
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective: The aim of this study was to develop a novel tablet formulation of amorphous candesartan cilexetil (CAN) solid dispersion involving effective P-gp inhibition for optimal drug delivery by direct compression (DC) method.
Methods: To accomplish DC, formulation blends were evaluated for micromeritic properties. The Carr index, Hausner ratio, flow rate and cotangent of the angle α were determined. The tablets with and without naringin prepared by DC technique were evaluated for average weight, hardness, disintegration time and friability assessments. The drug release profiles were determined to study the dissolution kinetics. In vivo pharmacokinetic studies were conducted in rabbits. Accelerated stability studies were performed for tablets at 40 ± 2 °C/75% RH ± 5% for 6 months.
Results: FTIR studies confirmed no discoloration, liquefaction and physical interaction between naringin and drug. The results indicated that tablets prepared from naringin presented a dramatic release (82%) in 30 min with a similarity factor (76.18), which is most likely due to the amorphous nature of drug and the higher micromeritic properties of blends. Our findings noticed 1.7-fold increase in oral bioavailability of tablet prepared from naringin with mean Cmax and AUC0–12 h values as 35.81 ± 0.13 μg/mL and 0.14 ± 0.09 μg h/mL, respectively. The tablets with and without naringin prepared by DC technique were physically and chemically stable under accelerated stability conditions upon storage for 6 months.
Conclusion: These results are attractive for further development of an oral tablet formulation of CAN through P-gp inhibition using naringin, a natural flavonoid as a pharmaceutical excipient.
Introduction
Candesartan cilexetil (CAN) is an esterified prodrug of candesartan and an angiotensin II receptor antagonist. Ester hydrolysis during absorption from gastrointestinal tract rapidly and completely bio-activates CAN to its active parent drug, candesartan. A very low aqueous solubility (0.0003 mg/mL) is the major drawback of CAN to develop an oral dosage form for its therapeutic application and efficacy. Incomplete absorption from the gastrointestinal (GI) tract is reported due to the low solubility of CAN across the physiological pH range. CAN is classified as a class II drug in the Biopharmaceutics Classification System (BCS) based on its solubility across physiologically relevant pH conditions and absorption characteristics (Vijaykumar et al., Citation2009).
Solid dispersions have been the focus of growing interest during the last decades to develop poorly soluble compounds like CAN to increase dissolution rate and impact saturation solubility. In these dispersions, the drug is often present as partially amorphous or amorphous substance (Yonemochi et al., Citation1997, Citation1999; Mura et al., Citation2002; Dai et al., Citation2007). Previously, we reported that freeze-drying is the most suitable preparative technique among various pharmaceutical interventions for preparing amorphous solid dispersions of poorly water-soluble drug, CAN (Gurunath et al., Citation2014).
Together, we demonstrated that, improved aqueous solubility followed by effective P-gp efflux pumps inhibition in the GI tract, the CAN oral bioavailability can significantly be enhanced through freeze-dried solid dispersions using naringin, as a novel pharmaceutical excipient (Gurunath et al., Citation2013, Citation2014).
Recently, Li et al. (Citation2013, Citation2014) reported the acute and chronic oral toxicity studies of naringin at a single dose of 16 g/kg that did not produce any mortality, abnormal changes in body weights and food consumption, undesirable clinical signs, toxicologically related alterations in hematology, clinical biochemistry and macroscopic findings upon exposure of rats. It is rational to establish that LD50 of rats is greater than 16 g/kg when treated with single oral dose of naringin. Therefore, naringin could be considered as reasonably safe with Hodge and Sterner scale but non-toxic with Gosselin, Smith and Hodge scale. According to the study, the no observed adverse effect level (NOAEL) of naringin is anticipated to be more than 1250 mg/kg/day to Sprague dawley rats following daily oral administrations for 13 weeks or 6 months.
P-gp-mediated drug efflux significantly intervenes the delivery of the drugs that requires a very small dose for their pharmacological actions or the drugs that show extremely slow diffusion and dissolution rates. Those small amounts of drugs cannot reach the blood circulation in adequate quantity as it decreases drug absorption and sometimes, that can be life threatening (Amin, Citation2013).
This impact on the bioavailability has been recognized in research using mdr1a/1b knockout mice, where the absorption of paclitaxel, a substrate of P-gp, was greater than that in wild-type mice (Schinkel et al., Citation1997; Sparreboom et al., Citation1997; Woo et al., Citation2003). These findings established that about half of paclitaxel dose administered is expelled out into the gut lumen by P-gp and simply a small amount of drug is lost by gut wall and liver metabolism. Thus, the effective inhibition P-gp-mediated efflux can significantly improve the oral bioavailability of paclitaxel.
Traditionally, the oral route is the most frequently accepted route of drug administration. Tablets are considered the most attractive dosage form by patients and industry for drug delivery (Allen et al., Citation2005; Rudnic, Citation2005; Staniforth, Citation2007; Emery et al., Citation2009).
Manufacturing tablets, a chiefly directly compressed tablet is clear-cut manufacturing process that involves low cost, hence attractive to pharmaceutical laboratories (Strydom et al., Citation2011).
The term “direct compression” (DC) is employed to describe the process where tablets are compressed directly from the powder blends of active ingredients and suitable excipients. No pre-treatment by wet or dry granulation is involved for the powder blends. The DC is more economical, decreases the cycle time of the products and is simple in terms of the requirements of good manufacturing practices. The DC is environment-friendly with less number of steps, without involving water and temperature, and finally the stability of the final product can be improved (Gohel & Jogani et al., Citation2005; McCormick, Citation2005; Strydom et al., Citation2011).
To the best of our understanding, there are no available published reports or works demonstrating the effects of naringin as P-gp inhibitor as a novel concept of employing naturally occurring bio-flavonoid as a pharmaceutical excipient with absorption-improving potential formulated as compressed tablets with significant improvement in the dissolution rate of CAN through amorphous forms.
Consequently, our study involves CAN; an ester prodrug that rapidly de-esterifies in vivo during absorption from the GI tract to produce its active metabolite, candesartan (McClellan & Goa, Citation1998) was used as a model drug. CAN is not only an extremely poor lipophilic compound (Vijaykumar et al., Citation2009) but also its active metabolite, candesartan is a substrate of P-gp (also known as MDR1, ABCB1; Zhou et al., Citation2009). The bioflavonoid, called naringin, was identified as an extremely safe; naturally occurring novel pharmaceutical excipient to show effective P-gp-mediated cellular efflux inhibition of P-gp substrate, candesartan (Lassoued et al., Citation2012; Gurunath et al., Citation2013, Citation2014).
Hence, our present investigation aims:
To prepare novel tablet formulation by DC method with and without naringin to combine the two systems (improved poor water-solubility through amorphization and effective P-gp efflux pump inhibition) to one system.
To evaluate the formulation blends for micromeritic properties.
To evaluate the physical characteristics of directly compressed tablets with and without naringin.
Pharmacokinetic evaluation of finished tablets with and without naringin in rabbits.
To perform accelerated stability studies for tablets with and without naringin at 40 ± 2 °C/75% RH ± 5% upon storage for 6 months.
Materials and methods
Candesartan cilexetil (of purity more than 99%, melting temperature = 163 ± 0.5 °C) was kindly obtained as a gift sample from the Dr. Reddy’s Labs (Hyderabad, India). Polyvinyl pyrrolidone K30 (PVP-K30) was obtained from SD Fine Chemicals Ltd., Mumbai, India. Candesartan, paracetamol, naringin, spray-dried lactose, Avicel PH 102, mannitol, magnesium stearate, talc, Na2HPO4, KH2PO4, NaOH, KCl, citric acid NaCl, CaCl2, MgCl2 and NaHCO3 were obtained from Sigma Aldrich (Hyderabad, India). Acetonitrile, methanol of HPLC grade and orthophosphoric acid were supplied by E-Merck India Ltd., Mumbai. All other chemicals were of analytical grade. All drug solutions were freshly prepared before use.
Drug–excipient interaction studies
It was imperative to understand the compatibility between the drug under study and the novel excipient used within the system while designing a tablet formulation. As a result, it is necessary to conform that drug is not interacting with excipient for a period of 4 weeks under experimental conditions (40 ± 5 °C and 75 ± 5% RH). The infrared absorption spectra were run between 4000 and 400 cm−1 for the pure drug and the physical mixture of drug and excipient. The IR spectra of pure drug and a mixture of excipient and drug are shown in .
Figure 1. FT-IR spectrum of (a) Candesartan cilexetil (CAN), (b) Naringin (NGN) and (c) a physical mixture of pure candesartan cilexetil and Naringin.
Formulation of blends
Freeze-dried CAN-loaded (1:2 w/w, CAN: PVP-K30) solid dispersions (Gurunath et al., Citation2014) and excipients, like naringin, Avicel PH 102, spray-dried lactose, mannitol, were co-grounded in pestle mortar (without talc and magnesium stearate) and were passed through mesh no. 60. Finally, talc and magnesium stearate were added and mixed for 5 min to obtain formulation blends ().
Table 1. Composition of 100 mg tablets (FDS-NT and FDS-T).
Micromeritic studies of the formulation blends with novel excipient (naringin)
Bulk and tapped densities determination of the blends
Apparent density (ρb) and tapped density (ρt) were measured through the apparent volume indirectly (Qiu et al., Citation2009). The tests were performed with an automatic compactor (Tapped Density Volumeter, Sisco, India). The bulk density was determined by gradually pouring the samples into a 100 mL graduated glass cylinder up to 60% of the container volume. The mass associated with the filled volume (60 mL) was evaluated on an analytical balance. The bulk density was calculated using the relation between the weight (g) and the apparent volume (mL). The following formula was used to calculate the bulk density.
The tapped density was obtained by tapping a graduated glass cylinder containing a known amount of samples. The volume after 100 taps (V100) and 500 taps (V500) was recorded. In cases where the difference between V100 and V500 was more than 2%, an additional cycle of 500 taps was carried out until variations were less than 2%.
The tapped density (g/mL) was obtained from the mass (g) and the final volume of solids (mL). The following formula was used to calculate the tapped density.
Hausner ratio and Carr’s index determination
The Hausner ratio (HR) is the association between ρt and ρb and is linked to inter-particle resistance. It can be used to evaluate the powder flow properties. The Carr index (CI%) is expressed as a percentage which is the measure between the apparent and compact densities. The compressibility percentage indirectly provides a concept of the cohesion, size uniformity and surface area of powders or their mixtures (Staniforth, Citation2007). The compressibility index (I) is calculated as follows:
where ρt is the tapped density and ρb is the bulk density.
Hausner ratio (HR) is an indirect measure of ease of powder flow that is calculated as follows.
where ρt is tapped density and ρb is bulk density. Smaller Hausner ratio (<1.25) signifies improved flow properties than higher ones (>1.25).
Flow rate and the cotangent of angle α analysis
The blends flow rate was evaluated with a flow tester from the flow time of the sample. The blends (50 g) were poured through the funnel (12 mm) attached to the stiff equipment. The funnel was opened and the time taken to release the samples was noted (Schüssele & Bauer-Brandl, Citation2003).
Tablets preparation by direct compression method with FSD-NT and without FSD-T naringin
A preliminary assessment was made to test the ability of the blends to form tablets by DC with and without naringin. An optimized tablet formulations were prepared as listed in . Fifty tablets for each formulation were prepared. shows the compositions and sample designations of the tablets. The entire constituents as shown in were co-grounded for 5–10 min in a glass mortar pestle for each formulation separately and were passed through sieve no. 60. The mixed blends of excipients were compressed to produce convex faced tablets weighing 100 mg each, respectively, with 5 mm diameter by means of a single punch tablet machine (Cadmach, Ahmedabad). The compression force and time were 15 kN and varied from 30 to 34 s for both tablet formulations, respectively.
Physical evaluation of tablets
According to official methods, all tablets (FSD-NT and FSD-T) were evaluated for weight uniformity, friability, thickness and hardness. Twenty tablets were used for weight variation determination using an electronic balance (Sartorius, India). Twenty tablets (Jadhav et al., Citation2011) were weighed separately and collectively using digital balance.
The average weight of one tablet was obtained by dividing the total sum of 20 tablets. The weight variation test is an acceptable method for determining the drug content uniformity of tablets. For an average weight equal to 130 mg or less, the maximum of 10% weight variation is considered satisfactory. Ten tablets were used to determine the tablet hardness using a Monsanto tablet hardness tester (Campbell Electronics, Mumbai, India). About 40 N (1 kg = 10 N), tablet breaking force is considered adequate for mechanical stability. Tablet friability was calculated as the percentage weight loss of 10 tablets at 25 rpm for 4 min using a friabilator (Sisco, India). The weight loss should not be greater than 1% w/w. The disintegration time was determined at 37 ± 0.5 °C in purified water with a disintegration tester (Sisco, India) fitted with sintered disks. The disintegration time is recorded for an average of six measurements. Results are shown in and .
Table 2. Micromeritic properties of formulation blends (Mean ± SD, n = 3).
Table 3. Physical properties of FSD-NT and FSD-T tablets (Mean ± SD, n = 3/6).
Dissolution study
The USP II Paddle dissolution apparatus (TDT-08 L, Electrolab, India) was used to perform the dissolution studies. The tablets were placed in 900 mL phosphate buffer (pH 6.8) maintained at 37 ± 0.5 °C, stirring at 50 rpm. At periodical intervals of 0, 2, 4, 6, 10, 15, 20, 25 and 30 min, samples were collected, filtered through 0.45 μm and replaced with a fresh dissolution medium.
Samples withdrawn at predetermined time intervals, filtered in-line and assayed using an HPLC method. Chromatographic separation was accomplished using a Shimadzu HPLC system (LC-20 A series) equipped with a UV detector, micro-volume double plunger pump (10 μL/stroke) and manual injector type model 7725-port sample injection valve was employed. The system was controlled through SCL-10Avp or SCL-10 A software (Conquer Scientific Lab Equipment, San Diego, CA).
HPLC analysis was performed under the following conditions: a Shim-pack VP-ODS or equivalent ODS column (Particle size 5 μm, column dimension: I.D 4.6 mm × length 150 mm) maintained at room temperature. An isocratic mobile phase composed of acetonitrile:methanol (60:40% v/v) was adjusted for pH 6.0 with orthophosphoric acid at ambient temperature and the injection volume was 20 μL at a flow rate of 1 mL/min. The detective wavelength was 255 nm. The mobile phase was filtered through a 0.45 μm filter in a Millipore solvent-filtration apparatus before use.
HPLC method was validated prior to assay as previously reported (Gurunath et al., Citation2013), which was carried out for linearity, precision, accuracy, limit of detection and quantification according to US Pharmacopoeia (The United States Pharmacopoeia, Citation2007) and International Conference on Harmonization (ICH) guidelines [ICH (Q2R1), Citation2007]
The similarity factor (f2) was calculated to compare the release profiles of two formulations under similar conditions of dissolution media, agitation rate, pH and temperature employed, to rule out or understand the influence of naringin on drug release as novel excipient. The similarity between two dissolution profiles was considered significant when its value falls between 50 and 100. Drug release studies were carried out in triplicate.
Pharmacokinetic study
Drug administration
Six male (2.8–3.1 kg) healthy adult albino rabbits were divided into two groups. Each group consisting of three rabbits each. Each rabbit was housed individually in separate cages under environmentally controlled conditions (25 ± 2 °C, 12 h light/dark cycle). Prior to each experiment, the rabbits were starved for 24 h and were allowed free access to tap water. Tablets (FSD-NT and FSD-T, each of 100 mg with 5 mm diameter) were orally administered into the stomach of rabbits using a gastric intubation tube (made of silicone rubber) with one tablet placed on the tip of tube. Care was taken to prevent any airway obstruction. The study was conducted as two-periods, two treatments crossover design for 2 weeks as follows.
Group I: Received tablet (FSD-T) in a randomized sequence (100 mg each; p.o.; n = 3) in two occasions with wash out period.
Group II: Received tablet (FSD-NT) in a randomized sequence (100 mg each; p.o.; n = 3) in two occasions with wash out period.
The prior approval for conducting the experiments in rabbits was obtained from our Institutional Animal Committee with registration number 1533/PO/a/11/CPCSEA.
Sampling procedures
Blood samples (2 mL) were collected at 0.0, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8 and 12 h of drug administration from a marginal ear vein into K3 EDTA tubes (J. K. Diagnostics, Rajkot, India) and placed on ice, protected from light. Samples were stored at −20 °C until assayed.
Preparation of plasma samples
Liquid–liquid extraction technique was employed for all the plasma samples. An aliquot of 500 μL plasma mixed with 25 μL of internal standard (paracetamol, 10 μg/mL) was spiked with 3 mL of HPLC grade acetonitrile by vortex mixing for 3 min. The mixture was centrifuged at 10 500 rpm for 10 min using a Remi-Model RM 12 centrifugal device, the supernatant was filtered through 0.45 μm filter (Millipore) and the organic supernatant was evaporated to dryness at room temperature. The residue was reconstituted with 200 μL of mobile phase, then the solution was centrifuged at 15 000 rpm for 10 min; finally, an aliquot of 20 μL of the solution was injected into the HPLC (Shimadzu, LC- 20 A series) equipped with a UV detector at a flow rate of 1 mL/min and the run time was 5 min. The mobile phase comprised 60% acetonitrile: 40% methanol at pH 6.0 was determined at UV wavelength of 255 nm.
HPLC analysis
The chromatograms were acquired using LC solutions software (Shimadzu, Japan) from a Shimadzu HPLC system (LC-20 A series) equipped with a UV detector. The retention times for candesartan and internal standard (IS, paracetamol) were approximately 3.93 and 2.85 min, respectively. Linear regression analysis was employed for standard curves obtained from drug/internal standard peak area ratios as a function of plasma concentration versus time data was analyzed and the oral pharmacokinetic data were developed.
Accelerated stability studies
In this study, the accelerated stability studies were performed for tablets prepared with and without naringin because these studies are more convenient and less time-consuming over long-term stability studies, which will take longer time to evaluate the physical and chemical stability of the product under normal conditions of temperature and humidity.
Moreover, tablet formulations containing active pharmaceutical ingredient (API) in any high-energy amorphous form (including solid dispersions) are expected to enhance solubility, dissolution rate and consequently, oral bioavailability of poor water-soluble drugs. However, the amorphous regions of the dispersed drug in the carrier are thermodynamically unstable and are therefore susceptible to recrystallization upon storage (Hecq, Citation2005).
Hence, in this study, the prepared tablets (FDT-NT and FDT-T) were subjected to accelerated stability studies where the product is stored under extreme condition of temperature and humidity of 40 ± 2 °C/75% RH ± 5% RH for 6 months, respectively.
Tablets (FDT-NT and FDT-T; 100 mg each) were wrapped in aluminum foil to prevent the formulation from exposure to light to simulate the aluminum packaging, i.e. Alu–Alu packing, of drug products and stored in airtight containers which is impermeable to solid, liquid and gases. These packed samples of tablets in airtight containers were subjected to accelerated stability studies for a period of 6 months as per ICH guidelines [ICH-Q1A (R2) Citation2003] at the extreme temperature and relative humidity conditions of 40 ± 2 °C/75% RH ± 5% RH in a humidity-controlled oven (Thermolab, India). The samples from airtight containers were withdrawn at 1, 3 and 6 months and evaluated for physical appearance, drug content, hardness, disintegration time and in vitro drug release at 30 min against initial (0 month) values.
The physical stability was analyzed to monitor for potential re-crystallization of the solid dispersions in prepared tablets with and without naringin using XRPD studies, while the chemical stability was studied with HPLC to evaluate for the formation of any degradation product/s under storage conditions for 1, 3 and 6 months, respectively. Any physical and chemical changes observed during storage for 6 months were compared against initial (0-month) diffractrograms and the peak areas, respectively.
Drug content determination
The content uniformity was assessed according to USP requirements. The test is used to ensure that every tablet contains the amount of drug substance intended with a negligible variation among tablets within a batch upon storage. Three tablets from each formulation (FDT-NT and FDT-T; 100 mg each) at initial (0), 1, 3 and 6 months were tested randomly. Each tablet was weighed individually and crushed to a powder in a glass mortar. The powder blend equivalent to 10 mg of drug (CAN) was weighed and dissolved in 10 mL of methanol, filtered using 0.45 µm filter paper and an aliquot of 20 μL of the resulting filtrate was injected into HPLC to determine the concentration of CAN from tablet samples. All assays were carried out in triplicate.
Hardness testing
The hardness or crushing strength of the tablets was measured using a Monsanto Hardness Tester (Campbell Electronics, Mumbai, India). Three tablets from each formulation (FDT-NT and FDT-T; 100 mg each) were tested randomly at initial (0), 1, 3 and 6 months, respectively, and the average reading was noted. The hardness is measured in kg/cm2.
X-ray powder diffraction studies
X-ray powder diffraction (XRPD) analyses were performed for crushed powder samples of two tablets (FDT-NT and FDT-T; 100 mg each) at initial (0), 1, 3 and 6 months, respectively, using an X-ray diffractometer (PW 1729, Philips, Netherlands). The samples were irradiated with monochromatized Cu Kα radiation (1.5406 Å) and analyzed between 5° and 50° 2θ. The voltage and current used were 40 kV and 40 mA, respectively. The range and the chart speed were 2 × 103 CPS and 10 mm/degree 2θ, respectively.
Degradation studies using HPLC
Three tablets from each formulation (FDT-NT and FDT-T; 100 mg each) were tested for degradation studies at initial (0), 3 and 6 months, respectively. Each tablet was weighed individually and crushed to a powder in a glass mortar. The powder blend equivalent to 10 mg of pure drug (CAN) was weighed and dissolved in 10 ml of methanol, filtered using 0.45 µm filter paper and an aliquot of 20 μL of the resulting solution was injected into the HPLC to observe the peaks corresponding to the pure drug (CAN) and degradation products, respectively. All assays were carried out in triplicate.
In vitro dissolution studies
Drug release studies were carried out in triplicate for two tablets (FDT-NT and FDT-T; 100 mg each) at initial (0), 3 and 6 months, respectively, as described above (refer “Dissolution study” section). The similarity factor (f2) was calculated to compare the release profiles of two tablets under similar conditions of dissolution media, agitation rate, pH and temperature employed, to understand the influence of storage conditions on drug release profiles for a period of 6 months.
The similarity between two dissolution profiles at initial (0), 3 or 6 months was considered significant when its value falls between 50 and 100.
Data analysis
Pharmacokinetic analysis
Plasma concentrations of candesartan in rabbits were plotted against time and non-compartmental analysis was used to determine the pharmacokinetic parameters using Kinetica 5.0 Software (Adept Scientific Ltd., Amor Way, Letchworth, Garden City, Herts, United Kingdom). The area under the plasma concentration versus time curve up to the last quantifiable time point, AUC0–t was obtained by the linear trapezoidal method. The AUC0–t was extrapolated to infinity (AUC0–∞) by adding the quotient Clast/Kel, where Clast represents the last measured concentration and Kel represents the apparent terminal rate constant. The half-life of the terminal elimination phase was obtained using the relationship t1/2 = 0.693/Kel. The Cmax and Tmax were obtained directly from the data. Oral clearance (Cl) was calculated as dose divided by AUC0–∞. The apparent volume of distribution was obtained from the equation Vd = D/(AUC0–∞) × Kel. Mean residence time (MRT) was determined by division of area under the first moment curve (AUMC) by AUC0–∞.
Statistical analysis
Data was expressed as mean ± standard deviation (±SD). The statistical significance was determined using One-way ANOVA followed by Dunnett’s test or by a Student’s t-test. Results were considered statistically significant from the control when p < 0.05 and very significant when p < 0.01.
Results
FTIR spectroscopy
shows the FT-IR spectra of pure CAN, naringin and their physical mixture under study. The spectrum of CAN shows prominent peaks at 3410, 2941, 2860,1753, 1716, 1614, 1548 and 1282 cm−1 corresponding to N–H stretching, C–H aromatic stretching, C–H aliphatic stretching, C = O stretching in ester, C = O stretching in acid, C = N stretching, C = C stretching and –C–O–ester.
The IR spectrum of naringin exhibited characteristic IR bands at around 3408 (broad), 3387 (broad), 2920, 1691 and 1616 cm−1 due to more than one O–H aromatic stretching, C–H aliphatic stretching, C = O and C = C bonds, respectively.
The physical mixture showed the superimposed spectra of CAN crystals and naringin. The crystalline form of CAN and naringin has sharper and significant peaks at all above-mentioned prominent regions. No shifting of peaks was observed with either pure drug or naringin in the physical mixture spectra. It might be speculated that the no intermolecular bonding was formed between the molecules of two systems indicating absence of any chemical interaction between CAN and excipient (naringin).
Evaluation of micromeritic properties
Results of flowability, bulk and tapped density, Hauser’s ratio and Carr’s index
According to the results presented in , it is possible to conclude that the blended powder has flow properties and compaction performance appropriate for pharmaceutical applications. The results show that naringin as an excipient displayed a good flow, in view of the fact that the values of flow time and cotangent of angle α were less than 10 s and 0.1, respectively ().
Physical characterization of tablets
Results of average weight, hardness and friability
Direct compression (DC) method was used due to its ease of manufacture and lower cost. We optimized () the tablet formulation for 100 mg prepared from naringin for hardness value, 3.8 kg/cm2, which was significantly (p < 0.05) greater than the hardness value, 1.5 kg/cm2 for 85 mg tablet without naringin. This signifies that naringin formulates the tablet harder than the moderate value (1.5 kg/cm2) without naringin. The significant (p < 0.05) change in the hardness formulated with naringin may be attributed to the glycosidic linkages present in the flavonoid glycoside. However, the mechanism is unclear and needs further elucidation.
Since, there is no such official specification specified in any pharmacopoeia with reference to hardness of material and hardness test for uncoated tablets. Formulator is supposed to decide and maintain the range of hardness during preparation based upon requirements in addition to type of formulation. However, minimum acceptable hardness of uncoated tablets is 40 N (4.08 kg/cm2) approximately (Banker, Citation1987).
Therefore, to equilibrate the hardness (3.8 kg/cm2) of tablets prepared from naringin, hardness of the tablets without naringin was increased with increased concentration of β-cyclodextrin at constant compression force (15 kN) for both tablets (FSD-NT and FSD-T) in our preliminary trials.
Consequently, the tablets were assessed for their weight variation, thickness, disintegration time, hardness and friability. The results in reveal that the tablets at hardness 3.8 kg/cm2, the disintegration time, friability and thickness remained invariable for both tablets (FSD-NT and FSD-T) with no significant change in their weight variation.
Dissolution study
features the dissolution release profiles from tablet formulations (FSD-NT and FSD-T) in the dissolution medium. In vitro dissolution results showed that tablets (FSD-NT and FSD-T) provided an immediate release of the drug with no significant differences in their release profiles under similar conditions of pH medium, agitation rate, temperature and dissolution media, respectively.
Figure 2. Dissolution profiles of FSD-NT and FST-T tablets showing cumulative amount of drug release in 900 mL of phosphate buffer (pH 6.8). Mean ± SD (n = 3).
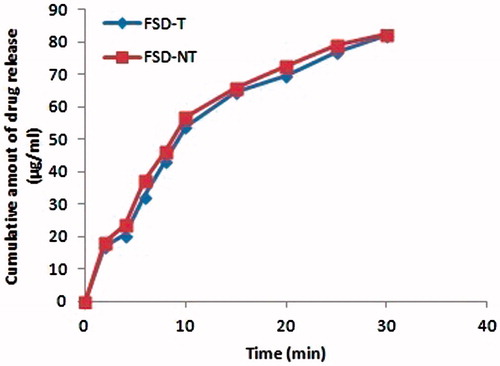
To describe the release behavior of CAN from the formulations, the data was fitted into a first-order kinetic model. The tablet formulations (FSD-NT and FSD-T) showed a good correlation to first-order kinetics equation (r2 = 0.992) suggesting that the existence of linear correlation between concentration and ratios of peak areas.
In reality, the release profiles were superimposable under analogous conditions tested. The similarity factor (f2) was used to compare the dissolution profiles that reached to a value of 76.18 in phosphate buffer (pH 6.8) at 50 rpm.
Pharmacokinetic study
In vivo studies in rabbits were conducted in order to evaluate whether an immediate plasma concentration profile of candesartan from the newly compressed tablet formulations (FSD-NT and FSD-T) as a solid dosage form could be achieved.
It has been reported that CAN must be transformed to candesartan to achieve antihypertensive effects (Vijaykumar et al., Citation2009). The plasma concentrations of candesartan were determined by HPLC method to evaluate the pharmacokinetic behavior of FSD-NT against FSD-T. The plasma concentration–time profiles of candesartan after single-dose oral administration of each tablet with and without naringin is presented in and the corresponding pharmacokinetic parameters are summarized in . The experimental results showed a significant (p < 0.001) difference occurred between the pharmacokinetic profiles of FSD-NT in comparison to that of FSD-T.
Figure 3. Plasma concentration–time profiles (mean ± SD, n = 6) of candesartan following oral administration of FSD-NT and FSD-T tablet (100 mg) formulations.
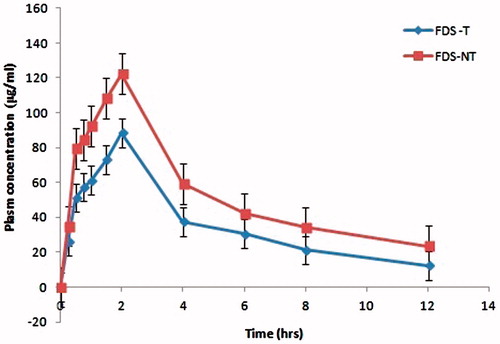
Table 4. Pharmacokinetic parameters (Mean ± SD, n = 6) of candesartan following oral administration of FSD-T and FSD-NT tablets.
Taken together, at each time point, the plasma concentration of candesartan from FSD-NT was higher than those measured for the plain FSD-T. These are evidenced by our previous reports (Gurunath et al., Citation2013, Citation2014), which showed high apparent permeability coefficient in vitro and also higher Cmax in vivo with freeze-dried CAN solid dispersions in the presence of naringin.
The peak concentrations (Cmax) of FSD-NT and FSD-T were found to be 88.44 ± 13.8 and 122.23 ± 34.5 µg/mL, respectively, which was significantly improved (p < 0.01) over 1.38 when compared to the peak concentration of FSD-T.
However, there was no change in the time to reach peak concentration (tmax) for both formulations (FSD-NT and FSD-T, 2 h), which was consistently similar to our previously demonstrated pharmacokinetic studies, indicating that candesartan could be absorbed more rapidly after its prodrug, CAN was formulated into freeze-dried solid dispersion.
In particular, areas under the concentration–time curves (AUC0–12 h) for FSD-NT (644.03 ± 78.6 µg h/mL, respectively) were enhanced by 1.5-fold over the area under the concentration–time profiles of FSD-T (428.51 ± 67.8 µg h/mL; p < 0.01) paralleled with a reduction in candesartan clearance from 3235.64 ± 231.7 to 1878.62 ± 115.7 μL/min.
In addition, a significant change (p < 0.01) in the elimination half life was observed between two tablet formulations (FSD-NT and FSD-T) from 7.15 ± 3.9 to 4.86 ± 2.5 h, respectively. This revealed that the oral bioavailability of candesartan could be dramatically increased in the presence of naringin. The significantly enhanced oral absorption of candesartan from FSD-NT was consistent after its direct delivery to the intestine, which indicated that the intestinal absorption was predominant in the improved oral absorption of CAN.
The therapeutic effect of CAN occurred at 4–6 h after the oral administration of free-CAN tablets (Israili, Citation2000; Gleiter & Mörike, Citation2002; Husain et al., Citation2011). Hence, the more rapid absorption of candesartan from FSD-NT tablet formulation could be helpful in treatment of hypertension or heart failure in clinical therapeutics.
Accelerated stability studies
The results of the accelerated stability studies showed, no significant differences in drug content uniformity, hardness, disintegration time and in vitro dissolution profiles before (0-month) and after the storage periods (1, 3 or 6 months) for tablets with and without naringin at extreme condition of temperature and relative humidity of 40 ± 2 °C/75% RH ± 5%.
The stability data of tablets (FDT-NT and FDT-T) are shown in for percentage of drug content, hardness and disintegration time to evaluate the effect of storage for 6 months. Percentage drug content of two tablets with and without naringin were found within the range of 97.1–99.7% which meets the USP guidelines as shown in . The results in reveal no significant (p > 0.05) difference in hardness values (3.8 kg/cm2) of tablets prepared from naringin (FSD-NT) as compared to initial 0-month. However, for tablets without naringin (FDT-T), there was significant difference in hardness (p < 0.01) values at temperature and relative humidity of 40 ± 2 °C/75% RH ± 5%, respectively, wherein hardness was increased with time, prolonging the disintegration time of the tablets (); but, in all cases, disintegration time was within the specified limit (within 15 min) of official monographs for uncoated tablets (Indian Pharmacopoeia, Citation2007).
Table 5. Percentage of drug content for FDT-T and FDT-NT tablets from accelerated stability studies (Mean ± SD, n = 3).
Table 6. Hardness of FSD-T and FSD-NT tablets from accelerated stability studies (Mean ± SD, n = 3).
Table 7. Disintegration time of FSD-T and FSD-NT tablets from accelerated stability studies (Mean ± SD, n = 3).
features the dissolution release profiles from tablets (FDT-NT and FDT-T) in the dissolution medium at initial (0), 3 and 6 months, respectively. In vitro dissolution results showed that tablets (FDT-NT and FDT-T) provided an immediate release of the drug with no significant differences in their release profiles upon storage for 6 months as compared to initial (0-month) release under the similar conditions of pH medium, agitation rate, temperature and dissolution media, respectively.
Figure 4. Dissolution profiles of (a) FSD-T and (b) FSD-NT tablets showing cumulative amount of drug release in 900 mL of phosphate buffer (pH 6.8) over storage for 3 and 6 months, respectively (Mean ± SD, n = 3).
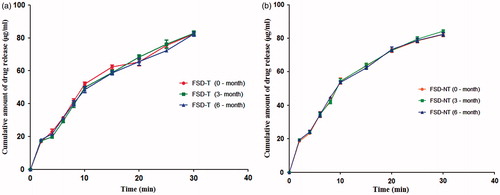
In fact, the release profiles of initial (0), 3 or 6 months were superimposable under analogous conditions tested. The similarity factor (f2) was used to compare the dissolution profiles for 3 and 6 months, respectively, that reached to a value of 65.23 (3-month; FDT-T); 71.43 (6-month; FDT-T) and 75.78 (3-month; FDT-NT); 76.86 (6-month; FDT-NT) as compared to initial (0-month) profiles in phosphate buffer (pH 6.8) at 50 rpm.
No significant effect on the dissolution profiles of tablets without naringin (FDT-T) was observed due to the changes in hardness and disintegration time upon storage for 6 months. This was clearly evidenced from the dissolution profiles of FDT-T tablets () with no significant (p > 0.05) changes as compared to initial (0-month) release profile with the similarity factors (f2) as 65.23 and 71.43 for 3 and 6 months, respectively.
In addition, the depicts the XRPD patterns of pure drug (CAN), hydrophilic polymer (PVP-K30), FDT-NT and FDT-T tablets, respectively, to evaluate the effect of storage at 40 ± 2 °C/75% RH ± 5% for different time periods. The diffraction pattern of pure drug (CAN) exhibited intensity peaks at 2θ values of 9.8°, 9.9°, 10°, 10.1°, 11.6°, 17.2°, 20.3°, 23.3°, 24.9°, 27.7° and 29.2° (). The spectrum of PVP-K30 exhibited complete absence of any diffraction peaks, which is characteristic of an amorphous compound showing the XRPD halo ().
Figure 5. (a) XRPD diffractograms of single components of CAN and PVP-K30 together with tablets prepared (a) without naringin–FSD-T at initial (0 month), FSD-T at 1st month, FSD-T at 3rd month and FSD-T at 6th month, respectively, upon storage for 6 months at 40 ± 2 °C/75% RH ± 5% and (b) with naringin–FSD-NT at initial (0 month), FSD-NT at 1st month, FSD-NT at 3rd month and FSD-NT at 6th month, respectively, upon storage for 6 months at 40 ± 2 °C/75% RH ± 5%.
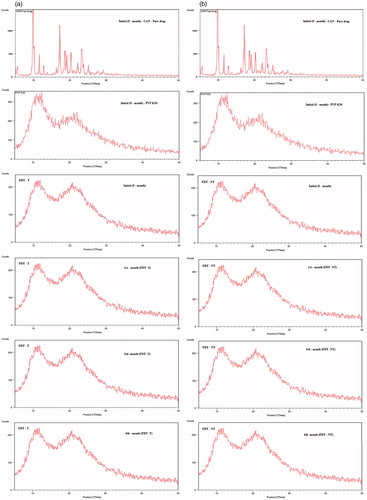
Neither FDT-NT nor FDT-T showed any signs of re-crystallization during the storage period as depicted in . The diffractrograms of tablets (FDT-NT and FDT-T) at initial (0-month), 1, 3 and 6 months showed peaks similar to PVP-K30 and absence of any major diffraction peaks corresponding to pure drug (CAN) indicating that the drug is present in an amorphous state, which is in agreement with our previously published report (Gurunath et al., Citation2014).
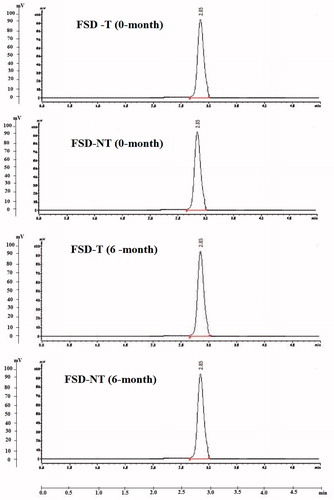
The HPLC results in showed no peaks other than those corresponding to pure drug (CAN) at retention time (3.95 ± 0.04 min) for tablets (FDT-NT and FDT-T) upon storage for 6 months. The observed peak areas of both tablets corresponding to pure drug (CAN) upon storage for 6 months were similar to those of their initial (0-month) peak areas with similar retention time values.
Figure 6. Typical HPLC chromatograms of FSD-T and FSD-NT upon storage for 6 months at 40 ± 2 °C/75% RH ± 5%.
Discussion
Candesartan cilexetil (CAN), an anti-hypertensive agent, is widely indicated for the treatment of hypertension or heart failure in clinical therapeutics. Data available on its poor solubility, limited intestinal absorption and disposition showed encouraging results with variable absorption, poor efficacy and bioavailability profiles. Numerous factors such as low aqueous solubility, poor dissolution properties and poor apparent permeability due to intrinsically low absorptive membrane permeability contribute to the low and erratic oral bioavailability of drugs. More recently, secretory membrane transporters (e.g. P-gp and BCRP) have also been implicated in controlling the oral bioavailability and variability of drug absorption. As a result, novel formulations of CAN exhibiting better oral absorption and bioavailability need to be developed.
Our FT-IR drug-excipient interaction studies showed no discoloration, liquefaction and physical interaction between the drug and the novel excipient used. No significant shift in the peak was observed which revealed that both the drug and excipient are compatible with each other. The FTIR spectra of drug, excipient and mixture of drug and excipient are shown in the , respectively.
The formulation blends illustrated good packing and flowability as indicated by Hausner’s ratio value and Carr’s index. The Carr’s compressibility index can be used as an indication to produce a uniform blend that predicts the flow properties. Compressibility index values that fall between 5 and 15% indicate excellent flow whereas those fall between 12 and 16% represent good flow. Conversely, values greater than 23% and up to 35% are acknowledged as poor flow materials.
The Hauser’s ratio (HR) obtained as a result of the ratio between the bulk density and compacted density of powders is correlated with the cohesion and adhesion forces of particles. Hausner demonstrated that powders such as coarse spheres, had ratios of approximately 1.25 with low inter-particle friction, whereas Hausner ratios greater than 1.5 showed more cohesive, less free-flowing powders such as flakes.
Therefore, it is established that values lower than 1.25 are related to a good flow, whereas values higher than 1.25 are related to a poor flow, in view of the fact that flow and cohesive properties are inversely proportional (Staniforth, Citation2007; Ofori-Kwakye et al., Citation2010).
The Carr’s index and Hausner ratio based on the cohesiveness between the particles only reflect the potential for consolidation of powders. To determine the ease and speed of flow, it is imperative to determine the flow rate by means of the time and cotangent of α angle.Since the values of flow time and cotangent of angle α were less than 10 s and 0.1, respectively, the formulation blends exhibited good flow properties.
Hardness is an element of interest to the compression process that can be used for equipment adjustment and calibration to obtain the quality of the finished product. In general, higher the compression force, greater will be the consolidation and deformation of the powder in the diet. This will results in the greater the contact points in the material being compacted with the low porosity of the tablet. There will be an inclination to increase the hardness and disintegration time.
Alternatively, low hardness will be an indirect measure of the inefficiency of the process of consolidation and compaction of the powders in tablets. Harder tablets will have inferior friability with a difficulty to be ejected from the press (Kuentz & Leuenberger, Citation2000; Armstrong, Citation2007; Staniforth, Citation2007; Niazi, Citation2009).
In our preliminary trials, the ability of β-cyclodextrin as effective diluent (Late & Banga, Citation2010) in place of naringin to formulate tablets containing freeze-dried CAN-loaded solid dispersions prepared by the DC was demonstrated in order to equilibrate the hardness of FSD-NT to that of FSD-T tablets at constant compression force and time to rule out or understand the influential role of naringin on drug release.
Besides, the increase in the oral bioavailability of hydrophobic drugs of low water solubility using water-soluble excipients, these diluents (naringin or β-cyclodextrin) can also act as fillers for such tablets.
The analogous disintegration time values for FSD-NT and FSD-T formulations may also attribute to the superior water solubility nature of naringin (i.e. 1 mg of naringin dissolved in mL of water) due to the hydroxyl groups and glycosidic linkages present in the flavonoid and hydrophilic nature of β-cyclodextrin, respectively.
Although hardness is not an attribute that can be directly correlated with drug release, a greater hardness value may be beneficial for maintaining tablet integrity and controlling drug delivery. From a comparison of the release profiles, increasing or equilibrating the hardness of the FSD-T to that of FSD-NT using β-cyclodextrin facilitated the release of CAN, most likely because the β-cyclodextrin is a hydrophilic polymer, allowing the penetration of the dissolution medium and thus showing similarity in the rate of drug release to that of tablet formulation with naringin (FSD-NT).
Both FSD-NT and FSD-T tablets offered dramatic improvements in the rate as well as the extent of drug release () and presented the highest (80%) drug release in 30 min. In the early 10-min period, there were about 54 and 57% increase of drug release from both tablets, respectively. This improved drug release attributed to the presence of amorphous form of CAN, as confirmed by our earlier reported DSC and PXRD studies (Gurunath et al., Citation2014). Other factors such as increased wettability, solubilization of the drug by the carrier at the diffusion layer, the reduction or absence of aggregation, agglomeration increased the rate of dissolution and higher micromeritic properties exerted by excipients might have been contributed to improved drug release from these tablet formulations. This can clearly be evidenced by superimposable drug release profiles with similarity factor value between 50 and 100 (76.18).
Based on all of the above findings, the possible mechanisms for an increased release rate from both tablets have been postulated and are summarized as follows: (i) reduction of crystallinity, (ii) increased water absorption by the hydrophilic carrier and excipients which leads to increased wettability, (iii) solubilization effect by the carrier and excipients, (iv) phase transition from crystalline to amorphous nature and (v) higher micromeritic properties exerted by naringin or β-cyclodextrin.
The results of the in vivo pharmacokinetic study demonstrated that rabbits exhibited the moderate inter-individual variation in the plasma concentrations of candesartan, which may be reflective of absorption, metabolism and elimination differences between individuals. As shown in , FSD-NT was absorbed significantly from the intestinal regions and reached a maximum peak plasma concentration at about 2 h after the drug treatment as compared to FSD-T indicating that the presence of naringin contributed significantly for the enhanced intestinal absorption of candesartan from its tablet formulation as a P-gp inhibitor.
All the Cmax and AUC0−t values of FSD-NT were much greater than the Cmax and AUC0−t values of FSD-T at the same dose of 100 mg, which may explain that the dissolution of CAN along with effective P-gp inhibition significantly contributed to improved intestinal absorption. This is also attributed to the presence of solid state (amorphous) of drug improved through wetting of drug particles and localized solubilization by hydrophilic carriers.
In addition, there was also concomitant decrease in plasma clearance and volume of distribution with increased the plasma concentration and AUC as shown in and , with FSD-NT tablet formulation. The small volume of distribution, low oral clearance and increased mean residence time obtained in this study indicated that the drug distributes primarily in the extracellular spaces.
The above-mentioned facts may attribute to high protein binding at physiological pH, as previously reported (Gleiter & Mörike, Citation2002; Husain et al., Citation2011). The significant change in MRT and Kel suggests that the increased time for which the drug remains in the body.
The relative absorption (RA) is defined as the AUC0–t values of FSD-NT compared to that of FSD-T, which was relatively higher for FSD-NT as depicted in . The increased rate of absorption could be attributed to the concomitant and synergistic inhibition of P-gp present in the intestine as a plausible explanation for prominent increase in oral bioavailability of CAN.
Accelerated stability studies for the tablet formulations (FDT-NT and FDT-T) did not show any significant changes in their physical appearance, drug contents, disintegration time and in vitro drug release profiles at 30 min (p > 0.01). Upon 6 months of storage at 40 ± 2 °C/75% RH ± 5% RH (), the tablets (FSD-NT and FSD-T) demonstrated similar dissolution profiles as compared to that of initial 0-month release profiles indicating that CAN was stable in the presence of PVP-K30 and other excipients like naringin.
Tablets prepared from naringin showed no significant changes in the hardness and disintegration time as observed for tablets without naringin. Although significant changes were noticed in the hardness and disintegration time of FDT-T tablets probably due to the loss of moisture, but no significant change was observed in their dissolution profiles upon storage for 6 months indicating that both formulations (FSD-NT and FSD-T) were fairly stable at storage time and conditions.
represents XRPD diffractrograms of tablet formulations (1, 3 and 6 months over storage at 40 ± 2 °C/75% RH ± 5% RH) showing no signs of re-crystallization in the samples. Hence, there was no influence of storage time as well as storage conditions on the XRPD patterns of the drug in the samples of tablets, exhibiting similar XRPD patterns that overlay the initial (0-month) patterns, thus retaining the amorphous nature of the drug.
Furthermore, the results of the chemical stability by HPLC analyses showed no peaks other than those corresponding to pure drug (CAN) at retention time (2.85 ± 0.04 min) for tablets (FDT-NT and FDT-T) upon storage for 6 months suggesting that the tablets did not undergo decomposition or degradation upon storage over 6 months’ study, indicating that the assay procedure is selective for the analysis of CAN without interference of the excipients. This could be attributed to the evolved interactions between CAN and PVP-K30 (Barmpalexis et al., Citation2013) or glycosidic linkages of naringin, which requires further elucidation.
Oral formulations of CAN having better efficacy and less toxicity could be developed using appropriate P-gp inhibitor. However, further studies are required to compare the effectiveness of naringin with other novel P-gp inhibitors to enable pharmacokinetic modulation of candesartan or its prodrug, CAN. Genetic differences mediated by P-gp in both hepatic metabolism and intestinal transport may be an important contributing factor in the wide inter-individual variability in the absorption and disposition of candesartan.
These results suggest that the administration of CAN in the form of tablet dosage form formulated with safe and naturally occurring P-gp inhibitor may improve the therapeutic index of CAN for the treatment of hypertension and other associated complications of hypertension.
Conclusion
The success of DC method in the design of oral dosage forms requires the study of the physical properties of the formulation blends. The blends formed more suitable tablets with and without naringin, as a result of improved flow and compressibility of the novel excipients. The compression time, force and amount of excipients used did not influence friability, hardness and drug release. The dissolution profiles demonstrated that the proposed formulations produced more than 80% of drug release within 30 min. No change in dissolution profiles and release kinetics were noticed between two formulations, most likely due to the similarity in nature of the excipients. Another improvement that can be associated with the developed tablets was that no other additional ingredients in the formulations were required since naringin acted as filler in the DC process. Consequently, the current results demonstrate that the intestine plays an important role in the net absorption and disposition of CAN, which showed that naringin increased significantly the oral bioavailability of candesartan, suggesting the involvement of P-glycoprotein in CAN disposition. It also revealed that naringin, a P-glycoprotein inhibitor can be used as pharmaceutical excipient for developing oral dosage forms to increase intestinal permeability and in vivo pharmacokinetic performance of CAN to enhance oral bioavailability. Also, under the accelerated stability conditions evaluated in this study, tablets prepared with and without naringin involving CAN solid dispersions prepared by DC process were physically and chemically stable over 6 months as compared to initial (0-month) physical and chemical parameters.
However, due to the complexity of the in vivo model, the role of P-gp and other putative secretory efflux transporters is not yet clear and needs to be further elucidated. Hence, further studies are required for comprehensive and multi-disciplinary evaluation of various claims to make effective use of these products.
Declaration of interest
Authors declare no conflicts of interest in the publication of this research work.
References
- Allen LV, Popovich NG, Ansel HC. (2005). Ansel’s pharmaceutical dosage forms, 8th ed. Baltimore, USA: Lippincot Williams & Wilkins
- Amin L. (2013). P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 7:27–34
- Armstrong NA. (2007). Tablet manufacture. In: Swarbrick J, ed. Encyclopedia of pharmaceutical technology, 3rd ed. New York, USA: Informa Healthcare, 3653–83
- Banker GS, Anderson NR. (1987). Tablets. In: Lachman L, Lieberman HA, Kanig JL, eds. The theory and practice of industrial pharmacy. 3rd ed. Philadelphia, PA: Lea & Febiger, 293–345
- Barmpalexis P, Koutsidis I, Karavas E, et al. (2013). Development of PVP/PEG mixtures as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique and optimization of dissolution using artificial neural networks. Eur J Pharm Biopharm 85:1219–31
- Dai WG, Dong LC, Song YQ. (2007). Nanosizing of a drug/carrageenan complex to increase solubility and dissolution rate. Int J Pharm 342:201–7
- Emery E, Oliver J, Pugsley T, et al. (2009). Flowability of moist pharmaceutical powders. Powder Technol 189:409–15
- Gleiter CH, Mörike KE. (2002). Clinical pharmacokinetics of candesartan. Clin Pharmacokinet 41:7–17
- Gohel MC, Jogani PD. (2005). Review of co-processed directly compressible excipients. J Pharm Pharm Sci 8:76–93
- Gurunath S, Baswaraj KN, Patil PA. (2013). Oral bioavailability and intestinal absorption of candesartan cilexetil: role of naringin as p-glycoprotein inhibitor. Drug Dev Ind Pharm (Early Online: 1–7). Available at http://www.informapharmascience.com/doi/abs/ 10.3109/03639045.2013.850716
- Gurunath S, Baswaraj KN, Patil PA. (2014). Enhanced solubility and intestinal absorption of candasartan cilexetil solid dispersions using everted rat intestinal sacs. Saudi Pharm J 22:246–57
- Hecq J, Deleers M, Fanara D, et al. (2005). Preparation and characterization of nanocrystals for solubility and dissolution rate enhancement of nifidipine. Int J Pharm 299:167–77
- Husain A, Azim MS, Mitra M, Bhasin PS. (2011). A review on candesartan: pharmacological and pharmaceutical profile. J Appl Pharm Sci 1:12–17
- ICH (Q2R1). (2007). Guideline on validation of analytical procedures: text and methodology. In: Proceedings of the International Conference on Harmonization, Geneva
- ICH-Q1A (R2). (2003). ICH harmonised tripartite guideline. Cover Note for Revision of Q1A(R) Stability Testing of New Drug Substances and Products
- Indian Pharmacopoeia. (2007). Tablet Disintegration Test Apparatus. Indian Pharmacopoeia Commission, 6th ed. Vol. I:187–9
- Israili ZH. (2000). Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J Hum Hypertens 14:S73–86
- Jadhav SB, Kaudewar DR, Kaminwar GS, et al. (2011). Formulation and evaluation of dispersible tablets of diltiazem hydrochloride. Int J Pharm Tech Res 3:1314–21
- Kuentz M, Leuenberger H. (2000). A new theoretical approach to tablet strength of a binary mixture consisting of a well and a poorly compactable substance. Eur J Pharm Biopharm 49:151–9
- Lassoued MA, Sfar S, Bouraoui A, Khemiss F. (2012). Absorption enhancement studies of clopidogrel hydrogen sulphate in rat everted gut sacs. J Pharm Pharmacol 64:541–52
- Late SG, Banga AK. (2010). Response surface methodology to optimize novel fast disintegrating tablets using β-cyclodextrin as diluent. AAPS PharmSciTech 11:1627–35
- Li P, Wang S, Guan X, et al. (2014). Six months chronic toxicological evaluation of naringin in Sprague–Dawley rats. Food Chem Toxicol 66:65–75
- Li P, Wang S, Guan X, et al. (2013). Acute and 13 weeks subchronic toxicological evaluation of naringin in Sprague-Dawley rats. Food Chem Toxicol 60:1–9
- McClellan KJ, Goa KL. (1998). Candesartan cilexetil: a review of its use in essential hypertension. Drugs 56:847–69
- McCormick D. (2005). Evolution in direct compression. Pharm Technol 4:52–62
- Mura P, Cirri M, Faucci MT, et al. (2002). Investigation of the effects of grinding and co-grinding on physicochemical properties of glisentide. J Pharm Biomed Anal 30:227–37
- Niazi SK. (2009). Handbook of pharmaceutical manufacturing formulations: compressed solid products, 2 ed. Vol. 1. New York, USA: Informa Health Care, 62–81
- Ofori-Kwakye K, Asantewaa Y, Kipo SL. (2010). Physicochemical and binding properties of cashew tree gum in metronidazole tablet formulations. Int J Pharm Pharm Sci 2:105–9
- Qiu Y, Chen Y, Zhang GGZ, et al. (2009). Developing solid oral dosage forms: pharmaceutical theory & practice. 1st ed. USA: Elsevier, 163–70
- Rudnic E. (2005). Oral solid dosage forms. In: Gennaro AR, ed. Remington: the science and practice of pharmacy. 21st ed. Philadelphia, PA: Lippincott Williams & Wilkins, 1615–49
- Schinkel AH, Mayer U, Wagenaar E, et al. (1997). Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA 94:4028–33
- Schüssele A, Bauer-Brandl A. (2003). Note on the measurement of flowability according to the European pharmacopoeia. Int J Pharm 257:301–4
- Sparreboom A, van Asperen J, Mayer U, et al. (1997). Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA 94:2031–5
- Staniforth J. (2007). Powder flow. In: Aulton M, ed. Aulton’s pharmaceutics: the design and manufacture of medicines. 3rd ed. Canada: Elsevier, 152–80
- Strydom SJ, Ottoa DP, Liebenberg W, et al. (2011). Preparation and characterization of directly compactible layer-by-layer nanocoated cellulose. Int J Pharm 404:57–65
- The United State Pharmacopoeia 30/NF 25. (2007). Asian Ed: the official compendia of standard. Rockville: The United States Pharmacopoeial Convection Inc., 621–720
- Vijaykumar N, Raviraj P, Venkateshwarlu V, Harisudhan T. (2009). Development and characterization of solid oral dosage form incorporating candesartan cilexetil. Pharm Dev Technol 14:290–8
- Woo JS, Lee CH, Shim CK, Hwang SJ. (2003). Enhanced oral bioavailability of paclitaxel by coadministration of the P-glycoprotein inhibitor KR30031. Pharm Res 20:24–30
- Yonemochi E, Kitahara S, Maeda S, et al. (1999). Physicochemical properties of amorphous clarithromycin obtained by grinding and spray drying. Eur J Pharm Sci 7:331–8
- Yonemochi E, Ueno Y, Ohmae T, et al. (1997). Evaluation of amorphous ursodeoxycholic acid by thermal methods. Pharm Res 14:798–803
- Zhou L, Chen X, Gu Y, Liang J. (2009). Transport characteristics of candesartan in human intestinal Caco-2 cell line. Biopharm Drug Dispos 30:259–64