat skin (Rizwan et al., Citation2008). Very recently, Ahad et al. (Citation2014b), developed transdermal carbopol gel formulation bearing VS and 1,8-cineole. The optimized formulation demonstrated highest transdermal flux, with an enhancement ratio of 4.53 when compared to control gel formulation. Incorporation of 1,8-cineole and ethanol in gel formulation enhance the permeation of VS significantly. Skin irritation study revealed that the developed formulation was safe, less irritant and well tolerable formulation for transdermal delivery and was successful in reverting the rat BP to normal values in experimental hypertensive rats.
Ahad et al. (Citation2011b) investigated the effectiveness and mechanism of seven novel terpenes, such as iso-eucalyptol, β-citronellene, valencene, rose oxide, safranal, lavandulol acetate and prenol, as potential penetration enhancers for improved skin permeation of VS through rat skin with reference to established terpene eucalyptol. It was reported that among all investigated terpenes, iso-eucalyptol produced the maximum enhancement via rat skin over control. Fourier transform infrared spectroscopy spectra and differential scanning calorimetry thermogram of skin treated with terpenes showed that permeation occurred due to the disruption of lipid bilayers. No apparent skin irritation (erythema, edema) was observed on treatment with terpenes except β-citronellene, safranal, lavandulol acetate and prenol, which caused mild irritation. In (Ahad et al., 2014) optimized a transdermal gel formulation of VS using the Box–Behnken design containing iso-eucalyptol as permeation enhancer and evaluated for pharmacokinetic study. The independent variables were carbopol 940, polyethylene glycol 400 and ethanol, while transdermal flux, Tlag, and gel viscosity were the reported dependent variables. It was described that the permeation rate of VS significantly increased in direct proportion to the ethanol concentration, but decreased on increasing the concentration of polymer. In vivo pharmacokinetic study showed a significant increase in the bioavailability (2.52 times) compared with oral formulation. Overall, it was accounted that iso-eucalyptol can be successfully used as a potential permeation enhancer for the enhancement of skin permeation and bioavailability of lipophilic drug (Ahad et al., Citation2014c). In a previous study, a monolithic drug-in-adhesive patch of VS was investigated (Nishida et al., Citation2010). Patch containing isopropyl myristate, n-octyl-β-d-thioglucoside and diisooctyl sodium sulfosuccinate as percutaneous promoters were investigated for in vitro permeation study across hairless mouse and Yucatan micro pig skin. A combination of isopropyl myristate/diisooctyl sodium sulfosuccinate was found to be the most effective permeation enhancer for the transdermal delivery of VS. It was demonstrated that the combination of isopropyl myristate/diisooctyl sodium sulfosuccinate may be useful for the development of a practical drug-in-adhesive patch for VS.
Bhosale & Avachat (Citation2013) developed TTS containing VS ethosomal formulations, amongst the various ethosomal formulations developed, ETC5 ethosomal suspension (phospholipon 90H (4%) and ethanol (40%) was found to have highest entrapment efficiency, transdermal flux as compared to the control. Authors claimed better and prolonged anti-hypertensive activity of VS ethosomes formulation as compared to oral administration in Wistar rats., Ahad et al. (Citation2012, Citation2013b) optimized and developed nanoethosomes and nanotransfersomes for transdermal VS delivery using the Box–Behnken experimental design and evaluated them for various parameters like entrapment efficiency, vesicles shape, size, size distribution and in vitro skin permeation. It was documented that both nano-formulations proved superior in terms of amount of drug permeated in the skin, entrapment efficiency and vesicles size as compared to rigid liposomes. In addition, nanoethosomes and nanotransfersomes showed better bioavailability enhancement and anti-hypertensive activity, respectively, in comparison to control formulation by virtue of better permeation through Wistar rat skin.
Olmesartan
Olmesartan is a selective AT1 subtype angiotensin II receptor antagonist. It exhibits more than 12 500-fold greater affinity for the angiotensin II receptor type 1 than for the angiotensin II receptor type 2, making it theoretically the second most potent agent (Destro et al., Citation2009). Oral OS 10–40 mg once daily is recommended for the treatment of adult patients with hypertension (Scott & McCormack, Citation2008). It is an effective and well-tolerated agent, with a long duration of action, and single daily dose may be used to treat hypertension (Manoria, Citation2006). Orally administered OS-medoxomil was rapidly absorbed from the gastrointestinal tract and converted during absorption to OS, the pharmacologically active metabolite that was subsequently excreted without further metabolism. The absolute bioavailability of OS from OS-medoxomil tablets was 28.6% (Laeis et al., Citation2001). Above properties of OS makes it a good candidate for the development of TTS. Recently, the colloidal soft nano-carrier system (microemulsion) for the transdermal delivery of OS-medoxomil was prepared using oleic acid as the oil phase, Labrasol® as a surfactant, Transcutol® as a co-surfactant, and water and examined for its in-vivo performance in human volunteers. The physicochemical and spectroscopic methods revealed the presence of water-in-oil and bicontinuous structures. Biophysical assessment demonstrated various stratum corneum changes. OS was delivered successfully across the skin and achieved improved bioavailability in comparison to commercial oral tablets with a more sustainment behavior. It was concluded that the developed microemulsion system considered proved potentially a useful vehicle for the transdermal delivery of lipophilic drugs like OS as demonstrated from the accomplished ex-vivo and in-vivo studies (Hathout & Elshafeey, Citation2012).
Telmisartan
Telmisartan is used in the management of hypertension. TS bind to the angiotensin II type 1 (AT1) receptors with high affinity, causing inhibition of the action of angiotensin II on vascular smooth muscle, ultimately leading to a reduction in arterial BP. It is used alone or in combination with other classes of anti-hypertensives for the treatment of hypertension (Sharpe et al., Citation2001). It is also indicated in the treatment of diabetic nephropathy in hypertensive patients with type 2 diabetes mellitus, as well as the treatment of congestive heart failure (only in patients who cannot tolerate ACEIs). TS exhibited bi-exponential decay kinetics with a terminal elimination half-life of approximately 24 h and absolute bioavailability of the TS is 42–58% (Wienen et al., Citation2000; Kausalya et al., Citation2011). Recently, Amin et al. (Citation2013) prepared transfersomes for enhanced skin absorption of TS. The prepared optimized transfersomes vesicles showed 81% entrapment efficiency, spherical in shape and size of 142 nm. The transfersomal gel showed 8-folds increase in transdermal flux as compared to control gel. Author accounted that the enhancement in transdermal permeation was attributed to hydration gradient built up across the skin as well as thinning of the epidermis due to formulation components.
Candesartan
Candesartan may be used alone or with other agents to treat hypertension. It is administered orally as the prodrug, CS-cilexetil, which is rapidly converted to its active metabolite during absorption in the gastrointestinal tract (Easthope & Jarvis, Citation2002). CS lowers BP by antagonizing the renin–angiotensin–aldosterone system; it competes with angiotensin II for binding to the type-1 angiotensin II receptor (AT1) subtype and prevents the BP increasing effects of angiotensin II. Unlike ACEIs drugs, CS does not have the adverse effect of dry cough. CS may be used to treat hypertension, isolated systolic hypertension, left ventricular hypertrophy and diabetic nephropathy. It may also be used as an alternative agent for the treatment of heart failure, systolic dysfunction, myocardial infarction and coronary artery disease (Detroja et al., Citation2011).
Recently, proniosomal formulation was prepared by using different surfactants, cholesterol and soya lecithin ratios. Authors claimed that, encapsulation efficiency of CS was found higher in proniosomes prepared with Span than formulated using Tweens. Vesicles formed with Spans were found smaller in size than vesicles prepared with Tweens. An optimized formulation prepared with 9:2:9 ratio of Span 60, cholesterol and lecithin gave maximum encapsulation efficiency and presented drug release in a controlled manner with a transdermal flux of 1.89 μg/cm2/h. It was demonstrated that proniosomes are promising and stable delivery system for CS cilexetil (Devi et al., Citation2014).
In 2013, Mittal et al. developed a proniosomal carrier system for enhanced transdermal CS delivery. Author claimed that the particle size of developed carrier system was found in the range of 175 to 277 nm with spherical and homogenous structure. It was concluded that the developed proniosomal gel are suitable for CS once a day controlled release formulation. Authors reported that the CS-loaded proniosomal formulation proved to be non-irritant to skin, exhibited enhanced BP lowering effects and reasonably well stability characteristics.
In another study, microemulsions containing olive oil, Tween 80 and isopropyl alcohol for transdermal CS delivery were prepared. Developed microemulsions formulations were evaluated for in vitro skin permeation and stability. It was mentioned in the study that the microemulsion containing olive oil (72%), water (8%), Tween 80 (15%) and isopropyl alcohol (5%) showed maximum viscosity (29.54 mPas), small droplet size (180.90 nm), and smaller polydispersity index (0.37), with enhanced CS transdermal flux across excised porcine skin (Malakar et al., Citation2013).
Previously, the TTS of CS were formulated using PVA with PVP K30 and E-RS 100 with HPMC as a blend of two in different concentrations of polymers by solvent casting method. Polyethylene glycol 300 in different concentration (8, 16 and 24%) was added as plasticizer. It was observed that the diffusion rate of CS is increase from the patches on increasing the concentration of plasticizers, while CS formulated patches comprising polyethylene glycol 300 (24%) demonstrated enhanced rate of diffusion than the patches prepared with polyethylene glycol 300 (16%) and polyethylene glycol 300 (8%). Among polymers, combination of E-RS and HPMC was found more suitable and had enhanced diffusion rate than the combination of PVA and PVPK30 in all formulations (Quadri et al., Citation2012).
Angiotensin-converting enzyme inhibitors
Briefly, ACEIs are medicines that are used to treat high BP. Most of these medicines have names that end in “pril”. ACEIs block the conversion of angiotensin I to angiotensin II, thereby decreasing the tension of blood vessels and blood volume, thus lowering BP (). They are particularly recommended for people who are under the age of 55. ACEIs are effective antihypertensive agents and they have cardio-protective benefits over and above those related to BP lowering. Clinical trials have consistently revealed that they are safe in the post–myocardial infarction period and, subsequently, with a very low risk of significant side effects. These records endorse the recommended role of ACEIs as first-line agents in cases with diabetes who are at a particularly high risk for cardiovascular consequences, including those peoples who previously had myocardial infarction, heart failure and kidney disease (Brown & Vaughan, Citation1998; Gerstein, Citation2000; Izzo & Weir, Citation2011; Helal & Lane, Citation2014).
Captopril
Captopril has been widely used for the treatment of hypertension and congestive heart failure. The drug is considered as a drug of choice in anti-hypertensive therapy due to its effectiveness and low toxicity (Nur & Zhang, Citation2000). It has a short half-life of 1 to 3 h and 65% oral bioavailability but presence of food reduces the oral absorption by 30–50% (Duchin et al., Citation1982; Desai et al., Citation2008). According to a previous research, the oxidation rate of CP in dermal homogenate is significantly lower than the intestinal homogenate because the oxidative product of CP, a CP disulfide shows poor absorption from the intestine (Zhou & Li Wan Po, Citation1994). These properties make it a suitable candidate to be delivered transdermally at a controlled rate.
In vivo skin permeation study in rats showed that iontophoresis could effectively enhance the transdermal transport of CP without producing any significant skin irritation. It was reported that no obvious skin irritation was observed after 9 h continuous iontophoresis treatment (Wang & Hou, Citation2000). It was concluded that both conventional direct current and pulsed-mode direct current constant-current iontophoresis of CP offer a safe and effective overture of mean arterial BP reduction in rabbits with induced hypertension (Zakzewski et al., Citation1992).
The potential of proniosomes as a TTS for CP was investigated by encapsulating the drug in various formulations of proniosomal gel composed of various ratios of sorbitan fatty acid esters, cholesterol, lecithin prepared by coacervation-phase separation method. The results concluded that in vitro studies showed prolonged release of entrapped CP. Encapsulation efficiency of prepared proniosomes was found between 66.7 and 78.7%. It was concluded that proniosomes are a promising prolonged delivery system for CP and have reasonably good stability characteristics (Gupta et al., Citation2007).
In a study, a wide range of transdermal drug-in-adhesive patches were prepared comprising ethyl ester of CP using two commercially available bioadhesive polymers to optimize drug release from prepared patches. Drug release profiles of suitable patches were obtained using both polydimethylsiloxane (silastic) and porcine skin in vitro. Diffusion results across silastic showed a gradual plateau in flux with an increased drug loading that may be attributable to intramolecular interactions, while flux across porcine skin was seen to increase with an increasing patch thickness and attained a therapeutic level. It was described that adhesion and drug loading are significant factors in optimizing a topical patch formulation for the delivery of CP (Gullick et al., Citation2010). In another study, authors claimed that TTS of CP prepared using a low temperature casting method performed better in comparison to those prepared using same method but at room temperature (Dubey et al., Citation1995).
Recently, TTS containing CP were developed by using synthetic and pH-independent polymers namely, E-RL 100 and E-RS 100. In vitro and ex vivo diffusion rate studies were performed through various synthetic membranes with different thickness, pore size and type and through human skin by using Franz diffusion cells. It was observed that the type of membrane and composition of the formulation affected the diffusion profiles of CP from the TTS. It was concluded that, controlled in vitro release for each developed formulation was observed. Amongst the formulations, especially two of the formulations (F15 and F16) are considered to be suitable to administer CP via skin (Kerimoglu et al., Citation2013).
In 2007, Jain and Joshi developed a matrix diffusion type TTS of CP employing different ratios of polymers, EC and HPMC as (3:1) and (2:2). The in vitro skin permeation and in vitro dissolution studies showed that CP release was more in matrices containing ratio of EC:HPMC as 2:2 compared to 3:1. CP from matrix containing EC:HPMC ratio of 2:2 was able to penetrate through rabbit abdominal skin. The in vivo study shows that the prepared matrices were free from any skin irritation potential and furthermore, prepared formulations found to be stable for 90 days (Jain & Joshi, Citation2007).
Park et al. (Citation2001) developed a monolithic adhesive matrix type patches containing penetration enhancers like dimethyl sulfoxide, N-methylpyrrolidone, oleic acid, transcutol, propylene glycol, polysorbate 20, oleyl alcohol and lauryl alcohol and CP (20%). Amongst the enhancers, fatty alcohols resulted in a marked enhancing effect on the skin permeation of CP, while dimethyl sulfoxide, N-methylpyrrolidone, oleic acid, transcutol and polysorbate 20 showed no significant enhancing effect. Decanol was also found as a penetration enhancer for improved delivery of CP via transdermal route (Wu et al., Citation1998). It was reported that the CP enhancement via rabbit skin by fatty alcohols and aliphatic esters were related to the chain length of enhancers. C6–C10 fatty alcohols, butyl acetate and N-dodecyl-Y-lactamat were enhancers presented highest penetration enhancement of CP (Wu et al., Citation1997).
In another study, the effect of the simultaneous use of l-menthol and ethanol on the skin permeation of hydrophilic (CP and nicardipine hydrochloride, atenolol) and lipophilic (nifedipine, vinpocetine and nilvadipine) cardiovascular agent was investigated. Pronounced enhancing effect by the l-menthol-ethanol system was found independent of drug lipophilicity, but the mode of action was dependent on the lipophilicity of the drug. The action of the system on lipophilic drugs (nifedipine, vinpocetine and nilvadipine) was mainly due to their increase in solubility in the system, while that on hydrophilic drugs (CP, nicardipine hydrochloride and atenolol) was the result of increase in their skin permeability coefficient (Kobayashi et al., Citation1993).
Enalapril
Enalapril is used in the treatment of hypertension, congestive heart failure and to alleviate strain on heart damaged by heart attack. EP has low log P of 0.19 and the effective half-life following multiple doses is 11–14 h. It has low bioavailability (about 40–60%) due to its hepatic first-pass metabolism. The administration of this antihypertensive drug via the transdermal route is needed to achieve controlled release in order to minimize adverse effects associated with oral administration and to improve its therapeutic efficacy and bioavailability (Ferguson et al., Citation1982; Riley et al., Citation1985). Aqil et al. (2008) investigated the feasibility of piperidine hydrochloride as penetration enhancer for improved EP transdermal delivery. The EP TTS containing piperidine hydrochloride (10% w/w) was prepared by solvent evaporation technique using polymers E-E100 and PVP K-30 in varying ratios. The optimized formulation provided 87.3% drug release in-vitro and a flux of 380 µg/cm2/h over a period of 48 h and there were no signs of skin irritation on application of patch. The optimized formulation was stable with a tentative shelf life of two years. There was 3-fold betterment in bioavailability was detected with developed TTS over marketed tablet. Significant fall in BP was also found in experimental hypertensive rats which were maintained for 48 h. These preclinical studies indicate the feasibility of matrix-type TTS of EP for 2-day management of hypertension (Aqil et al., Citation2008). In another study, different concentrations and polymeric grades of HPMC (K4M, K15M and K100M) for the development of transdermal patch of EP was investigated. It was reported that the thickness and weight of patch increase with the increase in polymeric grade and content. The in vitro drug release followed Higuchi kinetics as its coefficient of correlation values predominates over first-order kinetics (Gavali et al., Citation2010).
Lisinopril
Lisinopril is used against chronic conditions like hypertension, diabetes nephropathy and cardiac heart failure. It does not undergo metabolism and excreted unchanged in urine. The log P and half-life of LP is −1.2 and 12 h, respectively, thus effective as single daily dose medication, but severely suffers from average inter-subject bioavailability of 25% (Gomez et al., Citation1987; Kelly & O'Malley, Citation1990). Hence, transdermal delivery could be an alternative overture that will improve the bioavailability of drug and also sustained the drug release for long periods. To overcome the poor oral bioavailability of the LP, transdermal patches were prepared using HPMC and polyvinyl alcohol in 1:1 ratio as polymeric matrix using glycerol (6%) as plasticizer. Isopropyl alcohol and oleic acid alone and in combination were added as the penetration enhancers in different concentrations and ratios. It was described that blend of oleic acid and isopropyl alcohol produced more pronounced release via goat skin, which clearly dictates the synergistic effect of the enhancers if used in combination and the effect found to be dependent on the concentration of the enhancers (Banweer et al., Citation2010).
In (Gannu et al. Citation2010) optimized the iontophoretic important process parameter for enhanced delivery of LP using the statistical Box–Behnken design. While in another report (Gannu et al., Citation2009), the combined effect of three independent variables on the permeation kinetics of LP from transdermal hydrogels was optimized. A three-factor, three-level Box–Behnken design was used to optimize the independent variables, Carbopol 971 P, menthol and propylene glycol. The dependent variables selected were cumulative amount permeated across rat abdominal skin in 24 h (Q (24), flux, and lag time). The use of the Box–Behnken design approach helped in identifying the critical formulation parameters in the transdermal delivery of LP from hydrogels.
Ramipril
Ramipril is an antihypertensive drug, has log P of 0.92. Following oral administration it undergoes extensive first-pass metabolism and presented a bioavailability of 55–65%. Moreover, it has a half-life of 13 to 17 h. RP seems to be a potential candidate for transdermal delivery system (Shafiq et al., Citation2007).
In a study, the thin film hydration method used for the preparation of niosomes was found to be a good technique to encapsulate hydrophobic drug like RP in non-ionic surfactants. Non-ionic surfactant vesicles (niosomes) containing RP with different spans were prepared by thin film hydration method. The higher transdermal flux of approximately 18 µg/cm2/h was observed with niosomes having span 40. The prepared span 40 niosomes were found stable at 4°C for 12 weeks and could potentially be used for transdermal delivery of RP. The non-ionic surfactant prepared showed reasonable drug entrapment, suitable vesicles size and good permeation of the RP (Naveen et al., Citation2012). In a report, transdermal patch of RP using methyl acrylate, vinyl acetate was also prepared and evaluated (Reddy et al., Citation2010).
Thiazide-like diuretics
Thiazide-like diuretics drugs has formed the cornerstone of the management of hypertension for several decades. Control of BP may be difficult without the use of a diuretic. Most patients with hypertension, of which 90–95% have primary or essential hypertension, are effectively treated with diuretics. Anti-hypertensive therapy with diuretics is particularly effective when coupled with reduced dietary sodium intake. The efficacy of these drugs is derived from their ability to reduce blood volume, cardiac output and with long-term therapy, systemic vascular resistance (Tamargo et al., Citation2014).
Indapamide
Indapamide has an anti-hypertensive action causing a drop in systolic, diastolic and mean BP. This anti-hypertensive action is maximal at a dose of 2.5 mg/day and the diuretic effect is slight, usually without clinical manifestation (Tazri et al., Citation2014). It has log P of 2.52 and half-life of 14 h, possibly due to its high lipid solubility, absorption of IP from the gastrointestinal tract is rapid (within 0.5 to 1 h after an oral dose) and complete. Bioavailability of the tablet formulation is 100% and is virtually unchanged with food or antacids. It is defined by the 1999 WHO/ISH Hypertension Guidelines and JNC VII as a first-line drug for the treatment of hypertension (Jian-Liang et al., Citation2004). On oral administration it shows certain adverse drug reactions. Moreover, since IP is usually intended to be taken for a long period, patient compliance is also very important.
Transdermal monolithic system of IP using HPMC and EC was developed using the solvent casting method. Eight monolithic systems were prepared by using a drug polymer ratio of 1:4 with different vegetable oils, such as olive oil, linseed oil, castor oil sunflower oil, coconut oil and cottonseed oil as permeation enhancers. The in vitro release of drug across rat skin from HPMC and EC films showed only 53.63 and 36.50% at the end of 24 h, respectively. HPMC film has shown better release than that of EC film, which may attributed to high water vapor permeability of HPMC film and hydrophobic nature of EC. Among the systems, film comprising of olive oil 30% w/w in HPMC polymer (F3) has shown maximum release than that of systems containing other vegetable oils as permeation enhancers (Sanap et al., Citation2008).
An attempt has been made by Ren et al. (Citation2008) for the development of TTS of IP, authors disclosed that an effective TTS formulation for the delivery of IP may be possible. In the study, the efficacy of various permeation enhancers and organic acids with regard to the percutaneous absorption of IP was explored. It was indicated that N-dodecylazepan-2-one, N-methyl-2-pyrrolidone, menthol and oleic acid had a strong enhancing effect on the permeation of IP and N-dodecylazepan-2-one exhibited the most potent enhancing effect. Among the organic acids examined, lactic acid had the greatest enhancing effect. The formation of an ion-pair between IP and organic acids may be responsible for the enhanced skin permeation of IP. It was concluded that investigated approach of drug delivery should be further inquired so that the successful treatment of hypertension and other clinical conditions for which IP is indicated may become possible, resulting in increased benefit to patients.
In 2009, Ren and others developed the drug-in-adhesive type transdermal patches of IP and the effects of adhesives and permeation enhancers on the permeation of IP were studied. Various permeation enhancers were selected, and their effects on the permeation of IP through excised rat skin were investigated. The results indicated that DURO-TAK® (Bridgewater, NJ, US) adhesive is a suitable and compatible polymer for the development of TTS for IP. The study proved that N-dodecylazepan-2-one (4%) alone showed higher permeation enhancing effect than N-dodecylazepan-2-one (6%). It was predicted that at low concentration, N-dodecylazepan-2-one increase permeability by disordering or “fluidizing” the lipid structure of the stratum corneum, while at high concentration, N-dodecylazepan-2-one will hamper the permeation of drug because of its high hydrophobicity. The final formulation contained N-dodecylazepan-2-one (4%), l-menthol (6%) and isopropyl myristate (3%). The low Cmax and prolonged Tmax after transdermal administration are due to the barrier properties of the skin, while the sustained action observed was due to controlled and continuous release of drug into the systemic circulation over an extended period (Ren et al., 2009).
Potassium-channel openers
Potassium-channel openers are drugs that open ATP-sensitive K+-channels in vascular smooth muscle (Miura & Miki, Citation2003). Being effective arterial dilators, potassium-channel openers are used in the treatment of hypertension. These drugs are not considered as first-line therapy for hypertension because of their side effects, and therefore they are relegated to treating refractory, severe hypertension. They are generally used in conjunction with a beta-blocker and diuretic to attenuate the reflex tachycardia and retention of sodium and fluid, respectively.
Pinacidil
Pinacidil is a lipophilic drug used for the management of mild-to-moderate essential hypertension and has fewer side effects. It is belonging to the class of potassium channel openers. It acts by opening the potassium channels leading to hyperpolarization and peripheral vasodilation. It possesses low oral bioavailability (57%) due to hepatic first-pass metabolism after oral administration. It has a log P of 0.107 and has a short biological half-life of 1.6–2.9 h (McBurney et al., Citation1985; Ahnfelt-Ronne, Citation1988), which makes frequent dosing necessary to maintain the drug within the therapeutic blood levels for long periods. Hence, PD is an ideal drug candidate for transdermal drug delivery.
Aqil et al. (Citation2004) fabricated and evaluated E-RL100 and polyvinyl acetate films for TTS for PD (used monohydrate salt). The prepared polymeric films comprising of R-RL100 and polyvinyl acetate in 2:8, 4:6, 6:4, 8:2 ratios in films P-1, P-2, P-3, P-4, respectively, together with PD (5% w/w) and dibutyl phthalate (5% w/w) in all the films. The developed films were evaluated for physicochemical parameters, in vitro drug release and skin permeation profiles. It was reported that E-RL 100 has a major influence on the physicochemical profile of the films. The higher the quantity of E-RL100 in the film, better the film strength, flexibility as well as its higher drug release and skin permeation potential. The final optimized film was found to be the best in terms of drug release and skin permeation. The optimized film was found to be free of potentially hazardous skin irritation and was found to be stable and intact at ambient temperature and humidity conditions. The fabricated films hold promise for the development of a matrix type TTS for PD (Aqil et al., Citation2004). In another study, the monolithic matrix type TTS of PD (monohydrate salt) were prepared. Four formulations were developed which differed in the ratio of matrix forming polymers, E-RL-100 and PVP K-30. Reported cumulative percent of drug released in 48 h from the four formulations was 63.96, 55.95, 52.26 and 92.18%. The cumulative percent of PD permeated was again maximum for formulation coded as B-4 (carrying E-RL-100 and PVP K-30 in 6:4 ratio) with a value of 86.54%. On the basis of the in vitro characterization, it was concluded that PD remained intact and stable in the TTS on storage with no apparent chemical interaction between the drug and the excipients (Aqil & Ali, Citation2002). In 2006, Aqil et al. monitored the effect of the developed TTS on BP of methyl prednisolone acetate-induced hypertensive rats. The BP of rats was measured using a non-invasive rat BP instrument based on a cuff tail technique. A significant fall in rat BP was observed in the treatment of hypertensive rats with all the formulations, which was maintained for 48 h. Inter-formulation comparison revealed that formulation B-4 was the most effective with 37.96% reduction in BP. It was concluded that a single patch application of PD TTS (B-4) can effectively control hypertension in rats for 2 days (Aqil et al., Citation2006a).
Conclusion
The World Health Report 2002 identified hypertension, or high BP, as the third ranked factor for disability-adjusted life years. Hypertension is one of the leading risks for premature death and disability universally. Recent analyses have shown that the number of adults with hypertension in 2025 was predicted to increase by about 60% to a total of 1.56 billion (Kearney et al., Citation2005; Chockalingam et al., Citation2006; Campbell et al., Citation2012). Overall, hypertension is an important public-health challenge globally. Awareness, prevention, treatment and control of hypertension are a significant public health measure.
There are many categories of potentially anti-hypertensive drugs of which; ARBs, ACEIs, diuretics, calcium channel blockers and β-blockers are suitable for initial or single-drug therapy based on efficacy and tolerability. The conventional dosage forms of these medications have many disadvantages including higher frequency of administration, higher first-pass metabolism, serious adverse effects, and difficult dose regimens that leads to inadequate patient compliance. These findings indicated that in spite of the availableness of a surplus of therapeutically effective ARBs and ACEIs, poor patient benefit is found; this arguably demonstrates the chances to present these through a different route. Transdermal approach of drug delivery is more pertinent in case of chronic disorders, such as hypertension, which require long-term dosing to maintain therapeutic drug concentration. Transdermal drug delivery provides some desirable performances over conventional pharmaceutical dosage formulations, such as avoiding gut and hepatic first-pass metabolism, improving drug bioavailability, reducing dose frequency, lessen the side effects and thus improved patient compliance. However, its widespread application is constrained due to the significant penetration obstacle provided by the stratum corneum, the topmost skin layer. To overcome this barrier, several overtures have been assessed. Amongst the approaches used, chemical penetration enhancers have been intensively investigated over the years. Extensive research during the past two decades has revealed considerable information on several classes of chemical penetration enhancers. Efforts have been directed at identifying safe and effective enhancers from both natural products and synthetic chemicals. There have been noteworthy research attempts made globally to examine the percutaneous permeation and to develop TTS of various ARBs/ACEIs, nevertheless there is a further opportunity of transdermal delivery of ARBs/ACEIs other than those discussed above, including ARBs like eprosartan, irbesartan, olmesartan and ACEIs such as fosinopril, moexipril, perindopril, trandolapril and so on, depending on their physicochemical properties. There is a need to improve drug delivery devices for these actives because of the quantum of their utilization and short coming associated with their conventional dosage forms.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgment
This project was supported by NSTIP strategic technologies programs (No. 13-NAN1268-02) in the Kingdom of Saudi Arabia.
References
- Ahad A, Al-Jenoobi FI, Al-Mohizea AM, et al. (2013a). Transdermal delivery of calcium channel blockers for hypertension. Expert Opin Drug Deliv 10:1137–53
- Ahad A, Al-Jenoobi FI, Al-Mohizea AM, et al. (2014a). Systemic delivery of beta-blockers via transdermal route for hypertension. Saudi Pharm J (published online 3 January 2014; doi: 10.1016/j.jsps.2013.1012.1019)
- Ahad A, Aqil M, Kohli K, et al. (2009). Chemical penetration enhancers: a patent review. Expert Opin Ther Pat 19:969–88
- Ahad A, Aqil M, Kohli K, et al. (2011a). Interactions between novel terpenes and main components of rat and human skin: mechanistic view for transdermal delivery of propranolol hydrochloride. Curr Drug Deliv 8:213–24
- Ahad A, Aqil M, Kohli K, et al. (2011b). Role of novel terpenes in transcutaneous permeation of valsartan: effectiveness and mechanism of action. Drug Dev Ind Pharm 37:583–96
- Ahad A, Aqil M, Kohli K, et al. (2012). Formulation and optimization of nanotransfersomes using experimental design technique for accentuated transdermal delivery of valsartan. Nanomedicine 8:237–49
- Ahad A, Aqil M, Kohli K, et al. (2013b). Enhanced transdermal delivery of an anti-hypertensive agent via nanoethosomes: statistical optimization, characterization and pharmacokinetic assessment. Int J Pharm 443:26–38
- Ahad A, Aqil M, Ali A. (2014b). Investigation of antihypertensive activity of carbopol valsartan transdermal gel containing 1,8-cineole. Int J Biol Macromol 64:144–9
- Ahad A, Aqil M, Kohli K, et al. (2014c). Design, formulation and optimization of valsartan transdermal gel containing iso-eucalyptol as novel permeation enhancer: preclinical assessment of pharmacokinetic in wistar albino rats. Expert Opin Drug Deliv (Published online 15 May 2014; DOI:10.1517/17425247.2014.914027)
- Ahnfelt-Ronne I. (1988). Pinacidil: history, basic pharmacology, and therapeutic implications. J Cardiovasc Pharmacol 12:S1–4
- Amin S, Sarfenejad A, Ahmad J, et al. (2013). Nanovesicular transfersomes for enhanced systemic delivery of telmisartan. Adv Sci Eng Med 5:299–308
- Aqil M, Ahad A, Sultana Y, Ali A. (2007). Status of terpenes as skin penetration enhancers. Drug Discov Today 12:1061–7
- Aqil M, Ali A. (2002). Monolithic matrix type transdermal drug delivery systems of pinacidil monohydrate: in vitro characterisation. Eur J Pharm Biopharm 54:161–4
- Aqil M, Ali A, Sultana Y, et al. (2006a). In vivo characterization of monolithic matrix type transdermal drug delivery systems of pinacidil monohydrate: a technical note. AAPS Pharm Sci Tech 7:E38–E42
- Aqil M, Ali A, Sultana Y, Najmi AK. (2004). Fabrication and evaluation of polymeric films for transdermal delivery of pinacidil. Pharmazie 59:631–5
- Aqil M, Bhavna, Chowdhary I, et al. (2008). Transdermal therapeutic system of enalapril maleate using piperidine as penetration enhancer. Curr Drug Deliv 5:148–52
- Aqil M, Sultana Y, Ali A. (2006b). Transdermal delivery of beta-blockers. Expert Opin Drug Deliv 3:405–18
- Bairwa M, Pilania M, Gupta V, Yadav K. (2014). Hypertension vaccine may be a boon to millions in developing world. Hum Vaccin Immunother 10:708–13
- Banweer J, Pandey S, Pathak AK. (2010). Formulation, optimization and evaluation of matrix type transdermal system of lisinopril dihydrate using permeation enhancers. Drug Invent Today 2:134–7
- Barreras A, Gurk-Turner C. (2003). Angiotensin II receptor blockers. Proc (Bayl Univ Med Cent) 16:123–6
- Basile JN. (2003). Optimizing antihypertensive treatment in clinical practice. Am J Hypertens 16:13S–17S
- Baviskar DT, Parik VB, Gupta HN, et al. (2012). Design and evaluation of patches for transdermal delivery of losartan potassium. PDA J Pharm Sci Technol 66:126–35
- Beg S, Swain S, Singh HP, et al. (2012). Development, optimization, and characterization of solid self-nanoemulsifying drug delivery systems of valsartan using porous carriers. AAPS PharmSciTech 13:1416–27
- Berkow SE, Barnard ND. (2005). Blood pressure regulation and vegetarian diets. Nutr Rev 63:1–8
- Bhosale SS, Avachat AM. (2013). Design and development of ethosomal transdermal drug delivery system of valsartan with preclinical assessment in Wistar albino rats. J Liposome Res 23:119–25
- Brown NJ, Vaughan DE. (1998). Angiotensin-converting enzyme inhibitors. Circulation 97:1411–20
- Campbell N, Young ER, Drouin D, et al. (2012). A framework for discussion on how to improve prevention, management, and control of hypertension in Canada. Can J Cardiol 28:262–9
- Carr AA, Prisant LM. (1996). Losartan: first of a new class of angiotensin antagonists for the management of hypertension. J Clin Pharmacol 36:3–12
- Chockalingam A, Campbell NR, Fodor JG. (2006). Worldwide epidemic of hypertension. Can J Cardiol 22:553–5
- Cubillos-Garzon LA, Casas JP, Morillo CA, Bautista LE. (2004). Congestive heart failure in Latin America: the next epidemic. Am Heart J 147:412–17
- Desai BG, Annamalai AR, Divya B, Dinesh BM. (2008). Effect of enhancers on permeation kinetics of captopril for transdermal system. Asian J Pharm 2:35–7
- Destro M, Preti P, D'Ospina A, et al. (2009). Olmesartan medoxomil: recent clinical and experimental acquisitions. Expert Opin Drug Metab Toxicol 5:1149–57
- Detroja C, Chavhan S, Sawant K. (2011). Enhanced antihypertensive activity of candesartan cilexetil nanosuspension: formulation, characterization and pharmacodynamic study. Sci Pharm 79:635–51
- Devi AS, Pinnika A, Divya P. (2014). Formulation and evaluation of candesartan cilexetil transdermal proniosomal gel. J Drug Deliv Ther 4:90–8
- Dubey BK, Katare OP, Singh R, Jain SK. (1995). Lyophilized aqueous based polymer matrices for transdermal delivery of captopril. J Dermatol Sci 10:191–5
- Duchin KL, Singhvi SM, Willard DA, et al. (1982). Captopril kinetics. Clin Pharmacol Ther 31:452–8
- Easthope SE, Jarvis B. (2002). Candesartan cilexetil: an update of its use in essential hypertension. Drugs 62:1253–87
- Ferguson RK, Vlasses PH, Swanson BN, et al. (1982). Effects of enalapril, a new converting enzyme inhibitor, in hypertension. Clin Pharmacol Ther 32:48–53
- Gales BJ, Bailey EK, Reed AN, Gales MA. (2010). Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers for the prevention of migraines. Ann Pharmacother 44:360–6
- Gannu R, Yamsani VV, Palem CR, et al. (2010). Iontophoretic delivery of lisinopril: optimization of process variables by Box-Behnken statistical design. Pharm Dev Technol 15:169–77
- Gannu R, Yamsani VV, Yamsani SK, et al. (2009). Optimization of hydrogels for transdermal delivery of lisinopril by Box-Behnken statistical design. AAPS PharmSciTech 10:505–14
- Gavali P, Gaikwad A, Radhika PR, Sivakumar T. (2010). Design and development of hydroxypropyl methylcellulose (HPMC) based polymeric film of enalapril maleate. Int J Pharm Tech Res 2:274–82
- Gerstein HC. (2000). Cardiovascular and metabolic benefits of ACE inhibition: moving beyond blood pressure reduction. Diabetes Care 23:882–3
- Go AS, Mozaffarian D, Roger VL, et al. (2014). Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation 129:e28–292
- Goa KL, Wagstaff AJ. (1996). Losartan potassium: a review of its pharmacology, clinical efficacy and tolerability in the management of hypertension. Drugs 51:820–45
- Gomez HJ, Cirillo VJ, Moncloa F. (1987). The clinical pharmacology of lisinopril. J Cardiovasc Pharmacol 9:S27–34
- Gullick DR, Pugh WJ, Ingram MJ, et al. (2010). Formulation and characterization of a captopril ethyl ester drug-in-adhesive-type patch for percutaneous absorption. Drug Dev Ind Pharm 36:926–32
- Gungor S, Ozsoy Y. (2012). Systemic delivery of antihypertensive drugs via skin. Ther Deliv 3:1101–16
- Gupta A, Prajapati SK, Balamurugan M, et al. (2007). Design and development of a proniosomal transdermal drug delivery system for captopril. Trop J Pharm Res 6:687–93
- Harries C, Armstrong I. (2012). A review of the management of pulmonary arterial hypertension associated with congenital heart disease. Eur J Cardiovasc Nurs 11:239–47
- Hathout RM, Elshafeey AH. (2012). Development and characterization of colloidal soft nano-carriers for transdermal delivery and bioavailability enhancement of an angiotensin II receptor blocker. Eur J Pharm Biopharm 82:230–40
- Helal F, Lane ME. (2014). Transdermal delivery of angiotensin converting enzyme inhibitors. Eur J Pharm Biopharm (Published online 20 March 2014; doi: 10.1016/j.ejpb.2014.03.007)
- Israili ZH. (2000). Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J Hum Hypertens 14:S73–86
- Izzo Jr JL, Weir MR. (2011). Angiotensin-converting enzyme inhibitors. J Clin Hypertens (Greenwich) 13:667–75
- Jain R, Aqil M, Ahad A, et al. (2008). Basil oil is a promising skin penetration enhancer for transdermal delivery of labetolol hydrochloride. Drug Dev Ind Pharm 34:384–9
- Jain S, Joshi SC. (2007). Development of transdermal matrix system of captopril based on cellulose derivative. Pharmacolgyonline 1:379–90
- Jian-Liang Z, Yong-Wen Q, Xing Z, et al. (2004). Possible induction of diabetes by treatment of hypertension with indapamide (with four case reports). Diabetes Res Clin Pract 65:243–6
- Kausalya J, Suresh K, Padmapriya S, et al. (2011). Solubility and dissolution enhancement profile of telmisartan using various techniques. Int J Pharm Tech Res 3:1737–49
- Kearney PM, Whelton M, Reynolds K, et al. (2005). Global burden of hypertension: analysis of worldwide data. Lancet 365:217–23
- Kelly JG, O'Malley K. (1990). Clinical pharmacokinetics of the newer ACE inhibitors. A review. Clin Pharmacokinet 19:177–96
- Kerimoglu O, Keskin E, Dortunc B, Anah S. (2013). Matrix type transdermal therapeutic system containing captopril: formulation optimization, in vitro and ex vivo characterization. Acta Pol Pharm 70:291–300
- Kobayashi D, Matsuzawa T, Sugibayashi K, et al. (1993). Feasibility of use of several cardiovascular agents in transdermal therapeutic systems with l-menthol-ethanol system on hairless rat and human skin. Biol Pharm Bull 16:254–8
- Laeis P, Puchler K, Kirch W. (2001). The pharmacokinetic and metabolic profile of olmesartan medoxomil limits the risk of clinically relevant drug interaction. J Hypertens Suppl 19:S21–32
- Lake Y, Pinnock S. (2000). Improved patient acceptability with a transdermal drug-in-adhesive oestradiol patch. Aust N Z J Obstet Gynaecol 40:313–16
- Lewis EJ, Hunsicker LG, Clarke WR, et al. (2001). Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345:851–60
- Malakar J, Basu A, Nayak AK. (2013). Candesartan cilexetil microemulsions for transdermal delivery: formulation, in-vitro skin permeation and stability assessment. Curr Drug Deliv 10:1–9
- Manoria P. (2006). Olmesartan medoxomil: a clinical review. Indian Heart J 58:282–6
- McBurney A, Farrow PR, Ainsworth S, Ward JW. (1985). Serum concentrations and urinary excretion of pinacidil and its major metabolite, pinacidil pyridine-N-oxide following i.v. and oral administration in healthy volunteers. Br J Clin Pharmacol 19:91–4
- McInnes GT. (1999). Clinical advantage of valsartan. Cardiology 91:14–18
- Mittal S, Mittal A, Sharma K, Alam S. (2013). Proniosomes as a drug carrier for transdermal delivery of candesartan cilexetil. Int J Nano Studies Tech 2:201–7
- Miura T, Miki T. (2003). ATP-sensitive K+ channel openers: old drugs with new clinical benefits for the heart. Curr Vasc Pharmacol 1:251–8
- Mundargi RC, Patil SA, Agnihotri SA, Aminabhavi TM. (2007). Evaluation and controlled release characteristics of modified xanthan films for transdermal delivery of atenolol. Drug Dev Ind Pharm 33:79–90
- Naveen D, Rekha R, Kapil S, et al. (2012). Ramipril loaded non-ionic surfactant vesicles: characterization and in-vitro permeation studies. The Pharma Res 6:42–53
- Nishida N, Taniyama K, Sawabe T, Manome Y. (2010). Development and evaluation of a monolithic drug-in-adhesive patch for valsartan. Int J Pharm 402:103–9
- Nur AO, Zhang JS. (2000). Recent progress in sustained/controlled oral delivery of captopril: an overview. Int J Pharm 194:139–46
- Park ES, Chang SJ, Rhee YS, Chi SC. (2001). Effects of adhesives and permeation enhancers on the skin permeation of captopril. Drug Dev Ind Pharm 27:975–80
- Prausnitz MR, Langer R. (2008). Transdermal drug delivery. Nat Biotechnol 26:1261–8
- Prausnitz MR, Mitragotri S, Langer R. (2004). Current status and future potential of transdermal drug delivery. Nat Rev Drug Discov 3:115–24
- Quadri SA, Raga SR, Ahmed MZ, et al. (2012). Formulation and studies on candesartan transdermal patches. Int J Pharm Res Dev 4:234–41
- Reddy PD, Swarnalatha D, Dinakar A, et al. (2010). Formulation and evaluation of ramipril transdermal patch. Int J Chem Sci 8:681–6
- Ren C, Fang L, Li T, et al. (2008). Effect of permeation enhancers and organic acids on the skin permeation of indapamide. Int J Pharm 350:43–7
- Ren C, Fang L, Ling L, et al. (2009). Design and in vivo evaluation of an indapamide transdermal patch. Int J Pharm 370:129–35
- Riley Jr LJ, Vlasses PH, Ferguson RK. (1985). Clinical pharmacology and therapeutic applications of the new oral converting enzyme inhibitor, enalapril. Am Heart J 109:1085–9
- Rizwan M, Aqil M, Ahad A, et al. (2008). Transdermal delivery of valsartan: I. Effect of various terpenes. Drug Dev Ind Pharm 34:618–26
- Rokade MM, Thakare PR, Rupvate SR, et al. (2012). Formulation design and evaluation of transdermal film of losartan potassium using hydrophilic and hydrophobic polymers. Am J Pharm Tech Res 2:771–81
- Sanap GS, Dama GY, Hande AS, et al. (2008). Preparation of transdermal monolithic systems of indapamide by solvent casting method and the use of vegetable oils as permeation enhancer. Int J Green Pharm 2:129–33
- Saroha K, Yadav B, Sharma B. (2011). Transdermal patch: a discrete dosage form. Int J Curr Pharm Res 3:98–108
- Scott LJ, McCormack PL. (2008). Olmesartan medoxomil: a review of its use in the management of hypertension. Drugs 68:1239–72
- Selvam RP, Singh AK, Sivakumar T. (2010). Transdermal drug delivery systems for antihypertensive drugs – a review. Int J Pharm Biomed Res 1:1–8
- Shafiq S, Shakeel F, Talegaonkar S, et al. (2007). Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur J Pharm Biopharm 66:227–43
- Shams MS, Alam MI, Ali A, et al. (2010a). Pharmacodynamics of a losartan transdermal system for the treatment of hypertension. Drug Dev Ind Pharm 36:385–92
- Shams MS, Alam MI, Ali A, et al. (2010b). Pharmacokinetics of a losartan potassium released from a transdermal therapeutic system for the treatment of hypertension. Pharmazie 65:679–82
- Sharpe M, Jarvis B, Goa KL. (2001). Telmisartan: a review of its use in hypertension. Drugs 61:1501–29
- Shin SC, Choi JS. (2003). Enhanced bioavailability of atenolol by transdermal administration of the ethylene-vinyl acetate matrix in rabbits. Eur J Pharm Biopharm 56:439–43
- Tamargo J, Segura J, Ruilope LM. (2014). Diuretics in the treatment of hypertension. Part 1: thiazide and thiazide-like diuretics. Expert Opin Pharmacother 15:527–47
- Tazri J, Moghal MM, Dewan SM, et al. (2014). Formulation and quality determination of indapamide matrix tablet: a thiazide type antihypertensive drug. Adv Pharm Bull 4:191–5
- Terra SG. (2003). Cardiology patient page. Angiotensin receptor blockers. Circulation 107:e215–16
- Thakur R, Anwer MK, Shams MS, et al. (2009). Proniosomal transdermal therapeutic system of losartan potassium: development and pharmacokinetic evaluation. J Drug Target 17:442–9
- Tronvik E, Stovner LJ, Helde G, et al. (2003). Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. JAMA 289:65–9
- Vasan RS, Beiser A, Seshadri S, et al. (2002). Residual lifetime risk for developing hypertension in middle-aged women and men: the Framingham Heart Study. JAMA 287:1003–10
- Vasan RS, Larson MG, Leip EP, et al. (2001). Assessment of frequency of progression to hypertension in non-hypertensive participants in the Framingham Heart Study: a cohort study. Lancet 358:1682–6
- Velasquez MT. (1996). Angiotensin II receptor blockers. A new class of antihypertensive drugs. Arch Fam Med 5:351–6
- Wang H, Hou HM. (2000). Improvement of transdermal permeation of captopril by iontophoresis. Acta Pharmacol Sin 21:591–5
- Wienen W, Entzeroth M, Meel JCA, et al. (2000). A review on telmisartan: a novel, long-acting angiotensin II-receptor antagonist. Cardiovas Drug Rev 18:127–56
- Wu PC, Huang YB, Fang JY, Tsai YH. (1997). In vitro percutaneouus absorption of captopril. Int J Pharm 148:41–6
- Wu PC, Huang YB, Fang JY, Tsai YH. (1998). Percutaneous absorption of captopril from hydrophilic cellulose derivatives through excised rabbit skin and human skin. Drug Dev Ind Pharm 24:179–82
- Zakzewski CA, Amory DW, Jasaitis DK, Li JK. (1992). Iontophoretically enhanced transdermal delivery of an ACE inhibitor in induced hypertensive rabbits: preliminary report. Cardiovasc Drugs Ther 6:589–95
- Zhou XH, Li Wan Po A. (1994). Stability and in vitro absorption of captopril, enalapril and lisinopril across the rat intestine. Biochem Pharmacol 47:1121–6